

Abstract
Water molecules confined to pores with sub-nanometer diameters form single-file hydrogen-bonded chains. In such nanoscale confinement, water has unusual physical properties that are exploited in biology and hold promise for a wide range of biomimetic and nanotechnological applications. The latter can be realized by carbon and boron-nitride nanotubes which confine water in a relatively non-specific way and lend themselves to the study of intrinsic properties of single-file water. As a consequence of strong water-water hydrogen bonds, many characteristics of single-file water are conserved in biological and synthetic pores despite differences in their atomistic structure. Charge transport and orientational order in water chains depend sensitively on and are mainly determined by electrostatic effects. Thus, mimicking functions of biological pores with apolar pores and corresponding external fields gives insight into the structure-function relation of biological pores and allows the development of technical applications beyond the molecular devices found in living systems. In this Perspective, we revisit results for single-file water in apolar pores, and examine the similarities and the differences between these simple systems and water in more complex pores.
1 Introduction
Our everyday life, and life itself, relies on the transport of fluids through pipes, capillaries, and channels, controlled by pumps, valves, and gates. 1,2 At the molecular scale, water and ions are selectively and efficiently transported in and out of cells through the nanoscopic pores of transmembrane proteins. 3 The extraordinary transport properties of biological pores are due both to their specific and complex structures, 4–12 and to the unusual properties of fluids confined to such molecularly narrow pores. 13–17 Indeed, filling and flow behavior reminiscent of biological pores has also been observed in simpler, essentially structureless synthetic pores. Carbon nanotubes with nanometer-scale diameters, for instance, support water flow rates that exceed the expectations of macroscopic hydrodynamics by orders of magnitude. 13,18–20 Due to their unusual transport properties, nanopore membranes have great potential for technological applications ranging from water desalination devices to hydrogen fuel cells. 19,21–23 The length dependent dielectric constant of single-file water might be exploited in capacitors and in sensing devices. 24–26 In this Perspective, we provide an overview of the fundamental properties of water in nanopores, focusing on cylindrical channels with apolar walls and explore their significance for water in biological transmembrane pores as well as for biomimetic applications. Our discussion is based both on insights gleaned from computer simulations and on results from experimental investigations of these remarkable systems.
Carbon nanotubes, or their boron nitride analogues, provide exemplary systems to encapsulate water and realize nearly ideal 1d confinement. 18,27,28 As shown using molecular dynamics simulations, water penetrates the cavity of carbon nanotubes with pore diameters down to the nanometer regime, even though their graphitic walls are considered to be hydrophobic. 18 The filling of narrow carbon nanotubes with water has been confirmed with neutron diffraction experiments, 29 transmission electron microscopy, 30 nuclear magnetic resonance spectroscopy, 31 and density gradient centrifugation experiments. 32–34 Recently, Raman spectroscopy experiments based on the analysis of the radial breathing mode have been used to demonstrate that carbon nanotubes with diameters as small as 0.55 nm, just wide enough for single-file water, imbibe with water. 35 Due to their smooth walls, carbon nanotubes allow for high, nearly friction-less fluxes as observed in gas and water flow measurements. 18–20 In addition, these tubes can be functionalized with charges to mimic the special selectivity properties of transmembrane proteins, 36,37 making these systems particularly interesting for technological applications.
Inside the orifice of carbon nanotubes with sub-nanometer diameters, water forms continuous single-file chains, or wires, of hydrogen bonded molecules. 18 In contrast to bulk water, where each molecule is involved in four hydrogen bonds on the average, in single-file chains each water molecule participates in two hydrogen bonds, one donated to and one accepted from a neighboring water molecule. This particular hydrogen bonding topology is the origin of the unusual properties of single-file nanopore water, as discussed in depth in this Perspective. Along a short chain, hydrogen bonds point into the same direction leading to an orientationally and dipolarly ordered arrangement of molecules as illustrated in Fig. 1. 18 Flipping events, during which the dipole moment of the entire chain changes sign, are rare but occur occasionally via the diffusion of orientational defects from one end of the chain to the other. Such defects display atypical hydrogen bonding, analogous to the hydrogen bonding patterns of Bjerrum defects in hexagonal ice, 38 and form where ordered domains of water molecules of opposite direction meet. 10,39 Depending on the orientation of these domains, the defect molecule either donates two hydrogen bonds and accepts none, or accepts two hydrogen bonds without accepting any. In analogy to the hydrogen bonding defects occurring in hexagonal ice, these defects are named L defects and D defects, respectively. 39 The dynamics of hydrogen bonding defects is intimately connected to the response of water wires to time dependent electric fields and determines their dielectric relaxation behavior. 24 Thus, dielectric spectroscopy experiments can yield microscopic information on the particular hydrogen bonding structure and dynamics of 1d water. 24,25 Alternatively, vibrational spectroscopy has been suggested as a means to probe the molecular arrangements of nanopore water experimentally. 40 Due to the non-specific interactions of the water molecules with the carbon nanotube walls and the simple structure of the water wires, such systems provide an interesting way to investigate hydrogen bonding between water molecules in a simplified environment.
Fig. 1.

Snapshot of a molecular dynamics simulations of a membrane of carbon nanotubes immersed in water. 48 One of the tubes is sliced open to reveal the chain of hydrogen-bonded water molecules.
One-dimensional (1d) water chains are also of particular interest as wires for fast proton transport 41 via the so-called Grotthuss mechanism. 10,11,39,42–44 As indicated by computer simulations, the proton mobility in a 1d water wire inside a carbon nanotube exceeds that in bulk water by a factor of 40, 39 in accord with simulation data for hydrophobic model channels 45 and observations of gramicidin A, where the proton transport is considerably faster than the transport of other cations. 10,11,46 Proton translocation through membranes, however, is impeded by the desolvation penalty associated with removing the proton from bulk water and inserting it into the pore, where it experiences an environment of reduced polarization. 47,48 Also, rapid proton transport requires completely ordered, defect-free water wires 39. While for short water chains the strong dipolar interactions between the water molecules lead to long-lived perfect order, hydrogen bonding defects are bound to occur for longer wires. 49 In fact, as known from the statistical mechanics of 1d systems, dipole-dipole interactions are too short-ranged to support long ranged dipolar order, such that no ordered phase exists in the thermodynamic limit at finite temperature. 50,51 Nevertheless, the strong thermodynamic preference for filled carbon nanotube and the large energetic cost of hydrogen bonding defects counteracting the entropic gain of defect insertion leads to dipole-ordered chains of macroscopic lengths. 49
The remainder of this Perspective is structured as follows. We briefly describe the structural differences of synthetic nanotubes and biological pores in Sec. 2, and introduce the basic properties of single-file water in Sec. 3. Based on a coarse-grained model introduced in Sec. 4, we discuss the length dependence of these properties in Sec. 5, and the influence of external fields in Sec. 6. We then indicate how apolar pores can serve as model systems for more complex pore structures in Sec. 7, and conclude with a brief discussion in Sec. 8.
2 Nanopores
Water-carrying pores with diameters in the nanometer range occur naturally in biological systems and in minerals, and can be synthesized from a diverse range of materials. The most studied and structurally simplest containers for single-file water are carbon nanotubes (see Fig. 1). 52 Their electrical properties, ranging from semi-conducting to metallic, are determined by the chirality, i.e., the direction in which the graphene sheet has been “rolled up”. 53 The interactions of water molecules with the carbon nanotubes are relatively unspecific, primarily confining the water molecule to their interior. As shown by molecular dynamics simulations, water molecules experience the carbon nanotube walls as smooth, such that motion in the axial direction is largely unimpeded by water-wall interactions. Electronic structure calculations indicate that, at least for short carbon nanotubes, 27 the polarizability of the carbon nanotube has only a minor effect on the distribution of dipole moments of water molecules encapsulated by the tube, justifying the use of simple, non-polarizable models for the water carbon-interactions or computationally even less demanding effective potentials for the water-wall interactions. Remarkably, the effective dipole moment of water in 1d chains obtained from quantum chemical calculations was found to be close to that of rigid point-charge model developed originally for simulations of bulk water. 27,49
An alternative way to enclose water in a 1d cavity is provided by boron nitride (BN) nanotubes. 54,55 The structure of BN nanotubes is similar to that of carbon nanotubes, with the carbon atoms substituted by boron and nitride atoms such that boron and nitride atoms alternate. 56,57 Although isoelectronic to carbon nanotubes, the wider band gap of boron-nitride tubes renders them semi-conducting for all tube chiralities and sizes, 58 with electronic properties controlled primarily by covalent functionalization. 59,60 In contrast to water in carbon nanotubes, water molecules in boron nitride nanotubes form hydrogen bonds to the nitrogen atoms that slow down the diffusion. Nevertheless the properties of water in boron-nitride nanotube are qualitatively similar to those of water in carbon nanotubes such that these tubes have been suggested for similar applications. 54,55,61–64 In particular, dipole-ordered single file chains form in BN nanotubes as readily as in carbon nanotubes.
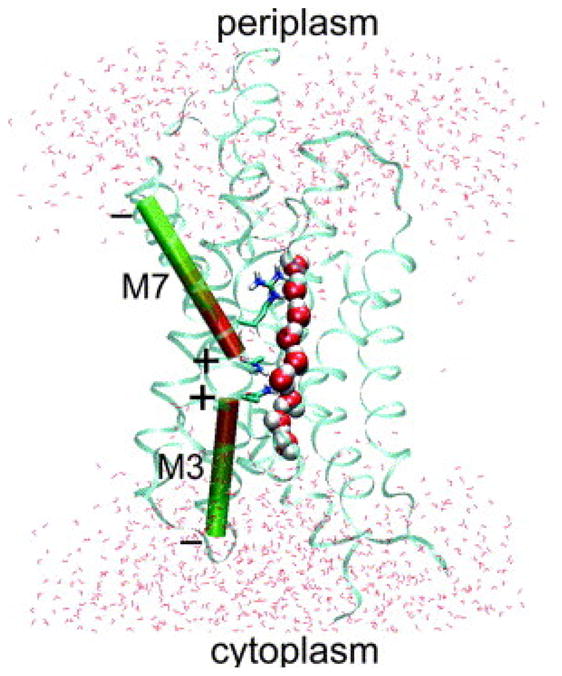
In contrast, biological nanopores spanned by 1d water wires form in many different ways. Their specific structures and geometries determine the selectivity and flux rates. In aquaporins, for example, the transmembrane helices forming the pore wall leave an hourglass-shaped water channel between them (see Fig. 2). 66 In gramicidin A the pore is located within two aligned β-helical polypeptide chains. Aquaporins transport water molecules through cell membranes but prevent the transport of ions including protons. Gramicidin A, in contrast, is a good conductor of protons and other monovalent cations. Both pores are cylindrical in the narrow single-file region, with the more polar gramicidin pore lined by backbone carbonyl groups that interact favorably with monovalent cations. The transport through both channels is highly selective and efficient. 4,67 We will discuss these transport properties in more detail in Sec. 7.
Fig. 2.
Snapshot of a molecular dynamics simulation of water in an aquaporin pore (monomeric GlpF channel). (Reprinted from Ref. 65, Copyright (2004), with permission from Elsevier.)
In addition to carbon and boron nitride nanotubes, several other containers for quasi-1d water can be manufactured, including solid state nanopores 68–71 such as silicon nitride sieves 72 and silanized channels in silicon wafers 73,74, silicon nanotubes 75, synthetic polypeptide channels 76, zeolites 77, and metal-organic nanotubes 78.
3 Water in apolar pores
Carbon nanotubes provide ideal systems to study single-file water and the translocation of water, ions and hydrogen bonding defects through nanopores. In this section, we will first discuss the filling of short pores immersed in water and then review the transport of water and ions through them. We will pay special attention to the transport of protons and defects along the water wire, because these modes of charge transport involve dipole reorientation effects specific to 1d nanopore water.
3.1 Filling of apolar pores
Molecular dynamics simulations showed that short (6,6) armchair carbon nanotubes, immersed in bulk water at ambient conditions, spontaneously fill with a single chain of water molecules. 18 In such chains, each water molecule donates a hydrogen bond to a neighboring molecule on one side and accepts one from a molecule on the other (Fig. 1). Water molecules in this single-file arrangement thus loose approximately one to two hydrogen bonds compared to bulk water. The associated loss in energy is compensated by (1) strengthening of the remaining hydrogen bonds relative to those in bulk, (2) favorable vander Waals (dispersion) interactions with the tube wall, and (3) residual rotational entropy of the dangling OH bond. 79–81
The hydrogen bonds in single-file water tend to be more directional than in bulk water because of the more ordered environment, a resulting lack of competition between water molecules for hydrogen bonding partners, and the collective orientation effect arising from long-range dipolar interactions. As a result of the strong hydrogen bonds, water molecules do not easily separate from each other to form a gap in the chain. Note, however, that also in single file water, the average life time of a hydrogen bond is in the picosecond range and exceeds that in bulk water only by a factor of 3 to 5. 18,40 The reason is that the short time dynamics of hydrogen bonds is dominated by a rotational exchange process, in which the hydrogen atom involved in a hydrogen bond and the dangling hydrogen atom of a particular water molecule change place. The barrier for this transition is low, leading to frequent flips between the two possible hydrogen bonding configurations that differ only by the identity of the hydrogen atom participating in the hydrogen bond donated to the neighboring water molecule.
The ordered water chains are further stabilized by long-range dipolar interactions between the water molecules, by van der Waals interactions of the water molecules with the pore, and by entropic contributions from dangling OH bonds. Dipolar interactions contribute about one quarter of the hydrogen bond energy. Vander-Waals interactions are important because of the high local density of carbon atoms. Despite the hydrophobic character of flat graphene sheets, wrapping them up into a nanotube increases the number of van der Waals interactions experienced by each water molecule. 82,83 Consequently, water molecules bind more strongly to the cylindrical tube walls than to the flat surface. In addition, an increase in rotational entropy due to the orientational freedom of the dangling hydrogen atoms not involved in hydrogen bonds contributes to the stabilization of the water molecules in the pore. As a result, the entropy of a water molecule within an ordered chain in the interior of the nanotube is comparable to the entropy of a molecule in bulk water. 79
The filling of short carbon nanotubes observed in computer simulations depends sensitively on the tube-water interactions and the chemical potential of the water bath applied in the simulations, 84 indicating that such pores in contact with bulk water at ambient conditions are close to the transition between the empty and filled states, reminiscent of a vapor-liquid equilibrium. 81 Consequently, a small modification of the tube-water interactions or the chemical potential can lead to a degeneracy of the free energy of the filled and the empty tube, 18,79,81,85–87 opening the possibility to control the filling transition of a carbon nanotube, for instance by chemical modification. Bistable filling behavior is captured by a 1d spin model, which was used to draw a qualitative map of bi-stability as a function of the energetic coupling between adjacent spins, which represent water molecules. 85 Note that this bistable behavior observed for short tubes depends on the boundary conditions of the tube, because the filled state of a tube immersed in water is more stable than that of an isolated tube. 84 Also, this behavior depends strongly on the pore length, as will be discussed later
3.2 Water and ion transport
Molecular simulations show that during tube filling, a single chain of water molecules pushes into the channel from one side and moves through the pore until it reaches the other opening. 84 An analysis using an Ising-type model suggested that the filling of the tube involves an energetic barrier in its initial stages, but once the first molecule has penetrated the pore, the filling proceeds rapidly. 85 In simulations, the rate of entry of the first water molecule was indeed found to be ≈5 times slower than the rates of subsequent entries. 84 Once the tube is filled, the water chain collectively moves back and forth as a result of thermal fluctuations. During this random walk, water molecules of the chain are released from one of its ends, while new ones are attached at the other end. Over time, this random motion leads to water transport 88 that becomes directional only under a free energy gradient, e.g., due to an osmotic imbalance. 21 Due to the smooth inner walls of the tube, the flow rate of water through the tube is high. In a filled tube that can hold at most five water molecules about 17 water molecules per nanosecond enter the tube on one side and exit it on the other side. 18 This collective diffusion of water is well described by a continuous time random walk model 88 or a collective diffusion model 89 for wider tubes. The random water transport through short nanotubes and gramicidin-like channels is nearly independent of the pore length. 90,91 In contrast to tube filling, where the water molecules entering the tube face an energetic barrier, model calculations suggest that the emptying of the tube has to overcome an entropic barrier related to the formation of an empty bubble at the pore entrance, a rare event in liquid water. 85
Narrow carbon nanotubes effectively suppress the transport of ions, while permitting high water fluxes through their smooth inner tube. 21,37,92,93 The suppression of ion flow can be exploited for desalination and osmotic water transport. 21,23,37 Ions are excluded because of the high energetic cost of partial desolvation in narrow pores, and by the electrostatic repulsion of tubes with charged rims. 37 Through pores with diameters exceeding ≈1.5 nm, typical ions can pass with an intact first hydration shell, and without significant barrier. 92,93 In contrast to the physical transport of water and ions, protons are transported via structural diffusion of ionic and hydrogen bonding defects, similar to the charge transport in water ice and quite distinct to that in bulk water. 38,43,94
3.3 Proton and hydroxide transport
In bulk water, an excess proton has a mobility that exceeds that of other small, monovalent ions by about an order of magnitude, an effect that can at least be partly explained with the so-called Grotthuss mechanism. 42–44 In this simple model, the excess proton exists in its hydrated form as hydronium ion that donates three hydrogen bonds to adjacent water molecules. Proton transfer occurs by a hop of one of the hydronium protons to a water molecule along a common hydrogen bond. The water molecule onto which the proton has hopped then forms the new hydronium ion, which will exist in this form until the next proton hop occurs. Consecutive proton hops lead to a rapid diffusion of the hydronium ion defect, which is thus formed by oxygen and hydrogen atoms whose identity constantly changes. While the Grotthuss-process captures the essential physics of proton transfer in bulk water, the true situation is more complicated as the hydronium ion is only the limiting form of a continuum of possible configurations of the hydrated proton and the fundamental proton hopping step involves the rearrangement of the hydrogen bond network around the charged complex, as shown in extensive molecular dynamics simulations based on a multi-state empirical valence bond model. 95–97 Similarly, auto-ionization of water, i.e., the formation of a hydroxide and hydronium ion and their subsequent separation, involves the collective rearrangement of water molecules to destabilize the dissociating water molecule, provide a proton wire, and then break the wire to prevent rapid recombination. 98 In single-file water, the structural diffusion of a proton along the 1d water wire is unencumbered by solvent reorganization and, therefore, follows more closely the picture envisioned by the Grotthuss mechanism. A similar picture applies to the transport of a hydroxide ion defect, which can be viewed as a proton hole moving along the water wire. Transport again occurs through a sequence of proton hops. 99
The diffusion of a proton, or a proton hole, along a water chain requires that the chain is dipole-ordered in the right direction, see Fig. 4. Transport of an excess proton, for instance, can occur only along a water chain, in which the hydrogen bonds, and consequently also the dipole moment of the water molecules, point away from the diffusing proton. In turn, hydroxide ions can diffuse along water chains, where the dipole moments point towards the hydroxide ion. These two situations are illustrated in Fig. 4B and C, respectively. Note that in Fig. 4B the hydrated proton is shown as hydronium ion, or Eigen cation, in which the excess proton is strongly attached to a particular water molecule. However, this is just one possible configuration of the protonic defect, which can also exist in a Zundelcation configuration where the excess proton is shared between two molecules, and in all intermediate configurations. 100 Note that once a defect is located within the chain it can diffuse in both directions.
Fig. 4.

A Transport mechanisms of a protonic charge. Models with different levels of coarse graining are shown from the left to the right. Molecular configurations are shown in the left column, simplified dipole configurations in the middle column, and corresponding effective charges in the right column. Ordered chains are depicted in rows A and D, protonic defects in rows B and C, and hydrogen bonding defects in rows E and F. Water molecules are colored according to their dipole orientation with respect to the tube axis (red-right, yellow-left, orange-defect). To transport a positive charge from the left to the right the dipole moments of the water molecules in the chain have to point to the right (A). This chain can either transport a proton (depicted as a hydronium ion) from the left to the right (B) or a proton hole (hydroxide ion) from the right to the left (C) via structural diffusion, with the same net effect. In both cases, the translocation of the charge leads to the re-orientation of the chain (D). To complete the transport process and return to the original state of the chain, the chain has to be re-oriented in the original direction via the diffusion of either a D defect from the left to the right (E) or an L defect from the right to the left (F).
The proton and the proton hole (hydroxide) move differently with respect to the direction of proton migration. In both cases the positive charge migrates in the direction of the proton hops. But while for the excess proton the protonic defect also moves in this direction, the hydroxide ion moves in the opposite direction. In both cases, the diffusion constant of the charge defects is considerably higher than that observed in bulk water. For the excess proton in a single file water chain, molecular dynamics simulations yielded a diffusion constant of approximately 0.17 nm2/ps, which is 40 times faster than proton diffusion in bulk water. 39 Hydroxide ions are expected to diffuse at a similar rate. 101 These fast motions and the similar diffusion constants of protons and proton holes along water wires result from the absence of solvent effects and the nearly barrier-less hop of the proton from one water molecule to the next along a hydrogen bond.
Proton diffusion is at least a factor 10 larger then the diffusion of water molecules through short pores 88. Consequently, the diffusion of the water chain has only a minor effect on the speed of proton diffusion through the pore.
While protons diffuse very rapidly along 1d water chains, the translocation rates of protons through water-filled carbon nanotube membranes are nevertheless very small. 48 The reason lies in the large desolvation penalty required to removing an excess proton from the bulk and introducing it into the pore interior where it is poorly solvated compared to the bulk. For example, a proton entering a membrane of short carbon nanotubes faces a barrier of about 10 kcal/mol, as shown by free energy calculations based on an empirical valence bond model to describe the water-proton interactions. 48 In these simulations it was demonstrated that protons are preferentially located at the tube entrance or in the spaces in-between nanotubes, where they can exist in the Eigen-like structure of a hydronium ion that donates three bonds to adjacent water molecules without accepting one. In the bulk such structures are less stable than close to the pore entrance because of the strain they exert on the hydrogen bond network of bulk water. These findings are consistent with the preference of the proton to be located at hydrophobic interfaces. 102,103 As the desolvation barrier creates an energetic bottleneck for proton entry into the pore, proton currents are small. At a pH of 7, about one proton per hour and pore crosses the membrane, but this rate is expected to speed up greatly at lower pH, in pores embedded in a dielectric medium, and under the influence of electric fields. The importance of desolvation is consistent with observations for simple model pores and for aquaporins. 47,104,105
3.4 Transport of orientational defects
As the proton or the hydroxide ion move along the water chain, they leave flipped water molecules behind with orientations opposite to those before ion passage. 106 Hence, the translocation of a proton or hydroxide through a membrane pore reorients the entire water the chain in the pore (see Fig. 4). As we will discuss later in detail, this reorientation of the water chain is a polarization effect that accounts for ≈40% of the effective charge transported, with the proton defect itself accounting for the remaining 60%. 39 To complete the transfer of the entire charge through the membrane and return to the initial water configuration before the charge transfer has started, a hydrogen bonding defect has to diffuse along the chain to re-orient the water molecules to their original direction, 38,46,107,108 as shown in Fig. 4E and F. Also, the hydrogen bonding defects carry effective charges arising from dipole flips accompanying defect migrations.
The origin of these effective charges, analogous to the effective charges of Bjerrum defects in hexagonal ice, 38,39 can be understood in detail in terms of the simple dipole model described in a later section. In the following, we will discuss the structure of hydrogen bonding defects and their transport along the water wires.
As depicted in Fig. 4E and F, hydrogen bonding (or orientational) defects are formed where two chains of ordered water molecules of opposite direction meet. At the defect site, an intermediate water molecule with anomalous orientation keeps the total number of hydrogen bonds constant with respect to the perfectly ordered chain. Depending on the orientation of the two water wires meeting at the defect, this defect molecule typically either accepts two bonds without donating any (see Fig. 4E) or donates two bonds without accepting any (see Fig. 4F). In allusion to the hydrogen bonding defects in ice, the former is called D defect and the latter L defect (Fig. 4F). 39,109
The basic step of defect motion involves the breaking and reformation of hydrogen bonds between the defect molecule and a next neighbor molecule. The two molecules rotate such that the defect molecule and the next neighbor molecule change roles as the old hydrogen bond acceptor becomes the new donor and vice versa. Experiments on water clusters suggest different mechanisms for this exchange. 110 The reorientation of the hydrogen bond network along the water wire renders the hydrogen bond defect motion slower than proton diffusion, which does not involve cleavage and formation of hydrogen bonds. In molecular dynamics simulations diffusion constants of DD ≈ 0.05 nm2/ps and DL ≈ 0.03 nm2/ps have been determined for D and L defects, respectively. 39
As mentioned earlier, the structural diffusion of orientational defects is necessary to complete the translocation of a proton (or proton hole) through a membrane and prepare the pore for the next translocation event. To re-orient the chain depicted in Fig. 4D, for instance, either a D defect must cross the wire from the left to the right or an L defect from the right to the left. In both cases, the chain is re-oriented after the passage of the defect, completing the charge translocation. Due to the effective charge of the defect (with a magnitude of 0.4e) and the reduced effective charge of the protonic defect (with magnitude 0.6e), a full elementary charge e has crossed the membrane only after both the protonic defect and the orientational defect have transversed the pore. Note that the sequence of migration of an ionic defect followed by the migration of a hydrogen bonding defect is arbitrary (but both the “hop” and the “turn” step are necessary for complete charge transport through the predominately ordered chain 10,111). We could have also started with the re-orientation of the chain depicted in Fig. 4D via migration of an orientational defect and subsequent migration of an ionic defect. Although orientational defects diffuse at a rate slower by a factor of 3–5 than the excess proton, 39 the reorientation of the chain due to the diffusion of a hydrogen bonding defect is not the rate limiting step for proton transport, because the latter face a larger desolvation barrier when entering the tube. For a short nanotube in water, chain reorientation was found to occur with a rate of ≈2/ns. 112
Finally, we point out an important conceptual relation between ionic defects and orientational defects. D defects and hydroxide defects are always located between ordered segments of water molecules with dipole moments pointing towards each other. Similarly, L defects and hydronium defects (or Zundel structures) are located between chains pointing away from each other. Thus, ionic defects can be viewed as deprotonated D defects and protonated L defects, respectively. In a single disordered chain, L and D defects have to alternate independently of the degree of ionization of these defects.
4 Coarse-grained view of single-file water
The molecular configurations depicted in Fig. 4 suggest a coarse-grained view of single-file water. Several coarse-grained models have been developed to study the thermodynamics and kinetics of tube filling and water transport, 49,49,85,113,114 charge transport and order properties, 36,39,49,81,115,116 and dielectric response of single-file water 24,25,117. In the following we discuss one of these models, the dipole lattice model, 49,81 to obtain physical insight into the chain energetics and effective interactions.
As observed in molecular dynamics simulations, water molecules in single-file arrangement are more or less evenly spaced, forming a 1d lattice with a spacing given by the average nearest neighbor distance. 10,39 Due to the strong hydrogen bonds acting between neighbors in the water chain, the dipole moments of the water molecules are essentially aligned with the tube axis with only small fluctuations about the parallel or antiparallel direction. The molecules associated with D and L defects experience reduced orientational freedom. Their dipole moment is oriented orthogonal to the pore axis due to the symmetric arrangements of hydrogen bonds to the neighboring molecules.
The effects of the configurational constraints imposed on the system by the hydrogen bonds, and the free energetics of the water chain, are captured by a 1d lattice model. 49,81 This reduced description provides a transparent picture of polarization effects induced by the flipping water molecules and the resulting effective charges carried by defects. Each water molecule is represented by a point dipole with orientation either parallel or antiparallel to the pore axis. The point dipoles are located on the sites of a 1d lattice with regular spacing, which can also be empty or occupied by ionic or hydrogen bonding defects. Ionic defects, i.e., protons and proton holes, are represented by Coulombic charges of magnitude ±e, and hydrogen bonding defects correspond to dipoles orthogonal to the pore axis. Since defects are located between dipole ordered segments of opposite direction, their positions determine the total dipole moment of the chain. To take into account the constraints imposed by tight hydrogen bonds on the configuration of the proton and the proton hole, the protonic defect is located between two ordered dipole segments that point away from the proton, and the proton hole between dipole segments that point toward the hole. These directions of the water chain segments near the proton and the proton hole coincide with the directions of possible proton hops. The center column of Fig. 4 shows exemplary dipole configurations corresponding to the molecular arrangements displayed in the left column.
The energy of a water chain is determined by the strong but short-ranged hydrogen-bond interactions of nearest neighbor molecules and by longer-ranged dipole-dipole interactions. The total Hamiltonian of the dipole model for a lattice of N sites and spacing a populated by n dipoles is given by
| (1) |
where the indices i and j number the lattice sites, ri j = a|i − j| is the distance between sites i and j, μ is the magnitude of the dipole moment and ε0 is the permittivity of the vacuum. Note that here, for simplicity, we have omitted ionic defects, which can be added to the Hamiltonian in a straightforward way. 81 The relative orientation of a pair of dipoles at sites i and j is encoded in the variable si j: if the dipoles are parallel si j = 1 and if they are antiparallel si j = −1. If one of the two sites is empty or occupied by a defect, si j = 0. In the equation above, the double sum runs over all pairs of dipoles that are at least two lattice spacings apart. Water molecules interact via their average total dipole moment μ along the tube axis which is about 80 % of the dipole moment of a free water molecule. 39,49 This estimate was obtained using an empirical, non-polarizable water model, with similar values obtained from density functional theory. 27 The dipole interaction of defect molecules can be neglected as their dipole moments are typically perpendicular to the tube axis. Short ranged nearest neighbor interactions are included in (n − nc)Ec, where Ec is the contact energy between two neighboring molecules connected by a hydrogen bond and nc is the number of continuous (gapless) chains in the system. The subtraction of nc from n in the contact energy term takes into account that a continuous dipole chain of m dipoles has only m − 1 hydrogen bonds. The total interaction energy of two hydrogen-bonded molecules Ec within an ordered segment ≈6kcal/mol, as estimated using empirical force fields. 39 The hydrogen bonds of defect molecules are slightly less stable, but part of the energy difference is compensated by an increase in entropy.
The entropy of single-file water can be separated in translational and rotational contributions, both of which are included in the dipole model. The translational contributions to the entropy are due to the fragmentation of chains, i.e., chains of hydrogen-bonded molecules with empty gaps between them, and the inner translational degrees of a chain, i.e., the configuration space explored by water molecules within a chain for fixed chain position. The latter is approximately constant for fixed particle number and acts like an effective chemical potential for varying particle numbers. The rotational entropy of a chain of water molecules is related to the number of dangling OH-bonds. 81 The latter can be considered to be a conserved quantity for a single chain as orientational defects always connect chains of opposite direction. For example, an L defect (Fig. 4F) donates two bonds without accepting any and thus the number of dangling OH-bonds is reduced by one. However, it connects two chains pointing away from each other. Consequently, the chain has two end molecules with two dangling OH-bonds instead of one molecule, such that the number of dangling OH-bonds is increased by one compensating for the lost bond at the defect. A similar argument can be made for the D defect. As a consequence, the number of hydrogen bonds, and thus the number of dangling OH-bonds, is conserved for an arbitrary number of defects in the chain. Only when a chain is split into two the number of dangling OH-bonds increases by one which results in higher entropy compared to the single chain which is included in ncSc of eqn (1). This entropic contribution to the free energy has been estimated to be Sc ≈ −2 kcal/mol. 81
The dipole model described above very naturally connects the reorientation process of the water molecules to the effective charges carried by the hydrogen bonding defects as well as the proton and the proton hole. In the mathematically equivalent charge picture of this model (Fig. 4 right), all long-range interactions in the sense of non-nearest neighbor interactions are expressed as Coulomb-like interactions between effective charges carried by orientational defects and by the endpoints of the water chains. Defects carry effective charges of magnitude 2μ/a ≈ 0.4e and chain ends carry charges with half this magnitude. While the charge of the D defect is positive, that of the L defect is negative. The sign of the charges at the chain endpoints depends on their orientation. Excess protons and proton holes can be viewed as L and D defects bearing an extra charge of +e and −e, respectively. Accordingly, the charge effectively transported along the water chain is e − 2μ/a ≈ 0.6e and that of the proton hole is −e + 2μ/a ≈ −0.6e.
The charge picture presented in Fig. 4 embodies the effective interactions of defects and of defects with the chain ends. 39,49,81 A single defect in a chain is always attracted by the charges at the chain ends, and likewise L and D defects attract each other. Similarly, a single ionic defect is always repelled by the chain ends, leading to an approximately quadratic free energy well for the total dipole moment of the chain as has been observed in molecular simulations. 10,39 Since L and D defects alternate within a chain, ionic defects are always repelled by hydrogen bonding defects next to them.
In the charge picture, the total energy of single-file water is given by
| (2) |
where the sum extends over all effective charges qk of the system. Note that the Coulomb 1/r-interaction in the above equation is not exact, but accurately approximates the true interaction of effective charges. The exact interaction between effective charges can be expressed in terms of polygamma functions. 81 The Hamiltonian of eqn (2) is linear in the number of defects nd, the number of chains nc, and the number of particles n. The defect excitation energy Ed is the free energy needed to introduce a defect at a specific position in an infinitely long chain and has been estimated as Ed ≈ 8kcal/mol. To split a long chain into two parts and separate the parts infinitely far from each other, a free energy of cc ≈ 6kcal/mol is needed. If we add a water molecule to an infinitely long chain we gain free energy of c ≈ −6kcal/mol. The dipole lattice provides explicit expressions for all of these quantities which are completely determined by the parameters μ, a, Ec, and Sc. 81 These parameters can be estimated in molecular simulations. To extend the dipole lattice model to ionic defects, the effective charges and the excitation energies of ionic defects have to be included.
The charge picture outlined above facilitates the understanding of the charge transport along a water wire from one side of a membrane to the other. In this picture, for instance, an ordered chain pointing to the right is represented by a positive charge of q = μ/a at the right end, and a negative charge −q at the left. After the proton has passed through the wire from left to right, the charge is e − q on the right and q − e on the left. Therefore, the proton motion carried an effective charge of e −2q across the wire. To complete the charge transport and bring the water wire back to its original orientation, a charge of magnitude 2q needs to be moved from the left to right. This additional charge transport is accomplished via the diffusion of a hydrogen bonding (orientational) defect. Only after this second step has an entire elementary charged been translocated across the membrane.
The dipole lattice model was developed for water in nanotubes in vacuum with no direct contact to a water bath. For short tubes, different boundary conditions can have large effects, and the dipole model has to be adapted accordingly. For example, in a free chain we observe a repulsive interaction of a protonated defect with the chain ends. However, contact of the tube with a water bath leads to an effective attraction with the chain ends due to desolvation and polarization effects. 48
The motion of defects along water chains is effectively diffusive 39,112 and can be modeled as a Markov process 25. In such a kinetic model, each defect hops with a certain transition rate to a neighboring position and subsequent hops are statistically uncorrelated with each other. The transition rates, defined such that they conserve the equilibrium distribution, depend on the diffusion constant of the defects as well as on the free energy surface experienced by them as they interact with other effective charges present in the chain. For short chains, defects are typically generated at the chain ends and no more than one defect at a time is typically observed in the system. Once a defect hops to the chain end, it is annihilated. If there are multiple defects in a chain, two defects recombine if they hop next to each other. The inverse process facilitates the generation a pair of defects, an L defect and a D defect separated by a single molecule, anywhere within an ordered segment of water molecules. This process becomes the predominant mechanism for defect generation in long tubes.
5 Phase behavior and its system size dependence
In the previous sections we have mainly discussed single-file water chain in short pores, whose properties are strongly influenced by the boundary conditions at the tube entrances and by their finite lengths. As synthetic pores up to essentially macroscopic lengths can be manufactured, 20 we now discuss the length dependence of various features of single-file water. Such an analysis not only yields insights into the fundamental nature of water under 1d confinement, but also points to ways to probe the peculiar properties of nanopore water experimentally.
The phase behavior of 1d systems is determined by the range of interactions. 50,51,118 For water molecules in single-file arrangement, the dipole-dipole interaction dominates at large distances, decaying as 1/r3 with distance r. For interactions decaying faster than 1/r2, the energy for creating an interface between an ordered “up” and an ordered “down” segment is finite. According to the Landau argument for 1d spin models, however, a finite interface energy leads to disorder for any sufficiently large system. 119 The reason for the non-existence of an ordered phase at finite temperatures is that the entropy due to the formation of a single kink increases like ln N with system size N. Consequently, if the system is large enough, the entropic gain of introducing a kink will become larger than the energetic penalty and the system becomes disordered. Note that the spins can represent both the up/down orientations of water molecules, and occupied/empty sites, thus ruling out both a vapor-liquid phase transition and an order-disorder phase transition (see Sec. 5.3). One might wonder how this argument translates to the dipole lattice model in the charge picture, where the long range interaction is Coulombic. Obviously, as this picture is mathematically equivalent to the dipole picture of the model, no phase transition occurs. From the point of view of the charge picture, the dipole character of the interaction between water molecules manifests itself in the fact that effective charges alternate in sign as one moves along the chain and that only neutral charge pairs can be created.
The Landau argument also rules out the existence of ordered states and consequently true phase transitions for water in wider nanotubes. 120,121 However, the apparent translational and orientational order of water in short tubes can exist to great, macroscopic lengths as we shall see in the following in the case of single-file water. In this sense, it is meaningful to discuss the “phase behavior” of nanopore water, including novel “ice phases” in cylindrical confinement, 122 but one must keep in mind that no true phase transitions exist in such systems.
5.1 Filling transition
The water filling of apolar pores with sub-nanometer diameters depends sensitively on the environmental conditions, the tube lengths, and the strength of the water-pore interactions. As a result of this sensitivity, water in short pores is strongly influenced by the boundary conditions and shows bistability of distinct filled and empty states over a range relative humidities (see Fig. 5). Using the dipole-lattice model, the dependence of this bi-stability on the tube length and the effective chemical potential has been studied with Monte Carlo simulations. 81 It was found that the bi-stability becomes less pronounced with increasing tube length and vanishes for tubes longer than ≈ 300 nm, i.e., tubes with a maximum occupancy number of at least ≈ 1000 molecules. As the bi-stability gets weaker, the transition from an empty tube to a completely filled tube as a function of the effective chemical potential, or, equivalently, of the fugacity, of water gets steeper with increasing tube length, as shown in Fig. 5. Although the filling transition is step-like for long tubes, the slope of the adsorption isotherm remains finite everywhere even in the thermodynamic limit, as expected for 1d-Ising behavior. When bistability is lost, the adsorption isotherms have essentially converged to their thermodynamic limit. The filling transition in this limit occurs at about 8.5 % relative humidity. 49
Fig. 5.

Adsorption isotherms: average number density of water in a carbon nanotube as a function of the relative fugacity for various system sizes N. 81 The latter approximately correspond to the maximum numbers of water molecules that can occupy the pore. Densities run from 0 to 1, corresponding to the completely empty and filled tube, respectively. The fugacity is relative to that of a bath of liquid water at ambient conditions, and thus essentially identical to the relative humidity. Results obtained from molecular simulations for system sizes N = 5,10,15 and 30 (dashed lines) agree well with results for the dipole lattice model (solid lines).
5.2 Order-disorder transition
According to the results discussed above, carbon nanotubes at ambient conditions are essentially completely filled for relative humidity > 10 %. Even for such filled tubes, however, empty gaps with sizes of the order of one lattice spacing occur as expected from the Landau argument. These gaps do not influence the ordering behavior of the water chain, as confirmed by investigations based on the dipole lattice model. 49,81 In the following we examine the dipolar order of single-file water chains and discuss how this order breaks down in the long chain limit.
In a short tube, the dipoles are typically completely ordered with all molecules pointing in the same direction, up or down. Transitions between these two possible ordered states with dipoles parallel or antiparallel to the tube axis occur rarely, but when they occur, they happen rapidly through the motion of a defect along the water chain. 112 Thus, single-file water chains in short pores form two-state systems, which spend nearly all of the time in one of the two dipole ordered states. This behavior of predominantly ordered chains is reflected in the free energy as a function of the total dipole moment, which shows two deep minima for the ordered states and a flat barrier in-between (see Fig. 6). With increasing tube length, the entropic gain of introducing orientational defects outweighs their energetic penalty. Consequently, the average defect number increases with chain length and destroys the orientational order. The order probability, i.e., the probability that the system is completely ordered and free of defects can be approximated by P0(N) = (1 + e−βEd)−N. This expression is a consequence of the low defect density, which renders the defect interactions, and therefore also their correlations, negligible for all statistically important configurations. The system size at which a single defect is present in the water chain with 50 % probability is given by ≈ 5 × 105 molecules, corresponding to a tube length of ≈ 0.1mm. This length is within the range of the diameters of human hair and we call the order macroscopic. 49 Beyond this size, the average number of defects grows linearly with system size like exp(−βEd)N. The reasons for the remarkable, almost macroscopic length over which 1d water chains are orientationally ordered are the strong thermodynamic preference for a filled tube at ambient conditions, the large energetic penalty of hydrogen bonding defects, and, finally, the relatively small number of molecules required to reach macroscopic lengths in one dimension.
Fig. 6.

Free energy as a function of the total dipole moment per molecule. 49 Top panel: comparison of results from molecular dynamics simulation and results from Monte Carlo simulations of the dipole lattice model for a chain length of N = 40 molecules (top). Bottom panel: simulations results obtained for the dipole model for different chain lengths.
5.3 Single-file water as a cold Ising Model
It is instructive to summarize the length dependence of the filling transition and the orientational order for single-file water at ambient conditions by pointing out the relation of nanopore water to the 1d Ising model at low temperatures. 25,117,123–125 Neglecting interactions between the effective charges carried by defects and chain endpoints, the dipole model can be reduced to the 1d Ising model with Hamiltonian, 117
| (3) |
where the N spins si = ±1 interact only with nearest neighbors and are exposed to the external field h. No periodic boundary conditions apply. For small systems, i.e., short tubes, at low temperatures, the Ising model shows two-state behavior with either all spins pointing “up” or “down”. The reason is that the formation of a kink where “up” and “down” spins are next to each other is energetically costly. As the tube length increases, however, the formation of defects becomes entropically favored and complete order is lost. The spins of the Ising model can represent either empty and occupied sites, such that the model can be used to study the filling of the pore, or molecules pointing to the left and the right, in which case the model mimics dipolar order. In both cases, the coupling constant and the external field can be readily expressed in terms of the model parameters of the effective Hamiltonian given in eqn (2). 117
In the case of tube filling, the total magnetization given by Σi si is related to the total occupancy number and the external field represents the effective chemical potential that includes both the effect of the water bath as well as the tube-water interaction. The coupling constant is then given by J ≈ c/2 ≈5ε, where ε = 2μ2/(4π ε0a3) is the energy scale of the dipole-dipole interactions. Here, the different possible orientations of the water molecules are neglected and configurations with all spins “up” and all spins “down” correspond to completely filled and empty states, respectively. The latter are predominant in short tubes, where we rarely observe states with a single kink. This behavior is in agreement with molecular dynamics simulations of single-file water, where during the filling or emptying of the tube typically only one interface is formed. For long tubes, the Ising-model estimate of the chemical potential at which filling takes place is in good agreement with simulations results based on the full Hamiltonian of eqn (2). 81
The order properties of a single-file chain are related to the 1d Ising model via the correspondence of the total dipole moment to the magnetization. In this case, the external field h represents an external electric field pointing along the tube axis. The coupling constant J is now system size dependent, i.e., J = (Ed − 6ε/N)/2 and converges to J = Ed/2 for long tubes. 25 This length dependence, important for the orientational dynamics and dielectric response of water in short tubes, is due to the Coulomb interactions of a kink (defect) with the chain ends.
6 Response to electric fields
Single-file water reacts sensitively to external electric fields. 79,126–129 Generally, one may differentiate between effects of fields parallel and orthogonal to the tube axis. Orthogonal fields can disrupt the hydrogen bond network, generate defects, and perturb proton transport 90,126. In contrast, small fields parallel to tube axis can have significant effects on the tube filling and on the order and transport properties of single-file water, whereas the orientations of individual water molecules within an ordered chain segment remain largely unaffected. An external homogeneous field E along the tube axis adds the energy −E Σi qizi to the Hamiltonian given by eqn (2). An inhomogeneous field couples to the individual dipoles instead.
6.1 Influence on filling
Molecular simulations of short pores, where the total dipole moment exhibits two-state behavior, show that an external electric field leads to a stabilization of the completely filled and ordered state, 79 because of the strong interaction of the aligned water dipoles with the field. External fields pointing along the tube axis favor the “up” state in which all water dipoles are aligned with the field. As a result, the total dipole moment responds sensitively to external electric fields. In the “up” state, the magnitude of the dipole moments of individual water molecules projected onto the tube axis was found to be unaffected by the external field up to strengths of ≈ 1V/nm. This is a consequence of the stabilizing effect of the strong hydrogen bonds between water molecules that remain largely undistorted by the field.
A similar stabilizing effect of an electric field on the filled state is expected for long tubes, where the average length of dipole ordered segments of molecules parallel to the field is increased while the length of segments anti-parallel to the field is decreased. Consequently, electric fields lower the energy of the filled state without necessarily changing the number of orientational defects.
6.2 Influence on order and proton transport
The coupling of the total dipole moment to the external electric field tilts the free energy profiles shown in Fig. 6 by −EM, where M is the total dipole moment. For predominantly ordered systems, the interaction energy of the ordered states with the electric field grows linearly with the tube length. The ordered state aligned with the field becomes more stable, and the free energy in the barrier region is slanted towards this state. A defect moving on this slanted free energy profile will drift faster than on the almost flat energy landscape in the absence of external fields (Fig. 6). The energy profile of a defect with effective charge q at position z incurs a linear perturbation at small fields, −qEz. As a result, electric fields lower the activation barriers for the transport of a proton through a pore, and for the subsequent transport of a defect restoring the original chain orientation. 48 Consequently, we expect a roughly exponential increase in the rate of proton transfer with small, linearly increasing fields.
For long tubes, the free energy curve for the total dipole moment shows a quadratic minimum that is skewed and shifted by the external field. It is important to note, that since the electric field couples to the total dipole moment, longer chains will generally react more sensitively to electric fields along the axis. This effect becomes clear, when we look at the linear response of single file water to a time-dependent homogeneous electric field.
6.3 Dielectric response
Due to the particular way single file water chains respond to external static and time-dependent fields, recent calculations suggest that dielectric spectroscopy offers a viable way to probe the remarkable properties of nanopore water experimentally. 24,25 Simulations of a kinetic version of the dipole model indicate that for all system sizes, single file water chains show Debye relaxation behavior, characterized by an exponentially decaying autocorrelation function of the total dipole moment. 24 Thus, the linear response to a time-dependent electric field is fully determined by the static susceptibility χ and the relaxation time τ. 130 The origin of the Debye behavior is different for short and for long chains. In short chains, the dipole autocorrelation function decays exponentially due to the their two-state behavior and the related first order kinetics between the two completely ordered states. Long chains, in contrast, are disordered, but the defect density is low such that they can be considered as uncorrelated. The dipole autocorrelation function then results from the diffusive motion of defects, which determine the time evolution of the total dipole moment. The combined effect of these defects leads to De-bye relaxation behavior as shown analytically for the kinetic 1d Ising model by Glauber. 131
The static susceptibility of a water-filled nanopore membrane, calculated under the assumption that chains in different tubes are uncorrelated, strongly depends on the length of the pore. For a pore density of ≈2,500μm−2, an area density of slightly wider tubes in a recent experimental study, 37 the static dielectric susceptibility spans five orders of magnitude, from χ ≈ 0.1 for nanometer long pores to χ ≈ 104 for millimeter long pores. 24,117 For short tubes, the static susceptibility increases linearly with tube length as expected for a two-state system, before it saturates at a constant value for longer tubes as defects arise. 25 From the slope of the susceptibility-vs.-size curve in the linear regime one can determine the average dipole moment of water molecules in the tube direction. The crossover between the linear and the constant regimes occurs at a length where one hydrogen bonding defect is present in the chain in the average, such that the crossover position provides information on the defect creation energy. The static susceptibility of long tubes exceeds that of bulk water (χ ≈ 80) by about a factor of 100, although the average water density in the nanopore membrane is 3000 times smaller than in the bulk liquid.
Similar size dependent behavior is observed for the relaxation time in kinetic simulations of the dipole lattice model, which varies from ≈ 10ns for short tubes holding ≈ 10 molecules to ≈ 0.1 s in the long tube limit, spanning seven orders of magnitude. 24,25 For short pores, the relaxation time increases sub-linearly due to the size dependence of the free energy barrier separating the two ordered states. The barrier grows with increasing length because a defect present at the center of the chain in the transition state configuration is separated from the effective charges of opposite sign at the chain ends. 25 Since for constant barrier height the relaxation time would increase linearly with system size, a curvature in the τ (N)-curve for small system sizes is a signature of the Coulombic interactions between the effective charges carried by defect and chain ends. Once the chains are long enough such that this interaction becomes negligible, the relaxation time increases linearly and then converges to a constant value for long tubes. By analyzing the size dependence of the relaxation time one can determine the diffusion constant for hydrogen bonding defects moving along the water chain. 24,25
6.4 Vibrational spectroscopy
Recently, vibrational spectroscopy has been suggested as another route to probe the structure of nanopore water experimentally. In particular, the vibrational line shape of the OH-stretching mode of an HDO molecule in a chain of D2O molecules carries information on the different local environments experienced by the OH bond. As shown by recent simulations 40 based on a mixed quantum/classical approach, 132–134 the shift of the OH-frequency from its value in the gas phase is considerably more pronounced in the hydrogen bonded configuration than for the OH-bond in its dangling configuration, in which the OH bond is less influenced by the neighboring water molecules. As a consequence, the infrared and Raman line shape of the OH-stretch is split into two peaks that can be assigned to these two configurations. These characteristic features also distinguish single-file water from the stacked-ring structures arising in wider nanopores, such that the vibrational spectroscopy may be used to verify the structure of 1d water chains, which have been investigated only theoretically to date.
7 Single-file water in apolar nanotubes as a model system
In the following, we compare single-file water in apolar pores to water in more complex confinement, such as metallic nanotubes and biological transmembrane water channels. As we will see, many properties of single-file water in apolar pores are conserved in more complex pores and pore surroundings.
7.1 Synthetic pores
In metallic nanotubes, screening charges lower the effective dipole moment of water molecules inside the tube. 135 These screening charges favor the filling of the tube and lower the desolvation penalty of a proton entering the pore. However, the water structure of the chain itself remains largely unaffected. For example, a proton in a chain shows similar repulsion by the chain ends as was observed for apolar tubes. For short tubes, we expect the metallic character to show up primarily as an enhanced axial polarizability.
Electronic structure calculations show that carbon and boron nitride nanotubes display curvature-induced polarization, 55,136 which leads to the preferential formation of L defects. 137 The polarization leads to electric fields at the pore ends pointing into the pore and the dipole moments tend to align with this field by generating an L defect. The effect of pore polarization on the water structure emphasizes the importance of the boundary conditions for short pores.
7.2 Biological pores
Aquaporins block proton transport but allow for water transport with conduction rates of 109 molecules/ns, comparable to transport through apolar pores. 21 The proton blockage is mainly due to electrostatic effects that enhance the desolvation penalty. 6,7,47,65,105,128,138 which is also observed in apolar nanotubes 39 A major contribution to the proton blockage is that proton transfer through the pore corresponds to transporting a charge through a medium with low dielectric constant as exemplified by a simple model channel. 47 Additionally, two conserved α -helices M3 and M7 form macro-dipoles parallel along the pore and lead to a bi-polar orientation of the water chain with a defect in the center (see Fig. 2). This effect is similar to what was observed for carbon nanotubes with curvature induced polarization 137 and can be mimicked with carbon nanotubes with additional charges acting as macro-dipoles 36 or using the electrostatic profile of aquaporins 139. In aquaporin, however, two asparagines in the center of the pore (NPA motif) donate hydrogen bonds to the L defect molecule. These hydrogen bonds compensate for part of the excitation energy needed to introduce a defect (which is ≈ 8kcal/mol for an infinitely long chain in an apolar pore 49, see Sec. 4) and additionally stabilizes the defect in place. Looking at the charge picture of Fig. 4F, we find that the L defect carries a negative charge of approximately 0.4e and thus screens partly the positive charges at the chain center. Similarly, the ends carry positive charges at the negative ends of the macro-dipoles.
In contrast to aquaporin, gramicidin transports not only water but also monovalent cations. 67 The pore of gramicidin A is formed by two aligned β-helices with their backbones on the inside of the channel. 140 Consequently, water within the pore can donate hydrogen bonds to the carbonyl oxygen atoms as depicted in Fig. 7. 11 The single-file region consists of eight water molecules. Water chains within the pore are predominantly ordered with the additional hydrogen bonds stabilizing the filled state. The dipole moments of the molecules point along the tube axis on average whereas the dipole moments of defects are orthogonal to the axis. The hydronium defect preferentially sits in the middle of the chain, as one would expect from its effective interactions with the chain ends. Also, as observed in aquaporin, additional hydrogen bonds are capable of stabilizing hydrogen bonding and ionic defects, which leads to localization of a defect within the pore. Single-file water plays an important role for the transport of ions, because ions in the pore are solvated by two single-file chains on either side. 141,142 Consequently, not only the desolvation penalty is reduced, but also the movement of the ion is directly coupled to water motion and thus fluctuations at the pore entrance.
Fig. 7.
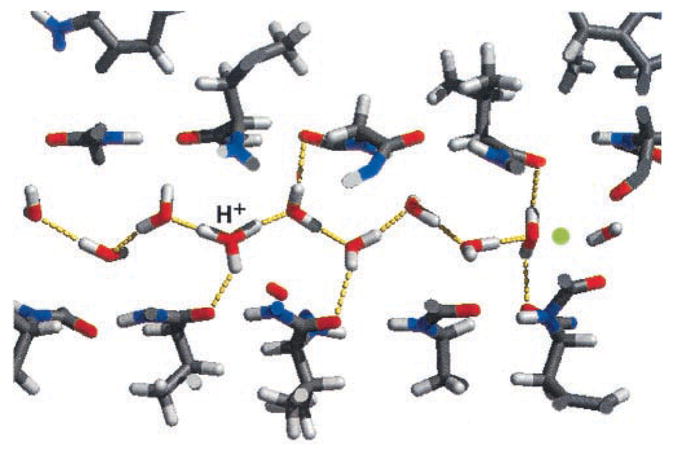
Snapshot of a molecular dynamics simulation of single-file water inside the channel of gramicidin A. Water molecules donate hydrogen bonds to the carbonyl groups of the channel. The configuration shows a hydronium ion and a D defect, each stabilized by three hydrogen bonds. (Reprinted from Ref. 11, Copyright (2002), with permission from Elsevier.)
In summary, the inner pore wall of gramicidin differs strongly from the smooth and apolar pore of carbon nanotubes. The most obvious difference are the hydrogen bonds donated by the water molecules to the pore wall. However, the water structural motifs found in apolar pores like defect structures and ordered segments are reminiscent of those in nanotubes. The water-filled state of the pore is stabilized and the desolvation barrier of ions entering the tube is reduced by the carbonyl groups while still taking advantage of the intriguing transport properties of single-file water.
8 Conclusions and perspectives
Single-file water in narrow pores such as carbon nanotubes has many unusual properties that arise from an interplay between the strong hydrogen bonds forming between water molecules and the 1d confinement inside the pore. These properties are reminiscent both of solids (e.g., the high dielectric susceptibility) and liquids (e.g., the high mobility of water molecules in the pore). However, the water wires, and similarly the ”quasi-1d” ice structures in nanopores, 122 should be considered an, albeit peculiar, 1d fluid in the 1d thermodynamic limit. The reason is that, as expected for 1d systems with short-ranged interactions, the orientational and translational order break down for sufficiently long chains. As a result, no true phase transitions exist in these systems.
For short pores, however, the strong hydrogen bonds in conjunction with dipole-dipole interactions lead to the formation of dipolarly ordered water wires that provide routes for rapid proton transfer occurring via a Grotthuss relay mechanism. Orientational order is ultimately destroyed by hydrogen bonding defects. As is the case for Bjerrum defects in ice, 109,143,144 the concentration and mobility of L and D defects in water chains are main factors in dielectric relaxation and charge transport.
The properties of nanopore water are strongly influenced by the electrostatic and dielectric properties of the pore and its surroundings as well as by boundary conditions. This sensitivity appears to be exploited in proteins that form single-file pores in biological cells to mediate the efficient, selective, and controlled transport of water, protons and other ions. At least some of the physical properties of biological water-filled channels can be mimicked by carbon or boron nitride nanotubes embedded in dielectric media and in contact with water reservoirs, and by applying corresponding external electrical fields and dielectric media. 36,139,145,146
Experimental studies of single-file water face difficult challenges. The synthesis of pristine nanotubes with specific chiralities, and thus defined geometric and electronic properties, remains difficult. Defects in the pore wall affect the transport properties. 147 In recent years, considerable progress in the fabrication of membranes of nanotubes with narrow distributions of diameters have been made. 19,20,148 With the desired pores available, experiments can be performed on nanotubes in different arrangements, such as nanotubes in solution 32–34, assembled in membranes 37, or even single nanotubes. 149 Since these different experimental setups lead to different electrostatic and dielectric surroundings of the pores, single file water is expected to show different behavior in each of them. For example, the interactions of single-file water chains in different nanotubes of a membrane might lead to collective effects 49,127,150 and to true phase transitions not seen in isolated nanotubes. Additionally, the transient phenomena on a molecular scale observed in molecular simulations are difficult to probe directly in experiments. 151 Therefore, quantitative theoretical frameworks for the transport properties of single-file water in apolar pores that include the dielectric and electrostatic effects of the surroundings on the intrinsic properties of single-file water are necessary to infer the microscopic properties of single-file water from experiments.
Many of the theoretical results for single-file water are based on simulations using empirical or semi-empirical interaction potentials or quantum-mechanical density functional calculations. By design, the rigid point-charge models applied do not include cooperative effects, i.e., many-body effects and polarization, which are known to cause strong variations in the hydrogen bond strengths for different molecular configurations 110,152,153. These variations are not only important for a microscopically detailed understanding of the energetics and dynamics of defect and charge transport, but can also facilitate cooperative pathways for charge transport. An example for such a cooperative pathway is multi-proton transport where a proton is transported via nearly simultaneous hops of protons involved in different hydrogen bonds. 154 Experimental and theoretical studies indicate that such cooperative pathways become especially prominent for short chains of water molecules connecting strong donor and acceptor groups. 154–156 To address these issues, quantum-chemical simulations of single-file water in nanopores are needed. 27,39,99,135,157
More complex structures made from nanotubes, such junctions and rings, offer exciting new possibilities to build molecular machines and other nanoscopic devices. 158,159 Such structures provide not only novel building blocks for nanofluidics applications, but also platforms to probe the fundamental properties of water in nanoconfinement.
Fig. 3.
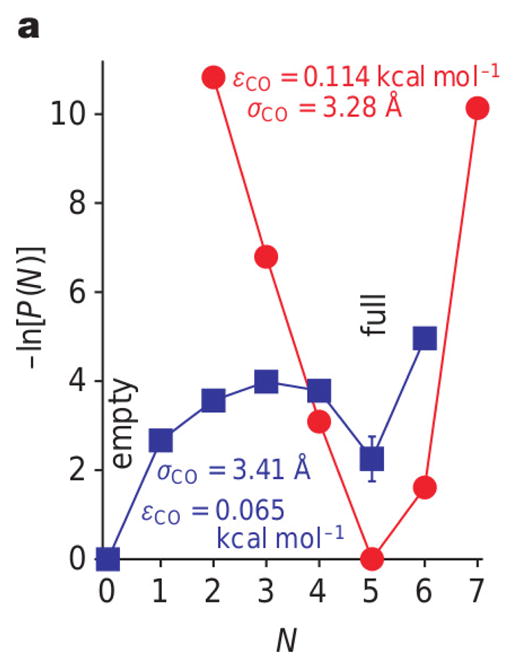
Sensitivity of filling probability, i.e., the probability P(N) to find N water molecules in the tube, on the parameters of the tube-water interaction energy for a (6,6) carbon nanotube immersed in a water bath. (Reprinted by permission from Macmillan Publishers Ltd: Nature Ref. 18, copyright (2001) ) While for one set of parameters (red curve), the free energy −ln P(N) has a single minimum at N = 5, corresponding to the completely filled state, for a slightly modified set of parameters (blue curve), the tube shows bi-stability with an empty and a full state of similar free energies. The numbers in the figure refer to the depth εCO and the range σCO of the Lennard-Jones potential acting between the oxygen atoms of the water molecules and the carbon atoms of the carbon nanotube.
Acknowledgments
J.K. and G.H. were supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health. C.D. gratefully acknowledges financially support by the Austrian Science Foundation (FWF) within the SFB ViCoM (F 41).
References
- 1.Tyree MT. Nature. 2003;423:923–923. doi: 10.1038/423923a. [DOI] [PubMed] [Google Scholar]
- 2.Ku DN. Annu Rev Fluid Mech. 1997;29:399–434. [Google Scholar]
- 3.Hille B. Ion Channels of Excitable Membranes. 3. Sinauer Associates, Inc; Sunderland: 2001. [Google Scholar]
- 4.Agre P. Angew Chem Int Ed. 2004;43:4278– 4290. doi: 10.1002/anie.200460804. [DOI] [PubMed] [Google Scholar]
- 5.Agre P, Kozono D. Aquaporin water channels: molecular mechanisms for human diseases. FEBS letters; Presented at the Nobel Symposium 126, Membrane Proteins: Structure, Function, and Assembly; Friibergh’s Herrgård, Örsundsbro, Sweden. August 23, 2003; Elsevier Science B.V; 2003. pp. 72–78. [Google Scholar]
- 6.Tajkhorshid E, Nollert P, Jensen MO, Miercke WLJ, O’Connell J, Stroud RM, Schulten K. Science. 2002;296:525–530. doi: 10.1126/science.1067778. [DOI] [PubMed] [Google Scholar]
- 7.de Groot BL, Grubmuller H. Science. 2001;294:2353–2357. doi: 10.1126/science.1066115. [DOI] [PubMed] [Google Scholar]
- 8.Parisi M, Dorr R, Ozu M, Toriano R. J Biol Phys. 2007;33:331–343. doi: 10.1007/s10867-008-9064-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Urban B, Hladky S, Haydon D. Biochim Biophys Acta. 1980;602:331–354. doi: 10.1016/0005-2736(80)90316-8. [DOI] [PubMed] [Google Scholar]
- 10.Pomès R, Roux B. Biophys J. 1998;75:33–40. doi: 10.1016/S0006-3495(98)77492-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pomès R, Roux B. Biophys J. 2002;82:2304–2316. doi: 10.1016/S0006-3495(02)75576-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sagnella DE, Voth GA. Biophys J. 1996;70:2043–2051. doi: 10.1016/S0006-3495(96)79773-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whitby M, Quirke N. Nature Nanotech. 2007;2:87–94. doi: 10.1038/nnano.2006.175. [DOI] [PubMed] [Google Scholar]
- 14.Pfahler J, Harley J, Bau H, Zemel J. Sensor Actuat A: Phys. 1989;22:431–434. [Google Scholar]
- 15.Miller SA, Young VY, Martin CR. J Am Chem Soc. 2001;123:12335–12342. doi: 10.1021/ja011926p. [DOI] [PubMed] [Google Scholar]
- 16.Thomas JA, McGaughey AJH. Phys Rev Lett. 2009;102:184502. doi: 10.1103/PhysRevLett.102.184502. [DOI] [PubMed] [Google Scholar]
- 17.Bocquet L, Charlaix E. Chem Soc Rev. 2010;39:1073–1095. doi: 10.1039/b909366b. [DOI] [PubMed] [Google Scholar]
- 18.Hummer G, Rasaiah JC, Noworyta JP. Nature. 2001;414:188–190. doi: 10.1038/35102535. [DOI] [PubMed] [Google Scholar]
- 19.Holt JK, Park HG, Wang Y, Stadermann M, Artyukhin AB, Grigoropoulos CP, Noy A, Bakajin O. Science. 2006;312:1034–1037. doi: 10.1126/science.1126298. [DOI] [PubMed] [Google Scholar]
- 20.Holt JK, Noy A, Huser T, Eaglesham D, Bakajin O. Nano Lett. 2004;4:2245–2250. [Google Scholar]
- 21.Kalra A, Garde S, Hummer G. Proc Natl Acad Sci USA. 2003;100:10175–10180. doi: 10.1073/pnas.1633354100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kreuer K-D, Paddison SJ, Spohr E, Schuster M. Chem Rev. 2004;104:4637–4678. doi: 10.1021/cr020715f. [DOI] [PubMed] [Google Scholar]
- 23.Corry B. J Phys Chem B. 2008;112:1427–1434. doi: 10.1021/jp709845u. [DOI] [PubMed] [Google Scholar]
- 24.Köfinger J, Dellago C. Phys Rev Lett. 2009;103:080601. doi: 10.1103/PhysRevLett.103.080601. [DOI] [PubMed] [Google Scholar]
- 25.Köfinger J, Dellago C. Phys Rev B. 2010;82:205416. doi: 10.1103/PhysRevB.82.205416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saha SK, Chakravorty D. Appl Phys Lett. 2006;89:043117. [Google Scholar]
- 27.Dellago C, Naor MM. Comput Phys Commun. 2005;169:36–39. [Google Scholar]
- 28.Striolo A, Chialvo AA, Gubbins KE, Cummings PT. J Chem Phys. 2005;122:234712. doi: 10.1063/1.1924697. [DOI] [PubMed] [Google Scholar]
- 29.Kolesnikov AI, Zanotti J-M, Loong C-K, Thiyagarajan P, Moravsky AP, Loutfy RO, Burnham CJ. Phys Rev Lett. 2004;93:035503. doi: 10.1103/PhysRevLett.93.035503. [DOI] [PubMed] [Google Scholar]
- 30.Naguib N, Ye H, Gogotsi Y, Yazicioglu AG, Megaridis CM, Yoshimura M. Nano Lett. 2004;4:2237–2243. [Google Scholar]
- 31.Wang HJ, Xi XK, Kleinhammes A, Wu Y. Science. 2008;322:80–83. doi: 10.1126/science.1162412. [DOI] [PubMed] [Google Scholar]
- 32.Hennrich F, Arnold K, Lebedkin S, Quintilla A, Wenzel W, Kappes MM. Phys Stat Sol. 2007;244:3896–3900. [Google Scholar]
- 33.Quintilla A, Hennrich F, Lebedkin S, Kappes MM, Wenzel W. Phys Chem Chem Phys. 2010;12:902–908. doi: 10.1039/b912847f. [DOI] [PubMed] [Google Scholar]
- 34.Cambré S, Wenseleers W. Angew Chem Int Ed. 2011;50:2764?2768. doi: 10.1002/anie.201007324. [DOI] [PubMed] [Google Scholar]
- 35.Cambré S, Schoeters B, Luyckx S, Goovaerts E, Wenseleers W. Phys Rev Lett. 2010;104:207401. doi: 10.1103/PhysRevLett.104.207401. [DOI] [PubMed] [Google Scholar]
- 36.Zhu F, Schulten K. Biophys J. 2003;85:236–244. doi: 10.1016/S0006-3495(03)74469-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fornasiero F, Park HG, Holt JK, Stadermann M, Grigoropoulos CP, Noy A, Bakajin O. Proc Natl Acad Sci USA. 2008;105:17250–17255. doi: 10.1073/pnas.0710437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petrenko VF, Whitworth RW. Physics of Ice. Oxford University Press; USA: 2002. [Google Scholar]
- 39.Dellago C, Naor MM, Hummer G. Phys Rev Lett. 2003;90:105902. doi: 10.1103/PhysRevLett.90.105902. [DOI] [PubMed] [Google Scholar]
- 40.Weinwurm M, Dellago C. J Phys Chem B. 2011;115:5268–5277. doi: 10.1021/jp109037q. [DOI] [PubMed] [Google Scholar]
- 41.Onsager L. In: The Neurosciences. Quarton GC, Melnechuk T, Schmitt FO, editors. Rockefeller University Press; New York: 1967. pp. 75–79. [Google Scholar]
- 42.de Grotthus CJT. Ann Chim. 1806;58:54–74. [Google Scholar]
- 43.Agmon N. Chem Phys Lett. 1995;244:456–462. [Google Scholar]
- 44.Marx D. Chem Phys Chem. 2006;7:1848– 1870. doi: 10.1002/cphc.200600128. [DOI] [PubMed] [Google Scholar]
- 45.Brewer ML, Schmitt UW, Voth GA. Biophys J. 2001;80:1691–1702. doi: 10.1016/S0006-3495(01)76140-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wraight CA. Biochim Biophys Acta. 2006;1757:886–912. doi: 10.1016/j.bbabio.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 47.Burykin A, Warshel A. Biophys J. 2003;85:3696–3706. doi: 10.1016/S0006-3495(03)74786-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dellago C, Hummer G. Phys Rev Lett. 2006;97:245901. doi: 10.1103/PhysRevLett.97.245901. [DOI] [PubMed] [Google Scholar]
- 49.Köfinger J, Hummer G, Dellago C. Proc Natl Acad Sci USA. 2008;105:13218–13222. doi: 10.1073/pnas.0801448105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ruelle D. Commun Math Phys. 1968;9:267–278. [Google Scholar]
- 51.Luijten E, Blöte HWJ. Phys Rev B. 1997;56:8945–8958. [Google Scholar]
- 52.Iijima S. Nature. 1991;354:56–58. [Google Scholar]
- 53.Jorio A, Dresselhaus G, Dresselhaus MS. Carbon Nanotubes: Advanced Topics in the Synthesis, Structure, Properties and Applications. Springer; 2008. [Google Scholar]
- 54.Won CY, Aluru NR. J Am Chem Soc. 2007;129:2748–2749. doi: 10.1021/ja0687318. [DOI] [PubMed] [Google Scholar]
- 55.Won CY, Joseph S, Aluru NR. J Chem Phys. 2006;125:114701. doi: 10.1063/1.2338305. [DOI] [PubMed] [Google Scholar]
- 56.Chopra NG, Luyken RJ, Cherrey K, Crespi VH, Cohen ML, Louie SG, Zettl A. Science. 1995;269:966–967. doi: 10.1126/science.269.5226.966. [DOI] [PubMed] [Google Scholar]
- 57.Zhi C, Bando Y, Tang C, Golberg D. Mater Scie Eng R. 2010;70:92–111. [Google Scholar]
- 58.Blase X, Rubio A, Louie SG, Cohen ML. Europhys Lett. 1994;28:335–340. [Google Scholar]
- 59.Zhi C, Bando Y, Tang C, Golberg D. Phys Rev B. 2006;74:153413. [Google Scholar]
- 60.Golberg D, Bando Y, Huang Y, Terao T, Mitome M, Tang C, Zhi C. ACS Nano. 2010;4:2979–2993. doi: 10.1021/nn1006495. [DOI] [PubMed] [Google Scholar]
- 61.Won CY, Aluru NR. J Phys Chem C. 2008;112:1812–1818. [Google Scholar]
- 62.Suk ME, Aluru NR. J Phys Chem Lett. 2010;1:1590–1594. [Google Scholar]
- 63.Hilder TA, Gordon D, Chung S-H. Small. 2009;5:2183–2190. doi: 10.1002/smll.200900349. [DOI] [PubMed] [Google Scholar]
- 64.Yuan Q, Zhao Y-P. Biomicrofluid. 2009;3:022411. [Google Scholar]
- 65.Chakrabarti N, Roux B, Pomès R. J Mol Biol. 2004;343:493–510. doi: 10.1016/j.jmb.2004.08.036. [DOI] [PubMed] [Google Scholar]
- 66.Fu D, Libson A, Miercke LJW, Weitzman C, Nollert P, Krucinski J, Stroud RM. Science. 2000;290:481–486. doi: 10.1126/science.290.5491.481. [DOI] [PubMed] [Google Scholar]
- 67.Andersen OS. Annu Rev Physiol. 1984;46:531–548. doi: 10.1146/annurev.ph.46.030184.002531. [DOI] [PubMed] [Google Scholar]
- 68.Storm AJ, Chen JH, Ling XS, Zandbergen HW, Dekker C. Nature Mat. 2003;2:537–540. doi: 10.1038/nmat941. [DOI] [PubMed] [Google Scholar]
- 69.Dekker C. Nature Nanotech. 2007;2:209–215. doi: 10.1038/nnano.2007.27. [DOI] [PubMed] [Google Scholar]
- 70.Kox R, Chen C, Maes G, Lagae L, Borghs G. Nanotech. 2009;20:115302. doi: 10.1088/0957-4484/20/11/115302. [DOI] [PubMed] [Google Scholar]
- 71.Dimitrov V, Mirsaidov U, Wang D, Sorsch T, Mansfield W, Miner J, Klemens F, Cirelli R, Yemenicioglu S, Timp G. Nanotech. 2010;21:065502. doi: 10.1088/0957-4484/21/6/065502. [DOI] [PubMed] [Google Scholar]
- 72.Tong HD, Jansen HV, Gadgil VJ, Bostan CG, Berenschot E, van Rijn CJM, Elwenspoek M. Nano Lett. 2004;4:283–287. [Google Scholar]
- 73.Xu D, Sun L, Li H, Zhang L, Guo G, Zhao X, Gui L. New J Chem. 2003;27:300–306. [Google Scholar]
- 74.Harrell CC, Lee SB, Martin CR. Analyt Chem. 2003;75:6861–6867. doi: 10.1021/ac034602n. [DOI] [PubMed] [Google Scholar]
- 75.Sha J, Niu J, Ma X, Xu J, Zhang X, Yang Q, Yang D. Adv Mater. 2002;14:1219–1221. [Google Scholar]
- 76.Raghavender US, Kantharaju, Aravinda S, Shamala N, Balaram P. J Am Chem Soc. 2010;132:1075–1086. doi: 10.1021/ja9083978. [DOI] [PubMed] [Google Scholar]
- 77.Fois E, Gamba A, Tabacchi G, Quartieri S, Vezzalini G. J Phys Chem B. 2001;105:3012–3016. [Google Scholar]
- 78.Otsubo K, Wakabayashi Y, Ohara J, Yamamoto S, Matsuzaki H, Okamoto H, Nitta K, Uruga T, Kitagawa H. Nature Mat. 2011;10:291–295. doi: 10.1038/nmat2963. [DOI] [PubMed] [Google Scholar]
- 79.Vaitheeswaran S, Rasaiah JC, Hummer G. J Chem Phys. 2004;121:7955–7965. doi: 10.1063/1.1796271. [DOI] [PubMed] [Google Scholar]
- 80.Rasaiah JC, Garde S, Hummer G. Annu Rev Phys Chem. 2008;59:713–740. doi: 10.1146/annurev.physchem.59.032607.093815. [DOI] [PubMed] [Google Scholar]
- 81.Köfinger J, Hummer G, Dellago C. J Chem Phys. 2009;130:154110. doi: 10.1063/1.3106223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gordillo MC, Martí J. J Phys: Cond Mat. 2010;22:284111. doi: 10.1088/0953-8984/22/28/284111. [DOI] [PubMed] [Google Scholar]
- 83.Leenaerts O, Partoens B, Peeters FM. Phys Rev B. 2009;79:235440. [Google Scholar]
- 84.Waghe A, Rasaiah JC, Hummer G. J Chem Phys. 2002;117:10789–10795. [Google Scholar]
- 85.Maibaum L, Chandler D. J Phys Chem B. 2003;107:1189–1193. [Google Scholar]
- 86.Beckstein O, Sansom MSP. Proc Natl Acad Sci USA. 2003;100:7063–7068. doi: 10.1073/pnas.1136844100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Saparov SM, Pohl P. Proc Natl Acad Sci USA. 2004;101:4805–4809. doi: 10.1073/pnas.0308309101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Berezhkovskii A, Hummer G. Phys Rev Lett. 2002;89:064503. doi: 10.1103/PhysRevLett.89.064503. [DOI] [PubMed] [Google Scholar]
- 89.Zhu F, Tajkhorshid E, Schulten K. Phys Rev Lett. 2004;93:224501. doi: 10.1103/PhysRevLett.93.224501. [DOI] [PubMed] [Google Scholar]
- 90.Portella G, Pohl P, de Groot BL. Biophys J. 2007;92:3930–3937. doi: 10.1529/biophysj.106.102921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Portella G, de Groot BL. Biophys J. 2009;96:925–938. doi: 10.1016/j.bpj.2008.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Beckstein O, Tai K, Sansom MSP. J Am Chem Soc. 2004;126:14694–14695. doi: 10.1021/ja045271e. [DOI] [PubMed] [Google Scholar]
- 93.Peter C, Hummer G. Biophys J. 2005;89:2222–2234. doi: 10.1529/biophysj.105.065946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nagle JF. J Phys Chem. 1983;87:4086–4088. [Google Scholar]
- 95.Lapid H, Agmon N, Petersen MK, Voth GA. J Chem Phys. 2005;122:014506. doi: 10.1063/1.1814973. [DOI] [PubMed] [Google Scholar]
- 96.Markovitch O, Chen H, Izvekov S, Paesani F, Voth GA, Agmon N. J Phys Chem B. 2008;112:9456–9466. doi: 10.1021/jp804018y. [DOI] [PubMed] [Google Scholar]
- 97.Chen H, Voth GA, Agmon N. J Phys Chem B. 2010;114:333–339. doi: 10.1021/jp908126a. [DOI] [PubMed] [Google Scholar]
- 98.Geissler PL, Dellago C, Chandler D, Hutter J, Parrinello M. Science. 2001;291:2121–2124. doi: 10.1126/science.1056991. [DOI] [PubMed] [Google Scholar]
- 99.ØJensen M, Röthlisberger U, Rovira C. Biophys J. 2005;89:1744–1759. doi: 10.1529/biophysj.104.058206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cao Z, Peng Y, Yan T, Li S, Li A, Voth GA. J Am Chem Soc. 2010;132:11395–11397. doi: 10.1021/ja1046704. [DOI] [PubMed] [Google Scholar]
- 101.Dellago C, Weissensteiner W. 2010 unpublished data. [Google Scholar]
- 102.Petersen MK, Iyengar SS, Day TJF, Voth GA. J Phys Chem B. 2004;108:14804–14806. [Google Scholar]
- 103.Köfinger J, Dellago C. J Phys Chem B. 2008;112:2349–2356. doi: 10.1021/jp0736185. [DOI] [PubMed] [Google Scholar]
- 104.Ilan B, Tajkhorshid E, Schulten K, Voth GA. Proteins. 2004;55:223–228. doi: 10.1002/prot.20038. [DOI] [PubMed] [Google Scholar]
- 105.Kato M, Pisliakov AV, Warshel A. Proteins. 2006;64:829–844. doi: 10.1002/prot.21012. [DOI] [PubMed] [Google Scholar]
- 106.Pomes R, Roux B. Biophys J. 1996;71:19–39. doi: 10.1016/S0006-3495(96)79211-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nagle JF, Morowitz HJ. Proc Natl Acad Sci USA. 1978;75:298–302. doi: 10.1073/pnas.75.1.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nagle JF. J Bioener Biomembr. 1987;19:413–426. doi: 10.1007/BF00770027. [DOI] [PubMed] [Google Scholar]
- 109.Bjerrum N. Science. 1952;115:385–390. doi: 10.1126/science.115.2989.385. [DOI] [PubMed] [Google Scholar]
- 110.Keutsch FN, Saykally RJ. Proc Natl Acad Sci USA. 2001;98:10533–10540. doi: 10.1073/pnas.191266498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nagle JF, Tristram-Nagle S. J Membr Biol. 1983;74:1–14. doi: 10.1007/BF01870590. [DOI] [PubMed] [Google Scholar]
- 112.Best RB, Hummer G. Proc Natl Acad Sci USA. 2005;102:6732–6737. doi: 10.1073/pnas.0408098102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chou T. Phys Rev Lett. 1998;80:85–88. [Google Scholar]
- 114.Zhou X, Li C-Q, Iwamoto M. J Chem Phys. 2004;121:7996–8002. doi: 10.1063/1.1799971. [DOI] [PubMed] [Google Scholar]
- 115.Chou T. Biophys J. 2004;86:2827–2836. doi: 10.1016/S0006-3495(04)74335-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chou T. J Phys A: Math and Gen. 2002;35:4515–4526. [Google Scholar]
- 117.Köfinger J, Dellago C. New J Phys. 2010;12:093044. doi: 10.1088/1367-2630/12/9/093044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Thouless DJ. Phys Rev. 1969;187:732–733. [Google Scholar]
- 119.Landau L, Lifshitz E. Statistical Physics. Pergamon Press; Oxford: 1980. [Google Scholar]
- 120.Wilms D, Winkler A, Virnau P, Binder K. Phys Rev Lett. 2010;105:045701. doi: 10.1103/PhysRevLett.105.045701. [DOI] [PubMed] [Google Scholar]
- 121.Winkler A, Wilms D, Virnau P, Binder K. J Chem Phys. 2010;133:164702. doi: 10.1063/1.3502684. [DOI] [PubMed] [Google Scholar]
- 122.Koga K, Gao GT, Tanaka H, Zeng XC. Nature. 2001;412:802–805. doi: 10.1038/35090532. [DOI] [PubMed] [Google Scholar]
- 123.Patria RK. Statistical Mechanics. Butterworth-Heinemann; Oxford: 1972. [Google Scholar]
- 124.Baxter RJ. Exactly Solved Models in Statistical Mechanics. Dover Publications; New York: 2007. [Google Scholar]
- 125.Antal T, Droz M, Rácz Z. J Phys A: Math and Gen. 2004;37:1465–1478. [Google Scholar]
- 126.Hassan SA, Hummer G, Lee Y-S. J Chem Phys. 2006;124:204510. doi: 10.1063/1.2198820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Garate J-A, English NJ, MacElroy JMD. J Chem Phys. 2009;131:114508. doi: 10.1063/1.3227042. [DOI] [PubMed] [Google Scholar]
- 128.Chakrabarti N, Tajkhorshid E, Roux B, Pomès R. Structure. 2004;12:65–74. doi: 10.1016/j.str.2003.11.017. [DOI] [PubMed] [Google Scholar]
- 129.Zhu F, Tajkhorshid E, Schulten K. Biophys J. 2004;86:50–57. doi: 10.1016/S0006-3495(04)74082-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.McQuarrie DA. Statistical Mechanics. 2. University Science Books; New York: 2000. [Google Scholar]
- 131.Glauber RJ. J Math Phys. 1963;4:294–307. [Google Scholar]
- 132.Eaves JD, Tokmakoff A, Geissler PL. J Phys Chem A. 2005;109:9424–9436. doi: 10.1021/jp051364m. [DOI] [PubMed] [Google Scholar]
- 133.Corcelli SA, Skinner JL. J Phys Chem A. 2005;109:6154–6165. doi: 10.1021/jp0506540. [DOI] [PubMed] [Google Scholar]
- 134.Bakker HJ, Skinner JL. Chem Rev. 2010;110:1498–1517. doi: 10.1021/cr9001879. [DOI] [PubMed] [Google Scholar]
- 135.Mann DJ, Halls MD. Phys Rev Lett. 2003;90:195503. doi: 10.1103/PhysRevLett.90.195503. [DOI] [PubMed] [Google Scholar]
- 136.Dumitrica T, Landis CM, Yakobson BI. Chem Phys Lett. 2002;360:182– 188. [Google Scholar]
- 137.Zimmerli U, Gonnet PG, Walther JH, Koumoutsakos P. Nano Lett. 2005;5:1017–1022. doi: 10.1021/nl0503126. [DOI] [PubMed] [Google Scholar]
- 138.de Groot BL, Grubmüller H. Curr Opin Struct Biol. 2005;15:176–183. doi: 10.1016/j.sbi.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 139.Oliva R, Calamita G, Thornton JM, Pellegrini-Calace M. Proc Natl Acad Sci USA. 2010;107:4135–4140. doi: 10.1073/pnas.0910632107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Allen TW, Andersen OS, Roux B. J Am Chem Soc. 2003;125:9868–9877. doi: 10.1021/ja029317k. [DOI] [PubMed] [Google Scholar]
- 141.Allen TW, Andersen OS, Roux B. Proc Natl Acad Sci USA. 2004;101:117–122. doi: 10.1073/pnas.2635314100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Allen TW, Andersen OS, Roux B. Biophys J. 2006;90:3447–3468. doi: 10.1529/biophysj.105.077073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Podeszwa R, Buch V. Phys Rev Lett. 1999;83:4570–4573. [Google Scholar]
- 144.Grishina N, Buch V. J Chem Phys. 2004;120:5217–5225. doi: 10.1063/1.1647523. [DOI] [PubMed] [Google Scholar]
- 145.ØJensen M, Tajkhorshid E, Schulten K. Biophys J. 2003;85:2884–2899. doi: 10.1016/S0006-3495(03)74711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Li J, Gong X, Lu H, Li D, Fang H, Zhou R. Proc Natl Acad Sci USA. 2007;104:3687–3692. doi: 10.1073/pnas.0604541104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Striolo A. Nanotech. 2007;18:475704. [Google Scholar]
- 148.Liu X, Bigioni TP, Xu Y, Cassell AM, Cruden BA. J Phys Chem B. 2006;110:20102–20106. doi: 10.1021/jp0647378. [DOI] [PubMed] [Google Scholar]
- 149.Qin X, Yuan Q, Zhao Y, Xie S, Liu Z. Nano Lett. 2011;11:2173–2177. doi: 10.1021/nl200843g. [DOI] [PubMed] [Google Scholar]
- 150.Radhakrishnan R, Gubbins KE. Phys Rev Lett. 1997;79:2847–2850. [Google Scholar]
- 151.Holt J. Microfluid Nanofluid. 2008;5:425–442. [Google Scholar]
- 152.Ojamaee L, Hermansson K. J Phys Chem. 1994;98:4271–4282. [Google Scholar]
- 153.Markovitch O, Agmon N. J Phys Chem A. 2007;111:2253–2256. doi: 10.1021/jp068960g. [DOI] [PubMed] [Google Scholar]
- 154.Cox MJ, Timmer RLA, Bakker HJ, Park S, Agmon N. J Phys Chem A. 2009;113:6599–6606. doi: 10.1021/jp9004778. [DOI] [PubMed] [Google Scholar]
- 155.Vendrell O, Gelabert R, Moreno M, Lluch JM. J Phys Chem B. 2008;112:5500–5511. doi: 10.1021/jp801169z. [DOI] [PubMed] [Google Scholar]
- 156.Maurer P, Thomas V, Iftimie R. J Chem Phys. 2011;134:094505. doi: 10.1063/1.3554654. [DOI] [PubMed] [Google Scholar]
- 157.Lu D, Li Y, Rotkin SV, Ravaioli U, Schulten K. Nano Lett. 2004;4:2383–2387. [Google Scholar]
- 158.Mukherjee B, Maiti PK, Dasgupta C, Sood AK. ACS Nano. 2008;2:1189–1196. doi: 10.1021/nn800182v. [DOI] [PubMed] [Google Scholar]
- 159.Tu Y, Xiu P, Wan R, Hu J, Zhou R, Fang H. Proc Natl Acad Sci USA. 2009;106:18120–18124. doi: 10.1073/pnas.0902676106. [DOI] [PMC free article] [PubMed] [Google Scholar]