

Abstract
Common problems hindering rapid progress in Plant Sciences include cellular, tissue and whole organism complexity, and notably the high level of genomic redundancy affecting simple genetics in higher plants. The novel model organism Ostreococcus tauri is the smallest free-living eukaryote known to date, and possesses a greatly reduced genome size and cellular complexity1,2, manifested by the presence of just one of most organelles (mitochondrion, chloroplast, golgi stack) per cell, and a genome containing only ~8000 genes. Furthermore, the combination of unicellularity and easy culture provides a platform amenable to chemical biology approaches. Recently, Ostreococcus has been successfully employed to study basic mechanisms underlying circadian timekeeping3-6. Results from this model organism have impacted not only plant science, but also mammalian biology7. This example highlights how rapid experimentation in a simple eukaryote from the green lineage can accelerate research in more complex organisms by generating testable hypotheses using methods technically feasible only in this background of reduced complexity. Knowledge of a genome and the possibility to modify genes are essential tools in any model species. Genomic1, Transcriptomic8, and Proteomic9 information for this species is freely available, whereas the previously reported methods6,10 to genetically transform Ostreococcus are known to few laboratories worldwide.
In this article, the experimental methods to genetically transform this novel model organism with an overexpression construct by means of electroporation are outlined in detail, as well as the method of inclusion of transformed cells in low percentage agarose to allow selection of transformed lines originating from a single transformed cell. Following the successful application of Ostreococcus to circadian research, growing interest in Ostreococcus can be expected from diverse research areas within and outside plant sciences, including biotechnological areas. Researchers from a broad range of biological and medical sciences that work on conserved biochemical pathways may consider pursuing research in Ostreococcus, free from the genomic and organismal complexity of larger model species.
Keywords: Microbiology, Issue 65, Plant Biology, Microbial Oceanography, Marine Biology, Genetics, Transformation, Electroporation, Marine algae, plankton, Cell biology, Ostreococcus tauri, Plant Science, Reduced complexity, Circadian
Protocol
1. Preparation of Algal Material
Ostreococcus cells are cultured in artificial seawater (ASW). Sea salts (typically about 40 grams per liter) are dissolved in deionised water to a salinity of 30 ppt, as measured using a salinity meter. Enrichment medium11,12, trace metal elements and vitamins are added as described in Tables 1-3. The media is then filter-sterilised through a 0.22 μm filter.
For maintenance, cells of Ostreococcus strain OTTH95 are sub-cultured aseptically at a dilution of 1 /100 in fresh ASW every 7 days and grown under constant light in a plant growth incubator fitted with Moonlight Blue filter. The light intensity should be close to 20 μmol m-2 s-1 and temperature is maintained at 20 °C. Cells do not require constant agitation, but are shaken once every 2 to 3 days to prevent aggregation.
For each transformation, 50 ml of cells are required at a cell density of 20-30 x 106 ml-1, which should be attained 5-7 days following sub-culturing. Approximate cell density and axeny can be determined using either flow cytometry or a haemocytometer at a minimum of x40 magnification.
2. Electroporation
Prepare DNA for the transformation. For each transformation, 5 μg of pure, linearised plasmid DNA is required at a concentration of 1 μg/μl in sterile deionised water. To obtain this DNA, the authors recommend using a Qiagen midi prep kit, though other methods might work equally well. Digest the product with an enzyme that cuts in the backbone of the vector used, but not in the transgene or selection gene. Further purify and concentrate the resulting linear DNA by ethanol precipitation, and resuspend the product in the appropriate volume of high-quality sterile deionised water.
Prepare microcentrifuge tubes containing 5 μg DNA for each transformation. A control with no DNA is necessary for each cell line to be transformed. Keep these tubes on ice, together with a 2 mm electroporation cuvette for each transformation.
Prepare 2.2 ml of resuspension buffer per transformation. Dissolve Sorbitol to a 1 M solution in ddH2O, add 0.1 % pluronic acid F68 and filter-sterilise.
Add pluronic acid F68, to a final concentration of 0.1% to the cells, and pellet them for 10 minutes at 8000x g at 10 °C in a 50 ml tube with conical bottom. Immediately resuspend the cells in 1 ml of resuspension buffer by pipetting up and down, and transfer to a microfuge tube. Spin down for 10 minutes at 8000 x g at 10 °C, and working quickly, repeat this wash step once more.
With a cut tip, resuspend each final pellet in 40 μl of resuspension buffer. Add 40 μl of the resuspended cells to every tube of linearised DNA on ice, mix gently and transfer to the electroporation cuvette.
Put the cuvette in the electroporation machine. Change the settings to 6 kV cm-1, 600 Ω, and 25 μF. Electroporate the cells.
Incubate the electoporated cells in the cuvettes at room temperature for 10 minutes, and use that time to prepare the tissue culture flasks. Label them and add 30 ml of fresh ASW to each. Take 1 ml out of each flask and gently add it to the corresponding cuvette. Incubate for 2 minutes and gently remove the ASW, now containing the globule of cells and gently and slowly pipette directly into the ASW in the culture flask.
Cells typically remain in a globule. Take care not to shake or disturb the aggregated cells at this moment. Allow the cells to recover in the incubator for 1-2 hours, then resuspend by shaking the flasks. At this time, the cells should resuspend freely and no clumps should be visible. Leave the cultures to recover overnight in the incubator.
3. Inclusion of Cells on Plates in Semi-solid Medium
The next day, autoclave a solution of 2.1% low melting point agarose in ddH2O in a bottle containing a stirring rod. Keep molten at 65-90 °C in a larger beaker containing water on a heating magnetic stirrer. For each transformation prepare 8 Petri dishes (55 mm diameter) and 8 x 15 ml tubes each containing 9 ml of ASW plus the required selection. If using Nourseothricin or G418, use 2 mg/ml.
Collect the transformed cells from the incubator. Working in a sterile flowhood, add 1 ml of boiling LMP agarose to the 9 ml in one of the tubes. Close the tube, and mix by inverting. Then add 0.5 ml of freshly transformed cells, quickly mix and pour into the plate. Repeat this process with the remaining 7 tubes and then proceed to the next transformation flask.
Leave the plates open in the flow hood for an hour, for the agarose to set. Then close the plates and transfer them to large square Petri dishes, which will hold 4 plates each. Seal the square plates with parafilm. Note that the plates will not set completely, and as a result, the gel is very fragile. Care should be taken not to break the gel when handling the plates. Place all square plates in the incubator.
4. Selection of Transformed Colonies
Colonies should appear after 10 to 21 days on the transformation plates, but not on the mock transformed plates. To pick colonies use a 200 μl pipette with cut-off tips. Simply select free colonies and suck out the green colony. Take care not to include any cells from neighbouring colonies.
Transfer the cells to 2 ml of liquid medium containing the selection, in a 24 wells plate. When using Nourseothricin, use 1.75 mg/ml. Mix by pipetting up and down. Select 24-50 colonies per transformation. Seal the plates with parafilm and transfer to the incubator.
After a week, transfer 100 μl of each well to 2 ml fresh ASW with selection in a 24 wells plate and grow for an additional 7 days. Surviving cell lines can be used for further studies. Stable integration into the genome and insertion number should be analysed using PCR and Southern Blot. Genomic DNA can be obtained for these purposes using any down-scaled standard method for plant DNA extraction.
5. Representative Results
Cells split 1 / 100 reached a density of 25 million cells per milliliter after growing for 7 days. Electroporation of the cells with the appropriate settings resulted in a time-constant between 10 and 14 milliseconds. When cells are transferred to medium in culture flasks, a globule of cells should form. When lightly shaken after an hour, the cells should easily resuspend. Inclusion of 1 ml transformed cells into 0.2% LMP agar should result in a consistent but only semi-solid gel. Colonies should appear after 10 to 21 days. When transforming 5 μg of linearised DNA using the protocol above, 50-100 colonies per transformation plate were typically expected, versus none on the negative control plates. ~80% of colonies picked were positively selected by antibiotic resistance in liquid medium and were used in subsequent studies.
| Final concentration | Stock solution | Volume for 1 liter | |
| NaNO3 | 8.83 x 10-4 M | 75 g/L dH2O | 1 mL |
| NH4Cl | 3.63 x 10-5 M | 2.68 g/L dH2O | 1 mL |
| β-glycerophosphate | 1 x 10-5 M | 2.16 g/L dH2O | 1 mL |
| H2SeO3 | 1 x 10-8 M | 1.29 mg/L dH2O | 1 mL |
| Tris-base(pH 7.2) | 1 x 10-3 M | 121.1 g/L dH2O | 1 mL |
| K trace metal solution | Table 2 | 1 mL | |
| f/2 vitamin solution | Table 3 | 0.5 mL |
Table 1. Medium constituents for growth of O. tauri11, 12. Make up a total volume of 1 liter of artificial seawater to a salinity of 30 ppt, and add the components above from stock solutions as indicated. Filter-sterilise the medium and use within a week. Stocks of all compounds except the vitamin solution can be pooled to add 6 ml of this solution for every liter of medium.
| Final concentration | Stock solution | Amount for 1 liter | |
| Na2EDTA • 2H2O | 1 x 10-4 M | 41.6 g | |
| FeCl3 • 6H2O | 1 x 10-5 M | 3.15 g | |
| Na2MoO4 • 2H2O | 1 x 10-8 M | 6.3 g/L dH2O | 1 mL |
| ZnSO4 • 7H2O | 1 x 10-9 M | 22.0 g/L dH2O | 1 mL |
| CoCl2• 6H2O | 1 x 10-9 M | 10.0 g/L dH2O | 1 mL |
| MnCl2 • 4H2O | 1 x 10-8 M | 180.0 g/L dH2O | 1 mL |
| CuSO4• 5H2O | 1 x 10-8 M | 9.8 g/L dH2O | 1 mL |
Table 2. Trace metal solution11,12. Make up to a total of 1 liter, in dH2O, and heat to dissolve. Aliquot the solution and freeze for storage. Final concentrations indicated refer to the final medium, not the trace metal solution.
| Final concentration | Stock solution | Amount for 1 liter | |
| Vitamin B12 | 1 x 10-10 M | 1 g/L dH2O | 1 mL |
| Biotin | 1 x 10-9 M | 0.1 g/L dH2O | 10 mL |
| Thiamine • HCl | 1 x 10-7 M | 200 mg |
Table 3. f/2 vitamin solution11. Make up to a total of 1 liter in dH2O, filter-sterilise, aliquot and freeze. Final concentrations indicated refer to the final medium, not the vitamin solution.
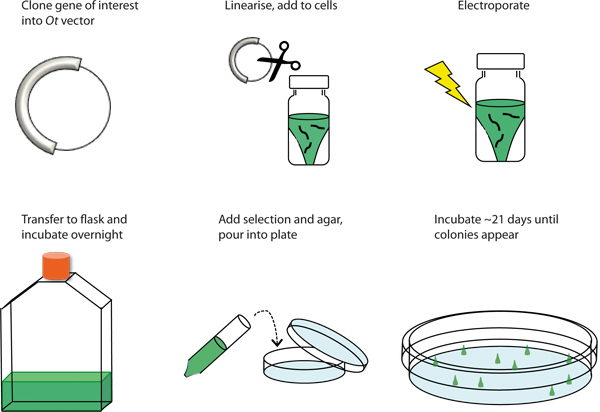 Figure 1. Graphical overview of the transformation procedure. Schematic representation of the procedure to genetically transform Ostreococcus tauri by electroporation.
Figure 1. Graphical overview of the transformation procedure. Schematic representation of the procedure to genetically transform Ostreococcus tauri by electroporation.
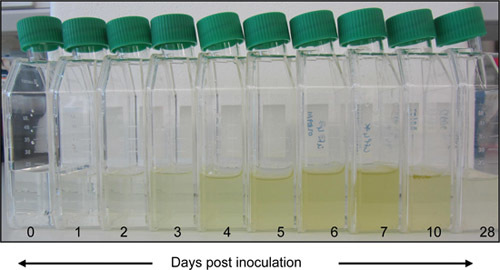 Figure 2. Culture growth. Cells are sub-cultured aseptically at a dilution of 1 /100 in fresh ASW every 7 days and grown under constant light in a plant growth incubator fitted with Moonlight Blue filter. The light intensity should be close to 20 μmol m-2s-1 and temperature is maintained at 20 °C Cells do not require constant agitation, but are shaken once every 2 to 3 days to prevent aggregation.
Figure 2. Culture growth. Cells are sub-cultured aseptically at a dilution of 1 /100 in fresh ASW every 7 days and grown under constant light in a plant growth incubator fitted with Moonlight Blue filter. The light intensity should be close to 20 μmol m-2s-1 and temperature is maintained at 20 °C Cells do not require constant agitation, but are shaken once every 2 to 3 days to prevent aggregation.
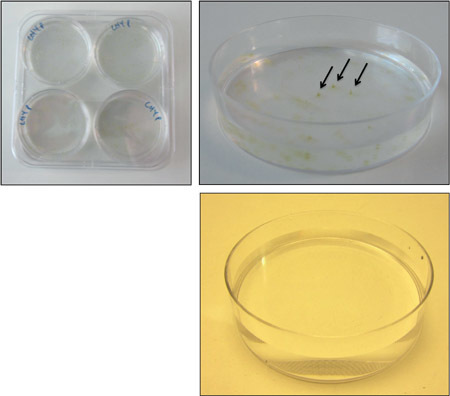 Figure 3. Colony formation on 0.2% LMP agarose. Transfer 4 plates to a large square Petri dish, and seal with parafilm (top left). When colonies have formed, simply select free (ideally conically shaped) colonies (top right) and suck them out of the plate with a p200 pipette. There should be no colonies on the negative control (bottom right).
Figure 3. Colony formation on 0.2% LMP agarose. Transfer 4 plates to a large square Petri dish, and seal with parafilm (top left). When colonies have formed, simply select free (ideally conically shaped) colonies (top right) and suck them out of the plate with a p200 pipette. There should be no colonies on the negative control (bottom right).
Discussion
Cellular state and culture axeny is of crucial importance for the transformation procedure. Although cells are usually maintained in cycles of 12 hours light, 12 hour dark, transformation efficiency in cells grown under these conditions were found to be insufficient. During the repeated pelleting / resuspension steps, cells should always resuspend easily. If cells aggregate, or a mucus-like substance is present, it is better to start the procedure from the beginning again. Typically, ~50 colonies can be expected on each transformation plate, versus none on the negative control plate. In case of low transformation efficiency, culturing conditions should be varied to ensure cells are in the optimal state. Variables that affect transformation efficiency include ambient temperature in the laboratory (should be close to 20 °C) and handling speed (Procedure from pelleting cells [2.3] to recovery [2.7] should take ~1 hour). It is also important that all solutions are made fresh before each transformation. For inclusion in plates, the concentration of LMP agarose is crucial. Ostreococcus cells will not grow in high agarose concentrations, and will diffuse in low concentrations.
Three transformation vectors for Ostreococcus have been published previously6, and more vectors are expected to be generated and published shortly. The existing vector Pot-Luc6 allows the use of a promotor of choice in combination with a C-terminal firefly luciferase tag allowing faster transgenic line selection plus tractable expression patterns, whereas the Potox vector6 carries the strong inducible13 promotor taken from the Ostreococcus High Affinity Phosphate Transporter (HAPT) gene for overexpression of the gene of interest. The Potox-Luc vector derives from Potox, carrying an additional luciferase marker that can be used for indirect selection of overexpression lines14. Successful selection markers on these vectors are Nourseothricin and G148. However, Ostreococcus cells are highly resistant to many antibiotics, and the concentrations of compounds necessary for selection of transgenic Ostreococcus cells is higher than for conventional model systems (2 mg/ml).
Although the process appears inefficient, the large number of cells and plasmid DNA needed for this procedure poses no significant obstacle. A larger limitation is that every generated line that is resistant to the selectable marker needs to be analysed individually to check that the entire construct is integrated in the DNA. We have observed examples in which successful expression of the selectable marker did not correspond to insertion of the actual gene of interest; i.e. partial integrations can occur. Evidently, random insertion into the genome also means that there is no control of the insertion site using this method, and methods based on homologous recombination are currently being developed. However, the method described here is, to our best knowledge, currently the only characterised method to generate genetically modified Ostreococcus cells.
As Ostreococcus tauri has only recently been used as an experimental model organism, most methods working with these cells is likely to evolve and be refined by the growing research community involved. This protocol is intended to help people initiate work with Ostreococcus, but by no means claims to describe all there is to know about genomic transformation of this microalga. The authors trust that readers will be able to adapt this protocol to their own needs as small differences in incubators (light levels or quality) and other parameters will exist and likely have to be optimised in every individual laboratory to obtain the healthy cultures needed for this protocol. After mastering the techniques described here, vectors can be constructed to generate luminescent, fluorescent, or other tagged fusion lines under the control of a range of promotors. This exciting novel experimental platform will be useful to study how complicated biochemical problems are solved in a eukaryote of reduced complexity, in which pharmacological approaches are highly facilitated3.
Disclosures
No conflicts of interest declared.
Acknowledgments
SynthSys a Centre for Integrative Systems Biology funded by BBSRC and EPSRC award D019621. EU FP6 Network of Excellence "Marine Genomics" grant to FYB has financed development of genetic transformation methods.
References
- Derelle E. Genome analysis of the smallest free-living eukaryote Ostreococcus tauri unveils many unique features. Proc. Natl. Acad. Sci. U.S.A. 2006;103:11647–11652. doi: 10.1073/pnas.0604795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson GP, Gan L, Jensen GJ. 3-D ultrastructure of O. tauri: electron cryotomography of an entire eukaryotic cell. PLoS One. 2007;2:e749. doi: 10.1371/journal.pone.0000749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill JS. Circadian rhythms persist without transcription in a eukaryote. Nature. 2011;469:554–558. doi: 10.1038/nature09654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ooijen G, Dixon LE, Troein C, Millar AJ. Proteasome function is required for biological timing throughout the twenty-four hour cycle. Curr. Biol. 2011;21:869–875. doi: 10.1016/j.cub.2011.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troein C. Multiple light inputs to a simple clock circuit allow complex biological rhythms. Plant. J. 2011;66:375–385. doi: 10.1111/j.1365-313X.2011.04489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corellou F. Clocks in the green lineage: comparative functional analysis of the circadian architecture of the picoeukaryote ostreococcus. Plant Cell. 2009;21:3436–3449. doi: 10.1105/tpc.109.068825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill JS, Reddy AB. Circadian clocks in human red blood cells. Nature. 2011;469:498–503. doi: 10.1038/nature09702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnier A. Orchestrated transcription of biological processes in the marine picoeukaryote Ostreococcus exposed to light/dark cycles. B.M.C. Genomics. 2010;11:192. doi: 10.1186/1471-2164-11-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bihan T. Shotgun proteomic analysis of the unicellular alga Ostreococcus tauri. J. Proteomics. 2011;74:2060–2070. doi: 10.1016/j.jprot.2011.05.028. [DOI] [PubMed] [Google Scholar]
- Corellou F, Bouget F-Y, inventors. Expression of polypeptides from the nuclear genome of Ostreococcus sp. 201001174050. US application patent. 2010
- Guillard RRL, Ryther JH. Studies of marine planktonic diatoms. I. Cyclotella nana Hustedt, and Detonula confervacea (cleve) Gran. Can. J. Microbiol. 1962;8:229–239. doi: 10.1139/m62-029. [DOI] [PubMed] [Google Scholar]
- Keller MD, Selvin RC, Claus W, Guillard RRL. Media for the culture of oceanic ultraphytoplankton. J. Phycol. 1987;23:633–638. [Google Scholar]
- Djouani-Tari elB. A phosphate-regulated promoter for fine-tuned and reversible overexpression in Ostreococcus: Application to circadian clock functional analysis. PLoS One. 2011;6:e28471. doi: 10.1371/journal.pone.0028471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulager M. Integration of light signals by the retinoblastoma pathway in the control of S phase entry in the picophytoplanktonic cell Ostreococcus. PLoS. Genet. 2010;6:e1000957. doi: 10.1371/journal.pgen.1000957. [DOI] [PMC free article] [PubMed] [Google Scholar]