

Background: Mapracorat is a drug candidate for ocular and skin inflammatory diseases. Its anti-inflammatory mechanism is not fully understood.
Results: Mapracorat augments MKP-1 expression, accelerates p38 deactivation, and inhibits the production of pro-inflammatory mediators.
Conclusion: Mapracorat exerts its anti-inflammatory effects, at least in part, by augmenting MKP-1.
Significance: This study highlights the therapeutic potential of augmenting the endogenous anti-inflammatory regulators.
Keywords: Cytokine, Glucocorticoid Receptor, Inflammation, MAP Kinases (MAPKs), Pharmacology, Signaling
Abstract
Mapracorat is a novel selective glucocorticoid receptor agonist (SEGRA), structurally distinct from corticosteroids. In preclinical studies, mapracorat potently inhibits the production of a variety of inflammatory mediators including cytokines and prostaglandin E2 (PGE2), with limited side effects associated with traditional corticosteroids. The objective of this study was to delineate the mechanisms underlying the anti-inflammatory properties of mapracorat. We found that mapracorat potently inhibited the production of GM-CSF and TNF-α in LPS-stimulated Raw 264.7 macrophages. Mapracorat also substantially attenuated the expression of COX-2 and the production of PGE2. The inhibition of mapracorat on the inflammatory response was dose-dependent, and substantially inhibitory effects were observed at concentrations in the 10–100 nm range. Examination of the activation kinetics of p38 and its downstream target MAPK-activated protein kinase-2 (MK-2) revealed a shortened activation course after LPS stimulation in cells pretreated with mapracorat. Supporting the notion that mapracorat augments a feedback control mechanism restraining the p38 pathway, we found that mapracorat enhanced the expression of MAPK phosphatase-1 (MKP-1), a critical negative regulator of MAPKs that drive the production of cytokines and other inflammatory mediators. While mapracorat alone did not stimulate MKP-1 expression, it markedly enhanced the expression of MKP-1 in cells stimulated by LPS, in a similar manner and potency to the augmenting effect of dexamethasone. Blocking MKP-1 expression by triptolide also abolished the accelerating effects of mapracorat on p38 and MK-2 deactivation, further supporting a role of MKP-1 in the anti-inflammatory mechanism of mapracorat. Taken together, these results indicate that mapracorat exerts its anti-inflammatory effects, at least in part, by augmenting MKP-1 expression.
Introduction
Inflammation is a common host defense response to external pathogens or tissue damage, and inflammatory processes have been implicated in a wide variety of diseases (1). While inflammation is an important physiological response for the immune defense against pathogens or tissue injury, chronic or excessive inflammation can lead to inflammatory diseases, organ damage, and in severe cases, mortality, such as in patients with severe sepsis (2). Inflammatory cytokines, such as TNF-α and GM-CSF, play a pivotal role in the inflammatory response and contribute to the induction of enzymes responsible for the physiology of the inflammatory cascade (3, 4). One of the enzymes induced during the inflammatory response is COX-2 (5–7), which is responsible for the production of prostaglandins, including PGE2, another important pro-inflammatory mediator. Signaling pathways mediating the inflammatory response during microbial infection have been studied extensively in antigen-presenting cells, such as macrophages (8–10). The conserved molecular patterns in microbes, like LPS in the cell wall of Gram-negative bacteria, are sensed by macrophages through pathogen recognition receptors, such as membrane-associated Toll-like receptors (TLRs)2 or the cytosol nucleotide binding and oligomerization domain (NOD)-like receptors. For example, LPS is detected by TLR4, which initiates a cascade of signaling events that ultimately result in activation of the transcription factor NF-κB and the MAPK pathways (8, 11). NF-κB is crucial for the transcription of numerous inflammatory cytokine genes, including TNF-α and GM-CSF. MAPKs, including ERK, JNK, and p38, can regulate the production of cytokines through both transcriptional and post-transcriptional mechanisms (12, 13). p38 was initially identified as the target of a class of small molecule inhibitors that block the production of inflammatory cytokines (14, 15), highlighting its critical function in the inflammatory response. The critical role of p38 in the inflammatory response is also illustrated by the phenotypes of the MK-2 knock-out mice (16, 17). MK-2 is the downstream target of p38, and knock-out of MK-2 abrogates the production of inflammatory cytokines and protects mice from endotoxemia (16).
It has been shown that the p38/MK-2 pathway can stimulate cytokine biosynthesis through at least three mechanisms. First, p38 stimulates the activity of the transcription factor AP-1, which plays a significant role in the induction of inflammatory genes (18, 19). Second, the 3′-untranslated region (UTR) of many pro-inflammatory mediator mRNAs contains AU-rich elements (AREs) that render these mRNAs susceptible to degradation (17, 20–22). Like the mRNA transcripts of many pro-inflammatory cytokines, COX-2 mRNA also contains several AREs in its 3′-UTR (23). The p38/MK-2 cascade enhances the inflammatory response by phosphorylating proteins that mediate the degradation of ARE-containing mRNA, such as tristetraprolin (17, 20–23). Third, the p38/MK-2 cascade also promotes the translation of ARE-containing mRNAs through the activation of the eukaryotic translation initiation factor 4E (24, 25).
The activity of p38 is regulated via reversible phosphorylation (12, 26). The activation of p38 is catalyzed by MKK3 and MKK6 through phosphorylation of the adjacent threonine and tyrosine residues on the TGY signature motif (27). The inactivation of p38 is mediated by dephosphorylation of the adjacent threonine and tyrosine residues catalyzed primarily by MAPK phosphatases (MKPs) (13, 28). It has been shown independently by several laboratories that MKP-1 plays an essential role in the deactivation of p38 and acts as a restraint for the production of inflammatory cytokines (29–33). MKP-1-deficient macrophages exhibit prolonged activation of p38 and MK-2 following LPS stimulation, and produce substantially greater quantities of pro-inflammatory cytokines, including TNF-α and GM-CSF (30–33). These cells also express substantially greater level of COX-2 (34, 35). While p38 and MK-2 inhibitors are still considered as promising anti-inflammatory agents, the development of such drugs has so far been hindered by their toxicities, particularly in the lymphoid and gastrointestinal system (36, 37).
Glucocorticoids are powerful anti-inflammatory drugs, which modulate the expression of genes involved in inflammation through both transrepression and transactivation mechanisms (38–42). While transcriptional repression of pro-inflammatory cytokines is widely accepted as the primary anti-inflammatory mechanisms (41, 42), such a mechanism occurs slowly, usually taking days or weeks to reach their maximal anti-inflammatory effects. In addition, glucocorticoids have also been shown to rapidly (within hours) inhibit the production of pro-inflammatory cytokines, associated with the inhibition of both p38 and JNK activities (41, 43, 44). Interestingly, several laboratories have shown that glucocorticoids enhance the expression of MKP-1 in a variety of cell types, including epithelial cells, mast cells, and macrophages (29, 43, 45). While glucocorticoids alone only modestly enhance the expression of MKP-1 (29, 46, 47), they potently enhance the expression of MKP-1 in LPS-stimulated macrophages (46, 47). Furthermore, deletion of MKP-1 abrogated the inhibitory effects of glucocorticoids on both p38 and JNK and substantially compromised the inhibitory effects on the production of inflammatory cytokines both in cultured macrophages and in mice after LPS challenge (46–48). While glucocorticoids as a class of potent anti-inflammatory drugs continue to be used for many inflammatory diseases, the application of glucocorticoids is limited by their side effects (41). Long-term glucocorticoid treatment can lead to severe and sometimes irreversible pathological consequences, including osteoporosis, diabetes, Cushing's syndrome, glaucoma, and muscle atrophy. Therefore, identification of small molecules that inhibit the p38/MK-2 pathway and yet are devoid of the undesirable side effects of current anti-inflammatory agents represents a major focus in the development of new anti-inflammatory drugs.
Mapracorat, also referred to as BOL-303242-X and ZK 245186 (49–51), is a small molecule compound in clinical trials for the treatment of inflammatory skin and eye diseases. Mapracorat exhibits potent anti-inflammatory activity both in vitro and in vivo (49, 50), yet the molecular mechanisms underlying the potent anti-inflammatory function has not been fully explored. In the present study, we demonstrated that mapracorat inhibited the production of inflammatory mediators including cytokines TNF-α and GM-CSF as well as PGE2 in LPS-stimulated macrophages. Examination of the effects of mapracorat on MAPK signaling revealed that mapracorat enhanced the expression of MKP-1, and inhibited p38 and its downstream kinase MK-2. Blockade of MKP-1 induction in LPS-stimulated macrophages by triptolide abolished MKP-1-mediated p38 deactivation, and sustained MK-2 activity. Finally, we showed that mapracorat enhanced MKP-1 induction in LPS-stimulated macrophages with a potency similar to dexamethasone. Our studies indicate that mapracorat exerts its anti-inflammatory effects not only through the inhibition of inflammatory mediators TNF-α, GM-CSF, COX-2, and PGE2, but also through augmentation of expression of anti-inflammatory mediators such as MKP-1.
EXPERIMENTAL PROCEDURES
Cell Culture and Reagents
Raw 264.7 murine macrophages were acquired from the American Type Culture Collection (ATCC, Manassas, VA) and were maintained in DMEM (Invitrogen, Carlsbad, CA) supplemented with 10% FBS (Hyclone, Logan, UT) and penicillin/streptomycin (Invitrogen). LPS (Escherichia coli, O55:B5) was purchased from EMD/Calbiochem and dissolved in PBS containing 0.1% BSA to a concentration of 10 mg/ml. Mapracorat (R-1,1,1-trifluoro-4-(5-fluoro-2,3-dihydrobenzofuran-7-yl)-4-methyl-2-{[(2-methyl-5-quinolyl)amino]methyl}pentan-2-ol) (51) was synthesized by Bayer Schering Pharma (Berlin, Germany). Both dexamethasone (Sigma-Aldrich) and mapracorat were dissolved in DMSO (Sigma-Aldrich) to a concentration of 100 μm. For the time course, cells were pretreated with 100 nm mapracorat for 4 h. Triptolide was acquired from Tocris Bioscience and prepared at a concentration of 10 mm DMSO. Cells were pretreated with triptolide for 60 min prior to treatment with LPS or mapracorat. The MKP-1 (M-18) and β-actin antibodies were both purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies specifically against phosphorylated p38 (T180/Y182), p38, phosphorylated MK-2, MK-2, COX-2, and HRP-conjugated anti-rabbit IgG secondary antibody were purchased from Cell Signaling (Danvers, MA). The GAPDH antibody was purchased from Invitrogen. HRP-conjugated goat anti-mouse IgG (H+L) secondary antibody was purchased from Bio-Rad.
Western Blotting
Following treatments, cell culture plates were aspirated of their media and washed twice with ice-cold PBS (Invitrogen). Cells were incubated on ice for 15 min with cold cell lysis buffer (Cell Signaling) supplemented with Proteinase Inhibitor Mixture (Sigma-Aldrich) and 1 mm PMSF (Sigma-Aldrich). Cell lysates were collected and centrifuged for 10 min at 15,000 × g at 4 °C. Following centrifugation, the supernatant was transferred to fresh microfuge tubes and assayed for protein concentration using a BCA protein assay kit (Pierce). Cell lysates were mixed with Laemmli Sample Buffer and boiled at 95 °C for 5 min, and the proteins were resolved by electrophoresis on 10% SDS-polyacrylamide gels. Proteins in the gels were wet-transferred to nitrocellulose. After blocking the membrane with either 5% blotting grade nonfat dry milk (Bio-Rad) or 5% BSA (EMD/Calbiochem) in TBS containing 0.1% Tween-20 (TBST), the membrane was incubated with primary antibody overnight at 4 °C. The blot was then washed, and incubated with HRP-conjugated secondary antibody. Following extensive washing, the blot was exposed to enhanced chemiluminescent substrate (Pierce). Digital images were collected using the VersaDoc 4000 MP imaging system and Quantity One software (Bio-Rad). The primary antibodies were removed from the blots by incubating with a stripping buffer (Pierce) and were re-probed with an antibody against β-actin or GAPDH, for loading controls. Digital images were processed using the Image Lab software (Bio-Rad).
Cytokine and PGE2 Detection
Cells (1 × 106 cells per well) were seeded into 6-well plates and grown to ∼80% confluence prior to treatment. Following treatments, cell culture medium was collected, centrifuged for 5 min at 800 × g, and stored at −70 °C. Cytokines in the medium were assayed by using multiplex bead technology, with a mouse cytokine/chemokine kit (Millipore, Bellerica, MA) according to the manufacturer's instructions. Briefly, samples were incubated overnight with antibody-immobilized beads at 4 °C on a 96-well filter plate. Samples were subsequently incubated with biotinylated detection antibodies followed by streptavidin-phycoerythrin conjugate, the reporter molecule. Data were acquired using the Luminex 200 (Luminex, Austin, TX), and converted to cytokine concentrations using the Milliplex Analyst software (Millipore) based on a standard curve.
PGE2 release was evaluated in the same set of samples assayed for cytokine contents. The concentration of PGE2 was determined using a highly sensitive PGE2 competitive ELISA kit (Enzo Life Sciences, Farmingdale, NY), according to the manufacturer's instructions. The colors were developed using p-nitrophenyl phosphate substrate, and the optical density was measured at 405 nm using a SpectraMAX2 plate reader (Molecular Devices, Sunnyvale, CA). The concentration of PGE2 in the samples was calculated based on a standard curve using StatLia workflow software (Brendan Technologies, Carlsbad, CA).
Statistical Analysis
Data were analyzed by two-way ANOVA, and differences were identified via contrast test using the JMP 8 statistical analysis software (SAS Institute, Cary, NC). A p value < 0.05 is considered as significant.
RESULTS
Mapracorat Inhibits the Production of Pro-inflammatory Mediators in LPS-stimulated Raw 264.7 Macrophages
Mapracorat is a novel small molecule with a potent anti-inflammatory activity, structurally distinct from glucocorticoids (Fig. 1A). Mapracorat, at a concentration of 100 nm, inhibited the production of pro-inflammatory cytokines, GM-CSF and TNF-α, in LPS-stimulated Raw 264.7 cells (Fig. 1, B and C). The inhibition occurred within 4 h. The marked attenuation of the production of both GM-CSF and TNF-α over a 16-h period indicated that the inhibitory effect of mapracorat was sustained within the period examined. Dose response studies indicated that the inhibition of mapracorat on the inflammatory cytokine production was dose-dependent (Fig. 1D). A significant inhibition of GM-CSF by mapracorat was seen at a dose as low as 10 nm, and it reached to its maximal inhibition at ∼100 nm (Fig. 1D).
FIGURE 1.

Mapracorat inhibits the production of TNF-α and GM-CSF in LPS-stimulated Raw 264. 7 cells. A, molecular structure of mapracorat. B, inhibitory effect of mapracorat on TNF-α production. Raw 264.7 cells were treated with LPS (100 ng/ml) or with LPS (100 ng/ml) and mapracorat (100 nm) for 4, 8, and 16 h. Medium was harvested and assayed for TNF-α concentration. C, inhibitory effect of mapracorat on GM-CSF production. Cells were treated as in B, and the medium was assayed for GM-CSF. D, dose-dependent inhibition of GM-CSF production by mapracorat in LPS-stimulated cells. Cells were treated with DMSO (control in the bar on the left), or 100 ng/ml LPS (bar graph on the left), 100 nm mapracorat (bar graph on the left), or LPS plus different concentration of mapracorat (points on the line graph on the right), for 18 h. The values are expressed as means ± S.E. of three independent experiments. *, p < 0.05, compared with cytokine production in the absence of mapracorat at the same time point. †, p < 0.05, compared with cytokine production by the control cells. ‡, p < 0.05, compared with cells treated with LPS.
COX-2 is responsible for the production of prostaglandins, including PGE2 (5). In unstimulated RAW 264.7 cells, COX-2 protein was not detectable. LPS stimulation resulted in a marked increase in COX-2 protein levels within 4 h (Fig. 2A). Mapracorat, at a concentration of 100 nm, significantly attenuated the protein levels of COX-2 in LPS-stimulated Raw 264.7 cells. Stimulation with LPS for 8 or 16 h further enhanced COX-2 expression (Fig. 3B). The potent inhibitory effects of mapracorat on COX-2 were reflected on the production of PGE2. The PGE2 concentration in the medium was undetectable prior to LPS stimulation. Upon LPS stimulation, PGE2 concentration was dramatically increased, reaching 1.3 and 4.6 ng/ml at 8 and 16 h, respectively (Fig. 3C). In the presence of mapracorat, PGE2 production in response to LPS stimulation was decreased by more than 70%.
FIGURE 2.
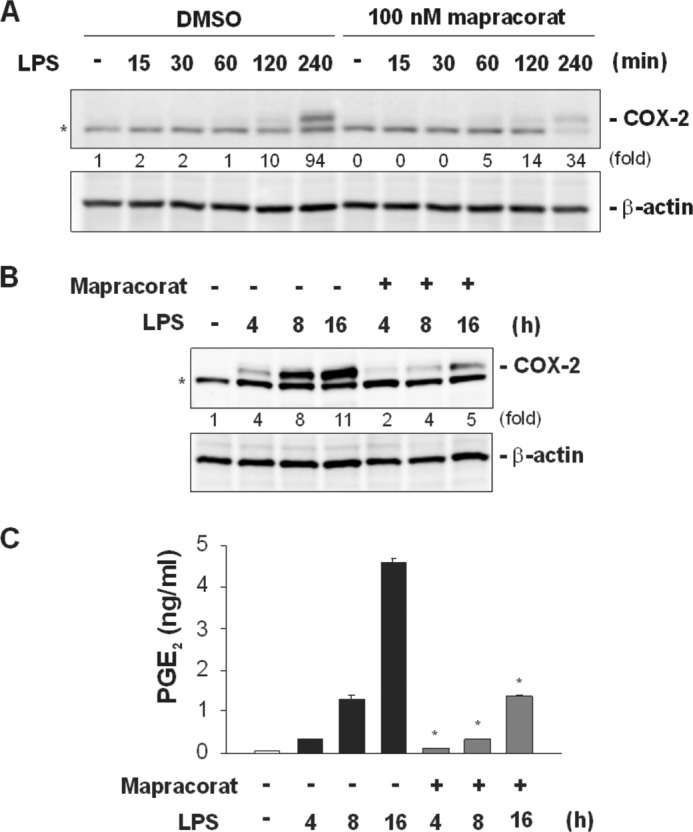
Mapracorat attenuates the induction of COX-2 and decreases PGE2 release in LPS-stimulated Raw 264. 7 cells. A, effects of mapracorat on the kinetics of early COX-2 induction in LPS-induced macrophages. Cells were pretreated for 4 h with either vehicle (DMSO) or mapracorat (100 nm), and then stimulated with LPS. Cells were harvested at the indicated time points. COX-2 protein in the cell lysates was detected by Western blotting, using a rabbit polyclonal antibody (upper panel). The membrane was then stripped and blotted with an antibody against β-actin (lower panel), to verify comparable protein loading. B, effects of mapracorat on the later COX-2 induction. Cells were treated with LPS or LPS and 100 nm mapracorat for 4, 8, and 16 h. COX-2 protein in the cells at different time points was detected by Western blotting (upper panel). The β-actin blot serves as a loading control (lower panel). In A and B, the intensity of the bands was quantified by densitometry. The signals of the bands of interest were normalized to the loading reference bands. Values presented below the blots represent fold-increase relative to the controls (DMSO treated, without LPS treatment, whose level was set equal to 1). C, inhibition of mapracorat on PGE2 production. Cells were treated as in B, medium was harvested and assayed for PGE2 contents, by using a competitive ELISA kit. Values in the graph are means ± S.E. of three independent experiments. *, p < 0.05, compared with PGE2 production in the absence of mapracorat at the same time point. Representative images are presented in A and B. The band marked by an asterisk in A and B is a nonspecific band.
FIGURE 3.

Mapracorat enhances LPS-induced MKP-1 expression and accelerates p38/MK-2 deactivation in Raw 264. 7 cells. A, effects of mapracorat on the kinetics of p38 and MK-2 activation in macrophages following LPS stimulation. Cells were pretreated with 100 nm mapracorat or vehicle (DMSO) for 4 h prior to stimulation with LPS (100 ng/ml). Cell lysates were collected at the indicated time points, and p38 and MK-2 activation was analyzed by Western blot, using antibodies against phospho-p38 (top panel) and phospho-MK-2 (third panel). The membranes were then stripped and blotted with antibodies against total p38 (second panel) or total MK-2 (bottom panel), to verify comparable sample loading. B, effect of mapracorat on the accumulation of MKP-1 shortly after LPS stimulation. Cells were treated as in A, MKP-1 accumulation were examined by Western blot, using an antibody against MKP-1 (upper panel). The membrane was stripped and reprobed with the β-actin antibody (lower panel), for confirming comparable sample loading. In A and B, the intensity of the bands was quantified by densitometry. The signals of the bands of interest were normalized to the loading reference bands. Values presented below the blots represent fold-increase relative to the controls (DMSO treated, without LPS treatment, whose level was set equal to 1). C, dampened oscillatory characteristics of phospho-p38 and MKP-1 levels in macrophages following LPS stimulation. Cells were stimulated with LPS (100 ng/ml) and harvested at the indicated time. Phospho-p38 and MKP-1 were detected by Western blot. Comparable loading is indicated by the similar intensities of the nonspecific band on the MKP-1 blot (marked by an asterisk, lower panel). The intensities of phospho-p38 and MKP-1 were quantified by densitometry. The reciprocal relationship between phospho-p38 and MKP-1 is depicted in the graph underneath the blots. D, effect of mapracorat on MKP-1 accumulation over a longer time frame. LPS (100 ng/ml) was added to the medium together with either vehicle (DMSO) or mapracorat (100 nm) in the cell culture. Cell lysates were prepared at the indicated time points. MKP-1 levels in the lysates were detected by Western blotting. Results presented are representative images.
Mapracorat Augments MKP-1 Induction and Shortens the Course of p38 Activation in LPS-stimulated Macrophages
MAPKs are critical for the production of pro-inflammatory cytokines, through both transcriptional and post-transcriptional mechanisms (12, 26). MAPKs, particularly p38, are also pivotal in the induction of COX-2 (23, 52, 53). Since both the cytokine production and COX-2 induction in LPS-stimulated macrophages were inhibited by mapracorat, we examined the effects of mapracorat on p38 and its downstream target MK-2 (Fig. 3A). Raw 264.7 cells were first pretreated with 100 nm mapracorat for 4 h, and then stimulated with LPS for different times. The activities of p38 MAPK and its downstream kinase MK-2 were assessed by Western blot analyses, using phosphospecific antibodies. In cells pretreated with vehicle (DMSO), LPS stimulation led to rapid p38 activation. p38 was activated within 15 min, indicated by the dramatic increase in phospho-p38 level. Maximal p38 activity persisted at 15–30 min, and substantially declined by 60 min, although considerable p38 activity was retained up to 120 min post LPS stimulation. By 4 h post-LPS addition, p38 activity returned to basal levels. Similarly, MK-2 activity was rapidly increased in response to LPS stimulation within 15 min, and then gradually declined after 30 min, in cells pretreated with vehicle. While pretreatment of Raw 264.7 cells with mapracorat did not change either the basal or maximal p38 activity (at 15 min), it accelerated the decline of p38 activity after the initial activation. The activity of p38 in cells pretreated with mapracorat was already lower than in cells pretreated with vehicle (DMSO) 30 min after LPS stimulation. By 60 min, p38 activity in mapracorat-pretreated cells already returned to the basal levels, while at this time point p38 activity in vehicle-pretreated cells was still markedly higher than the basal levels. Likewise, mapracorat also accelerated the deactivation of MK-2 following LPS stimulation.
In mammalian cells, MAPK activity is positively regulated by MAPKKs and negatively regulated by MKPs. It has been shown that MKP-1 plays a pivotal role in the deactivation of both p38 and JNK, and acts as a negative regulator of the production of inflammatory cytokines in macrophages (30–33, 54). Since mapracorat accelerated the deactivation of p38 in LPS-stimulated macrophages, we examined the effect of mapracorat on MKP-1 expression (Fig. 3B). In control cells, MKP-1 protein was not detectable. In the absence of mapracorat, LPS stimulation resulted in a robust increase in MKP-1 protein level by 60 min, which coincided with the substantial decline in p38 activity (Fig. 3A). The MKP-1 protein levels then markedly subsided by 120 min, and further declined at 240 min. While mapracorat alone did not elevate MKP-1 protein levels, it substantially accelerated the accumulation of MKP-1 protein in response to LPS stimulation (Fig. 3B). MKP-1 protein in mapracorat-pretreated cells was detectable at as early as 15 min after LPS stimulation. By 30 min, MKP-1 protein levels became substantial. MKP-1 protein also reached its maximal level at 60 min after LPS stimulation. It should be pointed out that the maximal MKP-1 protein level at 60 min post LPS addition in mapracorat-pretreated cells was substantially higher than in vehicle-pretreated cells. Most importantly, the substantial increase in MKP-1 protein level was associated with an accelerated dephosphorylation of p38, whose activity returned to basal level by 60 min. In contrast, in cells pretreated with the vehicle (DMSO), significant p38 activity remained at least up to 2 h post-LPS stimulation.
In cells treated with LPS alone, MKP-1 expression behaved in a dampened oscillation pattern: it reached its peak level first about 60 min post-LPS induction, and then declined (Fig. 3C). At a later time (6 h), MKP-1 levels were elevated again. Likewise, p38 activity also exhibited a pattern of oscillatory decay: p38 activity reached its peak 15–30 min following LPS stimulation, then substantially declined by 60 min, corresponding to the peak level of MKP-1. The p38 activity was slightly increased again at a later time (2–4 h) and plummeted again at 6 h. The perfect association between the increase in MKP-1 protein levels and the corresponding decrease in p38 activity depicted the critical role of MKP-1 in p38 regulation during the return to homeostasis. As the level of MKP-1 plays a critical role in shaping the course of p38 activation, we assessed whether the manner of mapracorat treatment also affects the MKP-1 level, particularly over a longer period of time. This is important since p38 also plays an important role in the regulation of many later inflammatory response genes, such as COX-2 (23, 52, 53). For these experiments Raw 264.7 cells were stimulated with LPS together with mapracorat or vehicle (DMSO) over 16 h. Mapracorat augmented MKP-1 expression not only when it was added into the cell culture 4 h prior to LPS stimulation, it also enhanced MKP-1 expression when mapracorat was added into the culture together with LPS (Fig. 3D). More importantly, mapracorat shifted the kinetics of MKP-1 induction. In cells stimulated by LPS, a second peak of MKP-1 was reached at about 8 h. However, in the presence of mapracorat, MKP-1 level at 4 h was higher than at 8 h, suggesting an accelerated process reaching the second peak. Since many later inflammatory events, including induction of COX-2, are mediated by p38 and MK-2 (23, 52, 53), alteration of MKP-1 induction would have profound effects on the inflammatory program.
Triptolide Abolishes the Accelerating Effects of Mapracorat on MAPK Deactivation in LPS-stimulated Macrophages
Previously, it has been shown that triptolide blocks the induction of MKP-1 in LPS-stimulated macrophages (29). To examine the importance of MKP-1 on the anti-inflammatory effects of mapracorat, we used triptolide to block MKP-1 induction by LPS in the presence of mapracorat, and assessed the effect of mapracorat on p38 deactivation (Fig. 4). If mapracorat exerts its effects by modulating MKP-1 expression, blockade of MKP-1 expression should abolish the accelerating effects of mapracorat on p38 deactivation. We treated cells with mapracorat or vehicle (DMSO) with LPS for 30 or 60 min. As expected, stimulation of Raw 264.7 cells with LPS for 30 min triggered a substantial increase in p38 activity. By 60 min post-LPS stimulation, p38 activity significantly declined, which was associated with a marked accumulation of MKP-1 protein. MKP-1 protein levels in cells treated with mapracorat and LPS became detectable at 30 min, and reached a substantially higher level by 60 min post-LPS stimulation than in cells treated with LPS alone. As expected, p38 activity at 60 min post-LPS stimulation in cells pretreated with mapracorat was considerably lower than in cells pretreated with DMSO. Pretreatment of cells with triptolide abolished the induction of MKP-1 by either LPS alone or LPS plus mapracorat. Abolishing MKP-1 induction by triptolide not only protected p38 from inactivation in LPS-stimulated macrophages but also in cells stimulated with both LPS and mapracorat, supporting a contributory role of MKP-1 augmentation in the anti-inflammatory activity of mapracorat. Likewise, pretreatment with triptolide also sustained the activity of MK-2 in cells treated with either LPS alone or with LPS plus mapracorat. These results suggest that the ability of mapracorat to accelerate p38 deactivation is dependent on its augmentative effects on MKP-1 induction.
FIGURE 4.
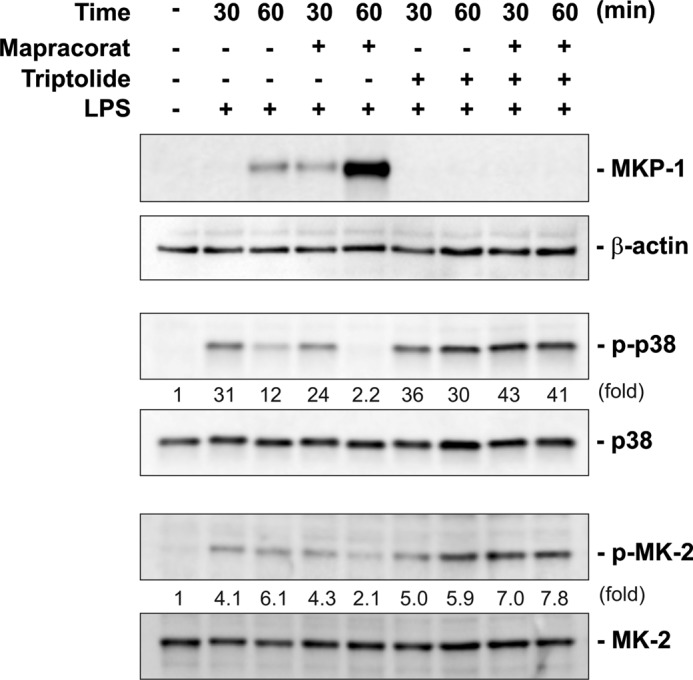
Triptolide blocks MKP-1 induction and abolishes the ability of mapracorat to accelerate p38 deactivation in LPS-stimulated Raw 264. 7 macrophages. Cells were pre-incubated with 1 μm triptolide or vehicle (DMSO) for 60 min and then treated with LPS or LPS and mapracorat simultaneously for either 30 or 60 min. The cell lysates were subjected to Western blot analyses, for detection of MKP-1, phospho-p38, and phospho-MK-2. The membranes were stripped and blotted with antibodies against β-actin, total p38, and total MK-2, to verify comparable protein loading. Results presented are representative images. The intensity of the bands was quantified by densitometry. The signals of the bands of interest were normalized to the loading reference bands. Values presented below the blots represent fold-increase relative to the controls (DMSO treated, without LPS treatment, whose level was set equal to 1).
Mapracorat Enhances MKP-1 Expression in LPS-stimulated Macrophages with Potency Similar to Dexamethasone
Glucocorticoids are powerful anti-inflammatory drugs currently used in the clinic for the treatment of many inflammatory diseases (41, 42). It has been shown that the immediate anti-inflammatory function of glucocorticoids is dependent on MKP-1 induction (46–48). Moreover, the anti-inflammatory potencies of glucocorticoids are found to be proportional to their ability to induce MKP-1 (54). Since both glucocorticoids and mapracorat induce MKP-1, we compared mapracorat with dexamethasone for the augmentation of MKP-1 expression. As expected, dexamethasone robustly enhanced MKP-1 protein levels in LPS-stimulated RAW 264.7 macrophages (Fig. 5). The enhancing effect of dexamethasone was observed at a concentration as low as 10 nm. Increasing dexamethasone to 100 nm further augmented MKP-1 expression. Mapracorat also strongly enhanced the induction of MKP-1, and the potency of mapracorat on MKP-1 induction is comparable to that observed for dexamethasone. At a concentration of 10 nm, mapracorat markedly enhanced MKP-1 expression in LPS-stimulated macrophages, and increasing mapracorat to 100 nm further augmented MKP-1 expression.
FIGURE 5.
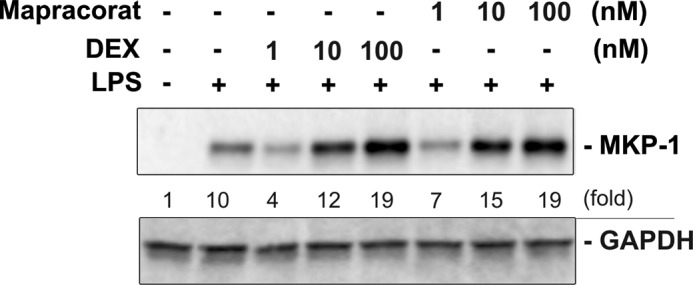
Mapracorat augments LPS-induced MKP-1 expression with potency similar to dexamethasone in Raw 264.7 macrophages. Cells were first pretreated with vehicle, different concentrations of dexamethasone, or different doses of mapracorat for 4 h, and then stimulated with LPS (100 ng/ml) for 60 min. Cell lysates were collected, and samples were assessed for protein expression by Western blotting using an antibody specific to MKP-1. The blots were stripped and re-probed using a GAPDH-specific antibody, to verify comparable loading. Results presented are representative images. The intensity of the bands was quantified by densitometry. The signals of the bands of interest were normalized to the loading reference bands. Values presented below the blots represent fold-increase relative to the controls (DMSO treated, without LPS treatment, whose level was set equal to 1).
DISCUSSION
Mapracorat is under development as a drug candidate for the treatment of inflammatory skin and eye diseases (49–51). A number of preclinical studies in both cultured cells and animal models have demonstrated that mapracorat is a potent anti-inflammatory agent with limited side effects associated with traditional corticosteroids such as elevation of intraocular pressure (IOP) in ophthalmology (50, 55, 56). In vitro quantitative assays of myocilin expression in trabecular meshwork (TM) cells can be used for characterizing anti-inflammatory drugs that are glucocorticoid receptor (GR) ligands. It has been reported previously that, compared with traditional steroids such as dexamethasone, therapeutic doses of mapracorat elicit a reduced myocilin expression in TM cells by virtue of the partial agonist properties of this compound (57). It has also been reported previously that mapracorat shows full anti-inflammatory efficacy (similar to dexamethasone) in experimental models of dry eye and postoperative inflammation in rabbits while demonstrating reduced effects in IOP and body weight (55). Taken together, these data indicate that mapracorat, a selective glucocorticoid receptor agonist (SEGRA), shows efficacy similar to that of traditional steroids while exhibiting an improved side effect profile.
Although the mechanism underlying the potent anti-inflammatory effects of mapracorat has not been fully explored, we have reported previously that incubation of human corneal epithelial cells with mapracorat inhibits hyperosmolar-induced cytokine release, and increased phosphorylation of p38 and JNK MAPKs caused by hyperosmolarity is inhibited by mapracorat. This inhibition is reversed by the glucocorticoid receptor antagonist mifepristone (RU-486) (50). Mapracorat also significantly decreases the hyperosmolar-induced increase in NFκB and AP-1 transcriptional activity (50). In the present study, we showed that mapracorat potently inhibited the production of inflammatory cytokines, TNF-α and GM-CSF, in macrophages stimulated with LPS (Fig. 1). The expression of COX-2 and the production of PGE2 were also substantially attenuated by mapracorat in LPS-stimulated macrophages (Fig. 2). Since p38 MAPK and its downstream target, MK-2, are critical for the production of pro-inflammatory cytokines and for the induction of COX-2 (12, 13, 23), we examined the effects of mapracorat on the activities of these kinases (Fig. 3). We found that although it did not inhibit the initial activation of p38 and MK-2 in LPS-stimulated macrophages, mapracorat accelerated the decline of p38 and MK-2 activity (Fig. 3). To understand the molecular mechanisms underlying the accelerated deactivation of p38, we examined the effects of mapracorat on the expression of MKP-1, which is induced in response to LPS. We found that mapracorat potently enhanced MKP-1 induction in LPS-stimulated macrophages, and the robust induction of MKP-1 coincided with the deactivation of p38 (Fig. 3). Previously, it has been demonstrated that the deactivation of p38 within 60 min in LPS-stimulated macrophages is mediated by MKP-1 (30–33), and knock-out of MKP-1 prolonged the activation of p38 and its downstream target, MK-2 (30). To evaluate the role of MKP-1 in the accelerated deactivation of p38, we took advantage of the fact that MKP-1 induction can be blocked by triptolide (29), a diterpene triepoxide. We found that blockade of the induction of MKP-1 by triptolide abrogated the inhibitory effects of mapracorat on p38 activity (Fig. 4), suggesting that the enhancing effect of mapracorat on p38 deactivation is dependent on MKP-1. Taken together, our results strongly support the idea that mapracorat exerts its anti-inflammatory action, at least in part, by augmenting MKP-1 expression. Joanny E. et al. have reported recently that two selective GR modulators (SGRMs), Cpd1 and Cpd2, are able to up-regulate dual specificity phosphatase 1 (DUSP1; MKP-1) in several cell types, and this response correlates with the ability of the compounds to suppress COX-2 expression. Several anti-inflammatory effects of SGRMs are ablated or significantly impaired in Dusp1−/− macrophages (58).
MAPKs are critical regulators of myriad cellular processes, including cell proliferation, metabolism, differentiation, and stress response (12, 59, 60). MAPK pathways are not only regulated by a linear kinase-mediated activation cascade, but also controlled by many feedback loops and a variety of crosstalks with other signaling pathways (13, 28, 61). Through sophisticated regulatory networks, MAPKs are tightly controlled to prevent dysregulation of the various cellular processes. Because of the complex function of MAPKs in the various cellular responses, it is not surprising that many MAPK inhibitors have failed in clinical trials as therapeutic drug candidates (36, 37, 61). The pharmacological inhibitors of p38 were initially considered as promising drug candidates for a variety of inflammatory diseases (14, 62, 63). However, the enthusiasm for p38 inhibitors as therapeutic drug candidates was dampened by their severe toxicity in the liver, gastrointestinal and lymphoid systems (36, 37). Likewise, similar toxicity was also observed with MK-2 inhibitors in preclinical investigations (37). Since MKPs play a critical role in determining both the magnitude and the duration of MAPKs, modulation of MKP offers a wide window for the manipulation of MAPK activities, and potentially avoids the side effects associated with MAPK inhibitors.
MKP-1, which prefers p38 and JNK as substrates (64, 65), is an endogenous feedback control regulator of p38. It acts to restrain the production of pro-inflammatory cytokines (30–33). MKP-1 is not expressed in unstimulated innate immune effector cells, such as macrophages, monocytes, and dendritic cells. Upon microbial infection p38 undergoes a robust activation, allowing the initiation of robust innate immune responses, including production of pro-inflammatory cytokines (13, 28, 66). MKP-1 is induced at a slower pace than p38 activation (Fig. 3). Unlike p38 activation, which relies on modification of pre-existing proteins, MKP-1 protein accumulation depends on de novo gene transcription and protein synthesis. For this reason, MKP-1-mediated p38 deactivation occurs in a delayed manner (Fig. 3). This brief delay likely sets the magnitude and temporal course of the inflammatory response that adequately facilitates the clearance of the invading pathogens yet avoids the collateral damage of excessive inflammation. The pivotal role of MKP-1 in shaping the p38 activation course in the innate immune system is consistent with the findings from mathematical modeling (67), which demonstrated that constrained MAPK signaling occurs by controlling MKP expression, rather than through MAPK activation. Using mathematical modeling in combination with growth factor-stimulated NIH 3T3 cells, Bhalla et al. have shown that at higher MKP levels, MAPK signaling system behaves as a proportional response system (67). If this mathematical model is applicable to the innate immune system, we would predict that in the presence of higher MKP-1 levels the p38-mediated inflammatory response will be elicited in proportion to the amount of pathogen detected. Such prediction is consistent with clinical observations that overwhelming inflammation only occurs in patients with severe sepsis. Because of the critical role of MKP-1 in the control of the inflammatory response, it is not surprising that some endogenous immune regulators modulate the innate immune response through altering the activity of MKP-1 (13). For example, the pro-inflammatory cytokines IFN-γ and macrophage migration inhibitory factor (MIF) inhibit MKP-1 induction (30, 68, 69), while the anti-inflammatory cytokine IL-10 and glucocorticoids booster MKP-1 expression (29, 47, 70). As MKP-1 expression level shapes the course of p38 activation, it is not surprising that agents capable of enhancing MKP-1 expression, such as mapracorat will substantially inhibit the production of proinflammatory cytokines, the expression of COX-2, and biosynthesis of PGE2 in LPS-stimulated macrophages (Figs. 1 and 2). Since MKP-1 expression levels vary substantially in different organs, enhancing MKP-1 in a selective manner may avoid the toxicity observed with p38 inhibitors, which block p38 activity in all organs. From this point, the profile of mapracorat is particularly favorable, since augmentation of MKP-1 induction by mapracorat is dependent on the LPS stimulation (Fig. 3). The synergistic nature of MKP-1 enhancement by mapracorat in a sense serves as a targeting mechanism to intervene on the most appropriate cells where MKP-1 is needed for the feedback control of MAPKs.
It is clear that MKP-1 plays a role in the anti-inflammatory function of mapracorat since abrogating MKP-1 expression using triptolide affects the inhibitory effects of mapracorat on p38 activity (Fig. 4). However, the underlying mechanism responsible for the augmenting effects of mapracorat on MKP-1 expression remains unclear. On the mouse MKP-1 promoter, there are at least three putative GR-binding elements (45, 71). We speculate that mapracorat-bound GR may interact with the GR-binding elements in the MKP-1 promoter to enhance MKP-1 expression. Like glucocorticoids, which only have a very weak effect on MKP-1 expression by themselves (29, 47), mapracorat alone also had little effect on MKP-1 expression under basal condition (Fig. 3B). Both mapracorat and dexamethasone (47) synergistically interact with LPS to induce MKP-1 expression (Fig. 5). This property is also somewhat similar to that of IL-10, a potent anti-inflammatory cytokine. Like mapracorat, IL-10 substantially enhances the expression of MKP-1 in LPS-stimulated macrophages, although IL-10 by itself does not alter MKP-1 expression (70).
MKP-1 expression can be regulated at several levels, including gene transcription, protein stability, and phosphatase activity. This multi-level regulation allows for tight control of MAPK activities (13). It has also been reported that dexamethasone increases the expression of MKP-1 gene at the promoter level, and attenuates proteasomal degradation of MKP-1 (45). However, it is not clear whether the effect of mapracorat on MKP-1 is through transcription or post-transcriptional events based on the results from the present study, and further study is needed.
At least 10 MKPs have been identified in mammalian cells so far, with MKP-1 being the archetype of the MKP family (13). We have focused on MKP-1, a well characterized anti-inflammatory member of MKP family in the present study. It will be interesting to find out if other MKPs are involved in the anti-inflammatory effects of mapracorat.
In summary, we demonstrated that mapracorat, a novel selective glucocorticoid receptor agonist under clinical development for the treatment of inflammatory diseases, robustly inhibits the production of inflammatory mediators such as inflammatory cytokines and PGE2 in macrophages. Our results suggest that mapracorat exerts its anti-inflammatory effects, at least in part, by inactivating MAPK pathways through augmenting the expression of MKP-1. Our studies also highlighted the great potential of compounds that augment the endogenous anti-inflammatory regulators as novel anti-inflammatory drugs.
Footnotes
- TLR
- Toll-like receptor
- MK
- MAPK-activated protein kinase
- MKP
- mitogen-activated protein kinase phosphatase
- ARE
- AU-rich element
- UTR
- untranslated region
- SEGRA
- selective glucocorticoid receptor agonist
- GR
- glucocorticoid receptor.
REFERENCES
- 1. Janeway C. A. J., Travers P., Walport M., Shlomchik M. J. (2001) Immunobiology: the Immune System in Health and Disease, 5th Ed., Garland Publishing, New York [Google Scholar]
- 2. Medzhitov R., Janeway C., Jr. (2000) The Toll receptor family and microbial recognition. Trends Microbiol. 8, 452–456 [DOI] [PubMed] [Google Scholar]
- 3. Beutler B., Bazzoni F. (1998) TNF, apoptosis and autoimmunity: a common thread? Blood Cells Mol. Dis. 24, 216–230 [DOI] [PubMed] [Google Scholar]
- 4. Beutler B. (1995) TNF, immunity and inflammatory disease: lessons of the past decade. J. Investig. Med. 43, 227–235 [PubMed] [Google Scholar]
- 5. Smith W. L., DeWitt D. L., Garavito R. M. (2000) Cyclooxygenases: structural, cellular, and molecular biology. Annu. Rev. Biochem. 69, 145–182 [DOI] [PubMed] [Google Scholar]
- 6. Eliopoulos A. G., Dumitru C. D., Wang C. C., Cho J., Tsichlis P. N. (2002) Induction of COX-2 by LPS in macrophages is regulated by Tpl2-dependent CREB activation signals. EMBO J. 21, 4831–4840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pratt P. F., Bokemeyer D., Foschi M., Sorokin A., Dunn M. J. (2003) Alterations in subcellular localization of p38 MAPK potentiates endothelin-stimulated COX-2 expression in glomerular mesangial cells. J. Biol. Chem. 278, 51928–51936 [DOI] [PubMed] [Google Scholar]
- 8. Beutler B. (2000) Tlr4: central component of the sole mammalian LPS sensor. Curr. Opin. Immunol. 12, 20–26 [DOI] [PubMed] [Google Scholar]
- 9. Akira S. (2003) Mammalian Toll-like receptors. Curr. Opin. Immunol. 15, 5–11 [DOI] [PubMed] [Google Scholar]
- 10. O'Neill L. (2000) The Toll/interleukin-1 receptor domain: a molecular switch for inflammation and host defense. Biochem. Soc. Trans. 28, 557–563 [DOI] [PubMed] [Google Scholar]
- 11. Kawai T., Akira S. (2007) TLR signaling. Semin. Immunol. 19, 24–32 [DOI] [PubMed] [Google Scholar]
- 12. Dong C., Davis R. J., Flavell R. A. (2002) MAP kinases in the immune response. Annu. Rev. Immunol. 20, 55–72 [DOI] [PubMed] [Google Scholar]
- 13. Liu Y., Shepherd E. G., Nelin L. D. (2007) MAPK phosphatases–regulating the immune response. Nat. Rev. Immunol. 7, 202–212 [DOI] [PubMed] [Google Scholar]
- 14. Lee J. C., Laydon J. T., McDonnell P. C., Gallagher T. F., Kumar S., Green D., McNulty D., Blumenthal M. J., Heys J. R., Landvatter S. W. (1994) A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 372, 739–746 [DOI] [PubMed] [Google Scholar]
- 15. Han J., Lee J. D., Bibbs L., Ulevitch R. J. (1994) A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 265, 808–811 [DOI] [PubMed] [Google Scholar]
- 16. Kotlyarov A., Neininger A., Schubert C., Eckert R., Birchmeier C., Volk H. D., Gaestel M. (1999) MAPKAP kinase 2 is essential for LPS-induced TNF-α biosynthesis. Nat. Cell Biol. 1, 94–97 [DOI] [PubMed] [Google Scholar]
- 17. Lehner M. D., Schwoebel F., Kotlyarov A., Leist M., Gaestel M., Hartung T. (2002) Mitogen-activated protein kinase-activated protein kinase 2-deficient mice show increased susceptibility to Listeria monocytogenes infection. J. Immunol. 168, 4667–4673 [DOI] [PubMed] [Google Scholar]
- 18. Karin M., Liu Z., Zandi E. (1997) AP-1 function and regulation. Curr. Opin. Cell Biol. 9, 240–246 [DOI] [PubMed] [Google Scholar]
- 19. Roebuck K. A., Carpenter L. R., Lakshminarayanan V., Page S. M., Moy J. N., Thomas L. L. (1999) Stimulus-specific regulation of chemokine expression involves differential activation of the redox-responsive transcription factors AP-1 and NF-κB. J. Leukoc. Biol. 65, 291–298 [DOI] [PubMed] [Google Scholar]
- 20. Kontoyiannis D., Pasparakis M., Pizarro T. T., Cominelli F., Kollias G. (1999) Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity 10, 387–398 [DOI] [PubMed] [Google Scholar]
- 21. Rigby W. F., Roy K., Collins J., Rigby S., Connolly J. E., Bloch D. B., Brooks S. A. (2005) Structure/function analysis of tristetraprolin (TTP): p38 stress-activated protein kinase and lipopolysaccharide stimulation do not alter TTP function. J. Immunol. 174, 7883–7893 [DOI] [PubMed] [Google Scholar]
- 22. Stoecklin G., Stubbs T., Kedersha N., Wax S., Rigby W. F., Blackwell T. K., Anderson P. (2004) MK2-induced tristetraprolin:14–3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J. 23, 1313–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lasa M., Mahtani K. R., Finch A., Brewer G., Saklatvala J., Clark A. R. (2000) Regulation of cyclooxygenase 2 mRNA stability by the mitogen-activated protein kinase p38 signaling cascade. Mol. Cell Biol. 20, 4265–4274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ueda T., Watanabe-Fukunaga R., Fukuyama H., Nagata S., Fukunaga R. (2004) Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Mol. Cell Biol. 24, 6539–6549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Andersson K., Sundler R. (2006) Post-transcriptional regulation of TNFα expression via eukaryotic initiation factor 4E (eIF4E) phosphorylation in mouse macrophages. Cytokine 33, 52–57 [DOI] [PubMed] [Google Scholar]
- 26. Rincón M., Davis R. J. (2009) Regulation of the immune response by stress-activated protein kinases. Immunol. Rev. 228, 212–224 [DOI] [PubMed] [Google Scholar]
- 27. Raingeaud J., Whitmarsh A. J., Barrett T., Dérijard B., Davis R. J. (1996) MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol. Cell Biol. 16, 1247–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lang R., Hammer M., Mages J. (2006) DUSP meet immunology: dual specificity MAPK phosphatases in control of the inflammatory response. J. Immunol. 177, 7497–7504 [DOI] [PubMed] [Google Scholar]
- 29. Chen P., Li J., Barnes J., Kokkonen G. C., Lee J. C., Liu Y. (2002) Restraint of proinflammatory cytokine biosynthesis by mitogen-activated protein kinase phosphatase-1 in lipopolysaccharide-stimulated macrophages. J. Immunol. 169, 6408–6416 [DOI] [PubMed] [Google Scholar]
- 30. Zhao Q., Wang X., Nelin L. D., Yao Y., Matta R., Manson M. E., Baliga R. S., Meng X., Smith C. V., Bauer J. A., Chang C. H., Liu Y. (2006) MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J. Exp. Med. 203, 131–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hammer M., Mages J., Dietrich H., Servatius A., Howells N., Cato A. C., Lang R. (2006) Dual specificity phosphatase 1 (DUSP1) regulates a subset of LPS-induced genes and protects mice from lethal endotoxin shock. J. Exp. Med. 203, 15–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Salojin K. V., Owusu I. B., Millerchip K. A., Potter M., Platt K. A., Oravecz T. (2006) Essential role of MAPK phosphatase-1 in the negative control of innate immune responses. J. Immunol. 176, 1899–1907 [DOI] [PubMed] [Google Scholar]
- 33. Chi H., Barry S. P., Roth R. J., Wu J. J., Jones E. A., Bennett A. M., Flavell R. A. (2006) Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc. Natl. Acad. Sci. U.S.A. 103, 2274–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang X., Zhao Q., Matta R., Meng X., Liu X., Liu C. G., Nelin L. D., Liu Y. (2009) Inducible nitric-oxide synthase expression is regulated by mitogen-activated protein kinase phosphatase-1. J. Biol. Chem. 284, 27123–27134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Frazier W. J., Wang X., Wancket L. M., Li X. A., Meng X., Nelin L. D., Cato A. C., Liu Y. (2009) Increased inflammation, impaired bacterial clearance, and metabolic disruption after gram-negative sepsis in Mkp-1-deficient mice. J. Immunol. 183, 7411–7419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sweeney S. E., Firestein G. S. (2006) Mitogen activated protein kinase inhibitors: where are we now and where are we going? Ann. Rheum. Dis. 65, iii83–iii88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Morris D. L., O'Neil S. P., Devraj R. V., Portanova J. P., Gilles R. W., Gross C. J., Curtiss S. W., Komocsar W. J., Garner D. S., Happa F. A., Kraus L. J., Nikula K. J., Monahan J. B., Selness S. R., Galluppi G. R., Shevlin K. M., Kramer J. A., Walker J. K., Messing D. M., Anderson D. R., Mourey R. J., Whiteley L. O., Daniels J. S., Yang J. Z., Rowlands P. C., Alden C. L., Davis J. W., 2nd, Sagartz J. E. (2010) Acute lymphoid and gastrointestinal toxicity induced by selective p38α map kinase and map kinase-activated protein kinase-2 (MK2) inhibitors in the dog. Toxicol. Pathol. 38, 606–618 [DOI] [PubMed] [Google Scholar]
- 38. Cato A. C., Wade E. (1996) Molecular mechanisms of anti-inflammatory action of glucocorticoids. Bioessays 18, 371–378 [DOI] [PubMed] [Google Scholar]
- 39. Barnes P. J. (1998) Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin. Sci. 94, 557–572 [DOI] [PubMed] [Google Scholar]
- 40. Joyce D. A., Gimblett G., Steer J. H. (2001) Targets of glucocorticoid action on TNF-alpha release by macrophages. Inflamm. Res. 50, 337–340 [DOI] [PubMed] [Google Scholar]
- 41. Rhen T., Cidlowski J. A. (2005) Antiinflammatory action of glucocorticoids–new mechanisms for old drugs. N. Engl. J. Med. 353, 1711–1723 [DOI] [PubMed] [Google Scholar]
- 42. Saklatvala J. (2002) Glucocorticoids: do we know how they work? Arthritis Research 4, 146–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lasa M., Abraham S. M., Boucheron C., Saklatvala J., Clark A. R. (2002) Dexamethasone causes sustained expression of mitogen-activated protein kinase (MAPK) phosphatase 1 and phosphatase-mediated inhibition of MAPK p38. Mol. Cell Biol. 22, 7802–7811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Swantek J. L., Cobb M. H., Geppert T. D. (1997) Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) is required for lipopolysaccharide stimulation of tumor necrosis factor α (TNF-α) translation: glucocorticoids inhibit TNF-α translation by blocking JNK/SAPK. Mol. Cell Biol. 17, 6274–6282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kassel O., Sancono A., Krätzschmar J., Kreft B., Stassen M., Cato A. C. (2001) Glucocorticoids inhibit MAP kinase via increased expression and decreased degradation of MKP-1. EMBO J. 20, 7108–7116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang X., Nelin L. D., Kuhlman J. R., Meng X., Welty S. E., Liu Y. (2008) The role of MAP kinase phosphatase-1 in the protective mechanism of dexamethasone against endotoxemia. Life Sciences 83, 671–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Abraham S. M., Lawrence T., Kleiman A., Warden P., Medghalchi M., Tuckermann J., Saklatvala J., Clark A. R. (2006) Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J. Exp. Med. 203, 1883–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Maier J. V., Brema S., Tuckermann J., Herzer U., Klein M., Stassen M., Moorthy A., Cato A. C. (2007) Dual specificity phosphatase 1 knockout mice show enhanced susceptibility to anaphylaxis but are sensitive to glucocorticoids. Mol. Endocrinol. 21, 2663–2671 [DOI] [PubMed] [Google Scholar]
- 49. Zhang J. Z., Cavet M. E., VanderMeid K. R., Salvador-Silva M., López F. J., Ward K. W. (2009) BOL-303242-X, a novel selective glucocorticoid receptor agonist, with full anti-inflammatory properties in human ocular cells. Mol. Vis. 15, 2606–2616 [PMC free article] [PubMed] [Google Scholar]
- 50. Cavet M. E., Harrington K. L., Ward K. W., Zhang J. Z. (2010) Mapracorat, a novel selective glucocorticoid receptor agonist, inhibits hyperosmolar-induced cytokine release and MAPK pathways in human corneal epithelial cells. Mol. Vis. 16, 1791–1800 [PMC free article] [PubMed] [Google Scholar]
- 51. Schäcke H., Zollner T. M., Döcke W. D., Rehwinkel H., Jaroch S., Skuballa W., Neuhaus R., May E., Zügel U., Asadullah K. (2009) Characterization of ZK 245186, a novel, selective glucocorticoid receptor agonist for the topical treatment of inflammatory skin diseases. Br. J. Pharmacol. 158, 1088–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lasa M., Brook M., Saklatvala J., Clark A. R. (2001) Dexamethasone destabilizes cyclooxygenase 2 mRNA by inhibiting mitogen-activated protein kinase p38. Mol. Cell Biol. 21, 771–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kang Y. J., Mbonye U. R., DeLong C. J., Wada M., Smith W. L. (2007) Regulation of intracellular cyclooxygenase levels by gene transcription and protein degradation. Prog. Lipid Res. 46, 108–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhao Q., Shepherd E. G., Manson M. E., Nelin L. D., Sorokin A., Liu Y. (2005) The role of mitogen-activated protein kinase phosphatase-1 in the response of alveolar macrophages to lipopolysaccharide: attenuation of proinflammatory cytokine biosynthesis via feedback control of p38. J. Biol. Chem. 280, 8101–8108 [DOI] [PubMed] [Google Scholar]
- 55. Shafiee A., Bucolo C., Budzynski E., Ward K. W., López F. J. (2011) In vivo ocular efficacy profile of mapracorat, a novel selective glucocorticoid receptor agonist, in rabbit models of ocular disease. Invest. Ophthalmol. Vis. Sci. 52, 1422–1430 [DOI] [PubMed] [Google Scholar]
- 56. Baiula M., Spartà A., Bedini A., Carbonari G., Bucolo C., Ward K. W., Zhang J. Z., Govoni P., Spampinato S. (2011) Eosinophil as a cellular target of the ocular anti-allergic action of mapracorat, a novel selective glucocorticoid receptor agonist. Mol. Vis. 17, 3208–3223 [PMC free article] [PubMed] [Google Scholar]
- 57. Pfeffer B. A., DeWitt C. A., Salvador-Silva M., Cavet M. E., López F. J., Ward K. W. (2010) Reduced myocilin expression in cultured monkey trabecular meshwork cells induced by a selective glucocorticoid receptor agonist: comparison with steroids. Invest. Ophthalmol. Vis. Sci. 51, 437–446 [DOI] [PubMed] [Google Scholar]
- 58. Joanny E., Ding Q., Gong L., Kong P., Saklatvala J., Clark A. R. (2012) Anti-inflammatory effects of selective glucocorticoid receptor modulators are partially dependent on up-regulation of dual specificity phosphatase 1. Br. J. Pharmacol. 165, 1124–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Johnson G. L., Lapadat R. (2002) Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298, 1911–1912 [DOI] [PubMed] [Google Scholar]
- 60. Hazzalin C. A., Mahadevan L. C. (2002) MAPK-regulated transcription: a continuously variable gene switch? Nat. Rev. Mol. Cell Biol. 3, 30–40 [DOI] [PubMed] [Google Scholar]
- 61. Jeffrey K. L., Camps M., Rommel C., Mackay C. R. (2007) Targeting dual-specificity phosphatases: manipulating MAP kinase signaling and immune responses. Nat. Rev. Drug Discov. 6, 391–403 [DOI] [PubMed] [Google Scholar]
- 62. Lee J. C., Young P. R. (1996) Role of CSB/p38/RK stress response kinase in LPS and cytokine signaling mechanisms. J. Leukoc. Biol. 59, 152–157 [DOI] [PubMed] [Google Scholar]
- 63. Lee J. C., Kassis S., Kumar S., Badger A., Adams J. L. (1999) p38 mitogen-activated protein kinase inhibitors–mechanisms and therapeutic potentials. Pharmacol. Ther. 82, 389–397 [DOI] [PubMed] [Google Scholar]
- 64. Franklin C. C., Kraft A. S. (1997) Conditional expression of the mitogen-activated protein kinase (MAPK) phosphatase MKP-1 preferentially inhibits p38 MAPK and stress-activated protein kinase in U937 cells. J. Biol. Chem. 272, 16917–16923 [DOI] [PubMed] [Google Scholar]
- 65. Franklin C. C., Srikanth S., Kraft A. S. (1998) Conditional expression of mitogen-activated protein kinase phosphatase-1, MKP-1, is cytoprotective against UV-induced apoptosis. Proc. Natl. Acad. Sci. U.S.A. 95, 3014–3019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Salojin K., Oravecz T. (2007) Regulation of innate immunity by MAPK dual-specificity phosphatases: knockout models reveal new tricks of old genes. J. Leukoc. Biol. 81, 860–869 [DOI] [PubMed] [Google Scholar]
- 67. Bhalla U. S., Ram P. T., Iyengar R. (2002) MAP kinase phosphatase as a locus of flexibility in a mitogen-activated protein kinase signaling network. Science 297, 1018–1023 [DOI] [PubMed] [Google Scholar]
- 68. Roger T., Chanson A. L., Knaup-Reymond M., Calandra T. (2005) Macrophage migration inhibitory factor promotes innate immune responses by suppressing glucocorticoid-induced expression of mitogen-activated protein kinase phosphatase-1. Eur. J. Immunol. 35, 3405–3413 [DOI] [PubMed] [Google Scholar]
- 69. Aeberli D., Yang Y., Mansell A., Santos L., Leech M., Morand E. F. (2006) Endogenous macrophage migration inhibitory factor modulates glucocorticoid sensitivity in macrophages via effects on MAP kinase phosphatase-1 and p38 MAP kinase. FEBS Lett. 580, 974–981 [DOI] [PubMed] [Google Scholar]
- 70. Hammer M., Mages J., Dietrich H., Schmitz F., Striebel F., Murray P. J., Wagner H., Lang R. (2005) Control of dual-specificity phosphatase-1 expression in activated macrophages by IL-10. Eur. J. Immunol. 35, 2991–3001 [DOI] [PubMed] [Google Scholar]
- 71. Noguchi T., Metz R., Chen L., Mattéi M. G., Carrasco D., Bravo R. (1993) Structure, mapping, and expression of erp, a growth factor-inducible gene encoding a nontransmembrane protein tyrosine phosphatase, and effect of ERP on cell growth. Mol. Cell Biol. 13, 5195–5205 [DOI] [PMC free article] [PubMed] [Google Scholar]