

Abstract
Like all negative-strand RNA viruses, the genome of influenza viruses is packaged in the form of viral ribonucleoprotein complexes (vRNP), in which the single-stranded genome is encapsidated by the nucleoprotein (NP), and associated with the trimeric polymerase complex consisting of the PA, PB1, and PB2 subunits. However, in contrast to most RNA viruses, influenza viruses perform viral RNA synthesis in the nuclei of infected cells. Interestingly, viral mRNA synthesis uses cellular pre-mRNAs as primers, and it has been proposed that this process takes place on chromatin1. Interactions between the viral polymerase and the host RNA polymerase II, as well as between NP and host nucleosomes have also been characterized1,2.
Recently, the generation of recombinant influenza viruses encoding a One-Strep-Tag genetically fused to the C-terminus of the PB2 subunit of the viral polymerase (rWSN-PB2-Strep3) has been described. These recombinant viruses allow the purification of PB2-containing complexes, including vRNPs, from infected cells. To obtain purified vRNPs, cell cultures are infected, and vRNPs are affinity purified from lysates derived from these cells. However, the lysis procedures used to date have been based on one-step detergent lysis, which, despite the presence of a general nuclease, often extract chromatin-bound material only inefficiently.
Our preliminary work suggested that a large portion of nuclear vRNPs were not extracted during traditional cell lysis, and therefore could not be affinity purified. To increase this extraction efficiency, and to separate chromatin-bound from non-chromatin-bound nuclear vRNPs, we adapted a step-wise subcellular extraction protocol to influenza virus-infected cells. Briefly, this procedure first separates the nuclei from the cell and then extracts soluble nuclear proteins (here termed the "nucleoplasmic" fraction). The remaining insoluble nuclear material is then digested with Benzonase, an unspecific DNA/RNA nuclease, followed by two salt extraction steps: first using 150 mM NaCl (termed "ch150"), then 500 mM NaCl ("ch500") (Fig. 1). These salt extraction steps were chosen based on our observation that 500 mM NaCl was sufficient to solubilize over 85% of nuclear vRNPs yet still allow binding of tagged vRNPs to the affinity matrix.
After subcellular fractionation of infected cells, it is possible to affinity purify PB2-tagged vRNPs from each individual fraction and analyze their protein and RNA components using Western Blot and primer extension, respectively. Recently, we utilized this method to discover that vRNP export complexes form during late points after infection on the chromatin fraction extracted with 500 mM NaCl (ch500)3.
Keywords: Virology, Issue 64, Immunology, Molecular Biology, Influenza A virus, affinity purification, subcellular fractionation, chromatin, vRNP complexes, polymerase
Protocol
A schematic flowchart of the protocol is shown in Fig. 1 and a table of reagents is presented below.
1. Infection (16 - 24 h)
Seed out HeLa cells in 5 150 mm plastic cell culture dishes in Dulbecco's modified Eagle's medium (DMEM)-high glucose medium, such that they will reach approximately 80% confluency the following day (approximately 5 x 108 cells). It is important that the cells do not reach 100% confluency.
On the next day, dilute the Strep-tagged influenza virus stock into phosphate-buffered saline (PBS) containing 0.3% bovine serum albumin (BSA) to a multiplicity of infection of 3.
Remove the growth medium from the cells, wash once with PBS, and add 10 ml PBS containing the virus to the cells. Incubate for 1 h at room temperature.
Replace the infection medium with DMEM-high glucose, and continue incubation at 37 °C.
The length of incubation depends on the phase of infection to be studied (see Discussion).
2. Subcellular Fractionation (3 h)
Wash cells once with cold PBS and then suspend cells in cold PBS with a rubber scraper and transfer to a 50 ml conical tube.
Pellet cells in a table-top centrifuge for 5 min at 1,000 rpm and then aspirate PBS.
Resuspend cell pellet in 10 ml cold sucrose buffer (10 mM HEPES pH 7.9, 10 mM KCl, 2 mM Mg Acetate (MgOAc), 3 mM CaCl2, 340 mM sucrose, 1 mM dithiothreitol (DTT), 1% protease inhibitor mix). After suspension, pass cells through a 200 μl pipette tip attached to a 10 ml plastic serum pipette to disrupt cell clumps.
Incubate 10 min on ice. Gently invert periodically to resuspend cells.
Add Nonidet P-40 (NP-40) to a final concentration of 0.5% and vortex at high speed for 15 sec. Immediately centrifuge for 10 min at 3,500 x g at 4 °C in a table-top centrifuge.
Remove supernatant and store at 4 °C. This is the cytoplasmic fraction. The pellet should be smaller and whiter than the original cell pellet. All fractions can be stored for at least 4 h at 4 °C.
Wash the pellet in 5 ml cold sucrose buffer, re-centrifuge as in step 2.5 and discard the supernatant.
Resuspend the pellet (containing nuclei) in 1.5 ml cold nucleoplasmic extraction buffer (50 mM HEPES pH 7.9, 150 mM KOAc, 1.5 mM MgCl2, 0.1% NP-40, 1 mM DTT, 1% protease inhibitor mix).
Transfer to a chilled, all-glass 4 ml Dounce homogenizer with a tight-fitting pestle and homogenize the nuclei with 20 strokes. Avoid foaming during homogenization. The resistance should increase after approximately 10 strokes. Efficient homogenization can be confirmed under a phase contrast microscope.
Transfer the homogenized nuclei to a 1.5 ml microcentrifuge tube and incubate 20 min on a rotating wheel at 4 °C.
Centrifuge at 16,000 x g in a microcentrifuge at 4 °C for 10 min.
Remove supernatant and store at 4 °C. This is the nucleoplasmic fraction.
Resuspend pellet in 1.5 ml digestion buffer (50 mM HEPES pH 7.9, 10 mM NaCl, 1.5 mM MgCl2, 1 mM DTT, 1% protease inhibitor mix, 100 U/ml Benzonase (for RNA analysis, substitute with 20 U/ml RNase-free DNase I)) pre-warmed to 37 °C, and incubate 10 min at 37 °C.
Add 42 μl of 5 M NaCl (for final concentration of 150 mM NaCl) and incubate 20 min further at 4 °C.
Centrifuge at 16,000 x g in a microcentrifuge at 4 °C for 10 min.
Remove supernatant and store at 4 °C. This is the ch150 fraction.
Resuspend pellet in 1.5 ml cold high-salt buffer (50 mM HEPES pH 7.9, 500 mM NaCl, 1.5 mM MgCl2, 0.1% NP-40, 1 mM DTT, 1% protease inhibitor mix) and incubate 20 min on ice.
Centrifuge at 16,000 x g in a microcentrifuge at 4 °C for 10 min.
Remove supernatant and store at 4 °C. This is the ch500 fraction.
3. Strep-tag Purification (2 h)
Add 300 μl of 5 M NaCl to the cytoplasmic fraction (for final concentration of 150 mM NaCl).
Aliquot 100 μl of packed Strep-Tactin Sepharose beads per fraction in a 15 ml conical tube.
Add 10 volumes of Strep wash buffer (20 mM HEPES pH 7.9, 150 mM NaCl (500 mM NaCl for ch500 samples), 1 mM EDTA) to beads, invert the tube to mix, and centrifuge 5 min at 1,000 x g in a tabletop centrifuge.
Discard the supernatant, repeat step 3.3 twice and then resuspend the bead pellet in an equal volume of Strep wash buffer.
Add 200 μl of washed Strep-Tactin bead slurry to each fraction (in 1.5 ml microcentrifuge tubes for nuceloplasmic, ch150, and ch500 fractions and a 15 ml conical tube for cytoplasmic fraction).
Rotate tubes 1h at 4 °C on a rotating wheel.
Centrifuge tubes 1 min at 1,000 x g at 4 °C. Discard supernatant.
Add 1 ml cold Strep wash buffer to each tube. Transfer beads from 15 mL tube (cytoplasmic fraction) to 1.5 mL tube. Wash all tubes 1 min at 4 °C, centrifuging after as in step 3.7. Repeat this wash step twice.
After the last wash step, remove all traces of wash buffer using a flat pipette tip. Add 100 μl elution buffer (Strep wash buffer containing 2.5 mM desthiobiotin) and gently mix.
Incubate 15 min on ice. Gently mix beads occasionally by tapping the bottom of the tubes.
Centrifuge tubes 1 min at 1000 x g at 4 °C in microcentrifuge. Collect supernatant containing eluted Strep-tagged vRNPs.
4. Representative Results
The efficiency of the fractionation is best judged by Western blot analysis of the fractionated lysates using antibodies specific for subcellular markers (Fig. 2). Specifically, successful subnuclear fractionation should show little or no cellular RNA polymerase II (Pol II) in the nucleoplasmic, or soluble nuclear fraction4, and most should be extracted with 150 mM NaCl5. A degradation of Pol II is also reproducibly observed in influenza virus-infected cells compared to uninfected cells, consistent with the literature6.
Purification of vRNPs is best judged by silver or Coomassie staining, as shown for a cytoplasmic eluate in Fig. 3. In our experience, most Strep-PB2 is purified in as part of a fully-formed vRNP, i.e. little soluble polymerase is captured. This is reflected in the high NP:PA/PB1/PB2 ratio observed by silver or Coomassie staining. A lower ratio suggests buffer contamination with RNases, as several ubiquitous RNases (such as RNase A) are known to cleave viral RNA in such a way as to dissociate the polymerase from NP multimers7. Interestingly, treatment with Benzonase alone, while sufficient to digest viral RNA, appears to have no effect on the stochiometric ratios of vRNP proteins. Although we cannot explain this phenomenon, we observed disruption of vRNPs after digestion with other RNases (data not shown), suggesting that Benzonase may leave certain structurally important RNA regions intact. After purification of vRNPs from the cytoplasm and nucleoplasm (performed without nuclease digestion), 8 faint but discrete bands at high molecular weights can be observed by silver staining, which correspond to the predicted molecular weights of the 8 influenza genome segments (see ref. 3).
The relative amounts of vRNPs purified from each fraction at 9 h post infection are shown in Fig. 4. The distribution of vRNPs varies over the course of infection, with strongest accumulation in the ch150 fraction at early time points, and increased accumulation in the nucleoplasm and cytoplasm at late time points.
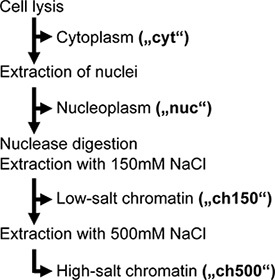 Figure 1. Flow chart of the subcellular fractionation. Adapted from ref. 3.
Figure 1. Flow chart of the subcellular fractionation. Adapted from ref. 3.
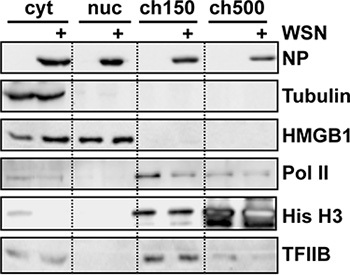 Figure 2. Western blot analysis of subcellular fractions from influenza virus-infected cells. 109 HeLa cells were infected with influenza virus strain WSN for 9 h prior to subcellular fractionation. Equal amounts of total protein were loaded in each lane and analyzed using the antibodies shown. RNA Polymerase II (Pol II) was detected using clone 8WG16, which recognizes all forms of the C-terminal domain (the hypophosphorylated band is most prominent). Adapted from ref. 3.
Figure 2. Western blot analysis of subcellular fractions from influenza virus-infected cells. 109 HeLa cells were infected with influenza virus strain WSN for 9 h prior to subcellular fractionation. Equal amounts of total protein were loaded in each lane and analyzed using the antibodies shown. RNA Polymerase II (Pol II) was detected using clone 8WG16, which recognizes all forms of the C-terminal domain (the hypophosphorylated band is most prominent). Adapted from ref. 3.
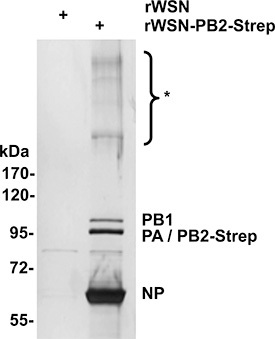 Figure 3. Silver stain analysis of purified vRNPs. HeLa cells were infected with rWSN (as a negative control) or rWSN-PB2-Strep for 9 h, followed by Strep purification from the cytoplasmic fraction. Asterisk denotes bands which are not visible after RNase digestion. Adapted from ref. 3.
Figure 3. Silver stain analysis of purified vRNPs. HeLa cells were infected with rWSN (as a negative control) or rWSN-PB2-Strep for 9 h, followed by Strep purification from the cytoplasmic fraction. Asterisk denotes bands which are not visible after RNase digestion. Adapted from ref. 3.
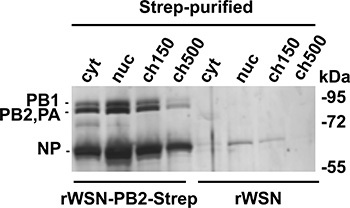 Figure 4. Silver stain analysis of vRNPs purified from different cellular fractions. 109 HeLa cells were infected with rWSN-PB2-Strep or rWSN for 9 h followed by subcellular fractionation and Strep purification. Equal amounts of eluate from each fraction were loaded. Adapted from ref. 3.
Figure 4. Silver stain analysis of vRNPs purified from different cellular fractions. 109 HeLa cells were infected with rWSN-PB2-Strep or rWSN for 9 h followed by subcellular fractionation and Strep purification. Equal amounts of eluate from each fraction were loaded. Adapted from ref. 3.
Discussion
While many studies have recently identified individual proteins or cellular networks involved in influenza virus infection8, the functional significance of the majority of these interactions remains unclear. Given the absolute dependence of chromatin-based functions for influenza virus RNA synthesis and the complex biophysical and biochemical nature of the nucleus9, new techniques will be required to elucidate these functions. The subnuclear fractionation we present here, coupled with affinity purification of vRNPs, allows the characterization of crucial viral RNA factories which were previously inaccessible.
Many subcellular fractionation protocols have been previously described in the literature. We observed that most of these traditional methods lead to premature leakage of vRNPs from the nucleus, a phenomenon also described for Pol II10. By using a short incubation of pre-swelled cells with a low concentration of a weak detergent, NP-40, efficient plasma membrane lysis occurred without significant nuclear leakage. Interestingly, performing our subcellular fractionation using A549 and HEK293T cells rather than HeLa cells led to strong nuclear leakage of vRNPs in both cell lines. Lowering the NP-40 concentration to approximately 0.2% reduced this issue; however, due to some variability of these cell lines, optimal conditions should be determined empirically. Another possible altermative is the use of non-human, influenza virus-susceptible cell lines such as MDCK or Vero cells.
Our salt extraction protocol differs from traditional chromatin extraction procedures which often use lower salt concentrations and/or high EDTA concentrations. Our protocol separates chromatin into two specific fractions, a Pol II-high/histone-low fraction and its reciprocal. However, other chromatin extraction methods may also be compatible with the affinity purification of vRNPs. Since most chromatin extraction procedures have focused on DNA-protein interactions as compared to protein-protein interactions, new techniques are required to extract tightly chromatin-bound multi-protein complexes11.
In general, the length of infection should be determined empirically due to variations among virus strains and cell lines. Using the rWSN-PB2-Strep strain at an MOI of 3, we have performed subcellular fractionation at time points varying from 3 to 10 h post infection. In our experience, less than 3 h of infection yields too few purified vRNPs for useful Western blot analysis, while infection longer than 3 h results in strong cytopathic effect and a subsequent mixing of fractions.
The ability to isolate intact, tightly chromatin-bound viral complexes opens up several possibilities. While we utilized a recombinant virus expressing a Strep-tagged PB2, other affinity tags can also be used, as well as traditional immunoprecipitation targeting other viral proteins. Furthermore, while our analysis of vRNPs was limited to the characterization of their protein and RNA components, the purified complexes could also be used to perform functional in vitro synthesis assays. Finally, the separation of vRNPs into coarse functional species through subnuclear fractionation may also allow the closer study of several recently-described post-translational modifications of viral proteins required for efficient viral RNA synthesis12,13.
Disclosures
No conflicts of interest declared.
Acknowledgments
The authors would like to thank Nada Naffakh and Marie-Anne Rameix-Welti (Institut Pasteur) for the rWSN-PB2-Strep virus.
References
- Engelhardt OG, Smith M, Fodor E. Association of the influenza A virus RNA-dependent RNA polymerase with cellular RNA polymerase II. J. Virol. 2005;79:5812–5812. doi: 10.1128/JVI.79.9.5812-5818.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Robles I, Akarsu H, Müller CW, Ruigrok R, Baudin F. Interaction of influenza virus proteins with nucleosomes. Virology. 2005;332:329. doi: 10.1016/j.virol.2004.09.036. [DOI] [PubMed] [Google Scholar]
- Chase GP, Rameix-Welti MA, Zvirbilene A, Zvirblis G, Götz V, Wolff T, Naffakh N, Schwemmle M. Influenza virus ribonucleoprotein complexes gain preferential access to cellular export machinery through chromatin targeting. PLoS Pathogens. 2011;7:e1002187. doi: 10.1371/journal.ppat.1002187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H, Tao Y, Roeder RG, Cook PR. Quantitation of RNA polymerase II and its transcription factors in an HeLa cell: little soluble holoenzyme but significant amounts of polymerases attached to the nuclear substructure. Mol. Cell. Biol. 1999;19:5383–5383. doi: 10.1128/mcb.19.8.5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Henikoff JG, Sakai A, Loeb GB, Ahmad K. Genome-wide profiling of salt fractions maps physical properties of chromatin. Genome Res. 2009;19:460. doi: 10.1101/gr.087619.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Perez-Gonzalez A, Nieto A. Influenza virus infection causes specific degradation of the largest subunit of cellular RNA polymerase II. J. Virol. 2007;81:5315–5315. doi: 10.1128/JVI.02129-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Z, Liu T, Offringa DP, McInnis J, Levandowski RA. Association of influenza virus matrix protein with ribonucleoproteins. J. Virol. 1999;73:7467–7467. doi: 10.1128/jvi.73.9.7467-7473.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Watanabe S, Kawaoka T, Y Cellular networks involved in the influenza virus life cycle. Cell Host Microbe. 2010;7:427. doi: 10.1016/j.chom.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt OG, Fodor E. Functional association between viral and cellular transcription during influenza virus infection. Rev. Med. Virol. 2006;16:329–345. doi: 10.1002/rmv.512. [DOI] [PubMed] [Google Scholar]
- Schwartz LB, Sklar VE, Jaehning JA, Weinmann R, Roeder RG. Isolation and partial characterization of the multiple forms of deoxyribonucleic acid-dependent ribonucleic acid polymerase in the mouse myeloma, MOPC 315. J. Biol. Chem. 1974;249:5889. [PubMed] [Google Scholar]
- Lambert JP, Baetz K, Figeys D. Of proteins and DNA--proteomic role in the field of chromatin research. Mol. Biosyst. 2010;6:30. doi: 10.1039/b907925b. [DOI] [PubMed] [Google Scholar]
- Wu CY, Jeng KS, Lai MM. The SUMOylation of matrix protein M1 modulates the assembly and morphogenesis of influenza A virus. J. Virol. 2011;85:6618. doi: 10.1128/JVI.02401-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao TL, Wu CY, Su WC, Jeng KS, Lai MM. Ubiquitination and deubiquitination of NP protein regulates influenza A virus RNA replication. EMBO J. 2010;29:3879. doi: 10.1038/emboj.2010.250. [DOI] [PMC free article] [PubMed] [Google Scholar]