

Abstract
Thalassemia has been recognized by the World Health Organization as important inherited disorders principally impacting on the populations of low income countries. In this report, the prevalence of common β-thalassemia mutations in India was defined in 126 β-thalassemia carrier subjects in a western Indian population mainly from the south-western Maharashtra. The six most common β-thalassemia mutations were detected, which included IVS I-5 (G–C), IVS I-1 (G–T), codon 8–9 (+G), codon 41/42 (–TCTT), Codon 15 (G–A), and 619 bp deletion at 3′ end of β-globin gene. These mutations accounted for 93.66 % in 126 β-thalassemia carrier subjects and 6.34 % remained uncharacterized. Out of 126, 82 (65.07 %) showed the most common (prevalent) type of mutation, IVS I-5 (G–C), followed by IVS I-1 (G–T) showed by 12 (9.52 %) subjects. Three (2.38 %) subjects showed 619 bp deletion, codon 8/9 (+G) and codon 15 (G–A) mutations were present in eight subjects each (6.34 %). Only five (3.96 %) subjects showed codon 41/42 (–TCTT). There were eight (6.34 %) subjects where mutation was not any of the six mutations studied. This study provides the pattern of β thalassemia mutations from south-western Maharashtra, which will help to prevent β-thalassemia using prenatal diagnosis and proper counseling.
Electronic supplementary material
The online version of this article (doi:10.1007/s12291-012-0230-y) contains supplementary material, which is available to authorized users.
Keywords: β-Thalassemia, South-western Maharashtra, Mutations, β-Thalassemia carrier, Prenatal diagnosis
Introduction
β-Thalassemia is a highly prevalent autosomal recessive disorder characterized by the reduced or absent expression of the β-globin gene, leading to an imbalance of α and β-globin chains [1]. It has been estimated that the prevalence of pathological hemoglobinopathies in India is 1.2/1,000 live births [2], and with approximately 27 million births per year this would suggest the annual birth of 32,400 babies with a serious hemoglobin disorders. Within this overall disease classification, 1989 WHO Working Group on, guidelines for the control of hemoglobin disorders estimated 3.9 % carrier frequency for β-thalassemia in India [3].
The β-globin gene families are clustered on chromosome 11 and are arranged over approximately 60,000 nucleotide bases. β-thalassemia is a quantitative deficiency of β-globin production, and are usually due to DNA mutations of the β-globin gene cluster and result from mutations affecting gene transcription, RNA processing, alter splice junctions or splice consensus sequences, mutations within exons and introns that create an alternative splice site. Majority of the mutations are point mutations and unlike α-thalassemia deletion mutations are relatively less frequent [4].
About the 3 % of the world’s population carries the β-thalassemia gene. The bulk of this disorder is reported from Mediterranean, African and south-east Asian populations where the incidence of gene carriage may be as high as 10 % [4]. In India, β-thalassemia is the most common monogenic disorder. The average incidence of β-thalassemia trait in India is 3.3 % with 1–2 per 1,000 couple being at risk of having an affected offspring each year. The disease is characterized by its genetic heterogeneity at the molecular level, and more than 300 mutations of the β-globin gene have been characterized all over the world. Most are small nucleotide substitutions within the cluster. All mutations result in either absence of synthesis of β-globin chains (βo-thalassemia) or a reduction in synthesis (β+-thalassemia) [5, 6].
India has multiple geographical, ethnic, religious, and language divisions, [7] though each population seems to harbor only a few of these mutations. As the ethnic composition of the Indian population is varied, complex [8] and each region of the country have its own distinct set of mutations [3, 9, 10].
There are few β-thalassemia mutations which are common in India. Twenty-two β-thalassemia mutations have been documented by various authors in Indian patients with β-thalassemia. Six mutations, codon 8–9 (+G), Codon 15 (G–A), codon 41/42 (–TCTT), IVS I-1 (G–T), IVS I-5 (G–C), and 619 bp deletion at 3′ end of β-globin gene, account for about 80 % of β-thalassemia mutation in Indian population [4].
Within each geographic population there are a few common mutations together with a few rare ones, which are responsible for over 90 % of β-thalassemia mutations. We undertook this study, because there was no study reporting the mutational spectrum from the south-western Maharashtra region. Identification of the exact mutation helps to define the severity of the phenotype, plan therapy and it was hoped that these observations might form the basis for a comprehensive diagnostic database that would be useful not only for genetic counseling but for prenatal diagnosis also.
Materials and Methods
A descriptive cross sectional study was carried out in the Department of Biochemistry, Government Medical College, Miraj from July 2010 to September 2011. A total of 126 β-thalassemia carrier subjects were included from south-western Maharashtra and adjoining region comprising Satara, Sangli, and Kolhapur districts from the state of Maharashtra and Belgaum & Hubali districts from the State of Karnataka. There are large number of patients who come from these places for medical treatment, to the Hospital(s) at Miraj and Sangli.
Subjects
This study was approved by the Institutional ethical committee, Government medical college, Miraj, Sangli, Maharashtra, India. The study sample consisted of 126 β-thalassemia carrier subjects. The parent of known β-thalassemia major patients were included in the present study and their β-thalassemia carrier status was confirmed by Hb A2 level, if Hb A2 was >3.5 % then the subjects were included for mutational analysis. Subjects with Hb A2 if <3.5 % were excluded from this study. Family members other than parents of known thalassemia major child, who were β-thalassemia trait, were excluded from mutational studies.
DNA Analysis
Genomic DNA was extracted from peripheral blood lymphocytes by phenol chloroform extraction method [11]. Mutational analysis was carried out using the amplification refractory mutation system polymerase chain reaction (ARMS-PCR) as described [12]. The mutation-specific ARMS primers, control primers, and common primers used to diagnose the six common β-thalassemia mutations in this study are listed in Table 1. For each reaction there were total four primers used, two of these four primers serve as internal control. Control C and control D primers serve as internal control primers, which amplifies a part of β-globin gene at the 3′ end. These internal control primers also encompasses the 619 bp deletion common in the Indian Subcontinent, if the 619 bp deletion mutation is present it forms 242 bp PCR product. The PCR was carried out in a total volume of 25 μl of reaction mixture, containing 100 ng of genomic DNA, 1.0 μM of each primer, 1.5 mM MgCl2; 200 μM deoxynucleotides triphosphates, and 2.5 U Taq DNA polymerase. Cycling was carried out on the thermal cycler with an initial 5 min denaturation step at 95 °C, followed by 30 cycles at 94 °C for 30 s, 1 min at 65 °C annealing temperature, and 72 °C for 1 min 30 s and the final extension at 72 °C for 5 min. Following amplification, 10 μl aliquots were mixed with gel loading buffer and electrophoresed on 2 % agarose gel in Tris–borate–EDTA buffer for 1 h at 100 volts, gel was stained with ethidium bromide and visualized using UV trans-illuminator. The 3′ region of the DNA is used as an internal control [13–15]. The experiment was performed twice for each group. The photograph below shows agarose gel electrophoresis pattern of ARMS-PCR for analysis of IVS I-5 mutation.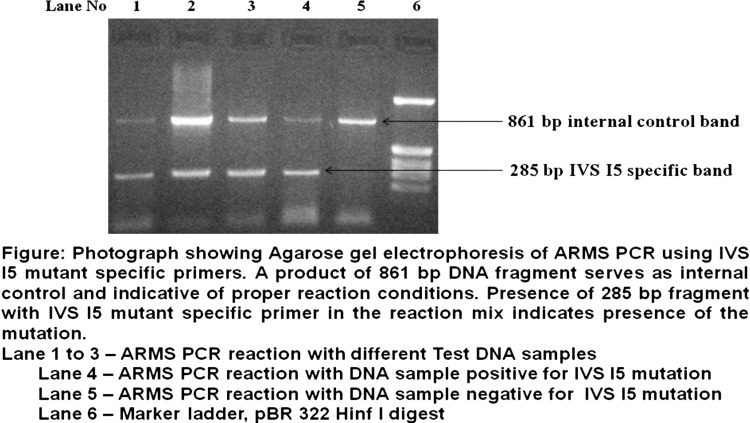
Table 1.
The mutation-specific ARMS primers, control primers, and common primers for six mutations included in the study
| Sr. No. | Mutation specific/common/control primer | Primer sequence (5′–3′) | Common primer to be used | Length of PCR product (bp) |
|---|---|---|---|---|
| 1 | Common A | CCCCTTCCTATGACATGAACTTAA | – | – |
| 2 | Common B | ACCTCACCCTGTGGAGCCAC | – | – |
| 3 | Control C | GAGTCAAGGCTGAGAGATGCAGGA | – | 861 |
| 4 | Control D | CAATGTATCATGCCTCTTTGCACC | – | |
| 5 | IVS I-5 (G → C) | CTCCTTAAACCTGTCTTGTAACCTTGTTAG | B | 285 |
| 6 | IVS I-1 (G → T) | TTAAACCTGTCTTGTAACCTTGATACGAAA | B | 281 |
| 7 | Cd 8/9 (+G) | CCTTGCCCCACAGGGCAGTAACGGCACACC | B | 225 |
| 8 | Cd 41/42 (–TCTT) | GAGTGGACAGATCCCCAAAGGACTCAACCT | B | 439 |
| 9 | Cd 15 (G → A) | TGAGGAGAAGTCTGCCGTTACTGCCCAGTA | A | 500 |
Results
In the present study, ARMS-PCR based analysis has been standardized for detection of common β-globin mutations. Mutational analysis was performed on 126 subjects who were proven carriers for β-thalassemia either during the screening or parents of known β-thalassemia major patients. DNA samples using PCR based techniques (ARMS) we studied six common mutations from India.
Blood samples of β-thalassemia carrier patients were collected and subjected to DNA isolation and mutational analysis. Out of 126 subjects of β-thalassemia trait, the G > C substitution at IVSI-5, a mutation of Asian-Indian origin, is the most common mutation reported in this study, a total of 82 (65.07 %) subjects were reported as IVS I-5 (G–C) mutation, a Mediterranean mutation IVS I-1(G–T) of lower frequency, was found in 12 (9.52 %) subjects. Asian-Indian mutation, Codon 8/9 (+G) was found at low frequency present in eight subjects (6.34 %). Codon 15 (G–A) an another Asian-Indian mutation, clustered around countries of the Gulf, but also found in Syria and Israeli Arabs, found in eight (6.34 %) subjects. There were five (3.96 %) subjects who showed mutation of codon 41/42 (–TCTT). 619 bp deletion of Indian origin was found in three subjects (2.38 %). There were eight (6.34 %) subjects who did not show any of the six mutations studied. Tables 2 shows mutational analysis of subjects, carrier for β-thalassemia. And Fig. 1 shows the graphical presentation of the six common mutations of β-thalassemia carrier subjects from south-western Maharashtra.
Table 2.
Mutational analysis of β-thalassemia carrier subjects
| Mutation | No. of subjects | Percentage (%) |
|---|---|---|
| IVS I-5 (G–C) | 82 | 65.07 |
| IVS I-1(G–T) | 12 | 9.52 |
| Codon 8/9 (+G) | 8 | 6.34 |
| Codon 15 (G–A) | 8 | 6.34 |
| Codon 41/42 (–TCTT) | 5 | 3.96 |
| 619 bp deletion | 3 | 2.38 |
| Uncharacterized | 8 | 6.34 |
| Total | 126 | 100 |
Fig. 1.

The graphical presentation of the six common mutations of β-thalassemia carrier subjects from south-western Maharashtra
Discussion
β-Thalassemia is one of the most common single gene disorder in India and in number of other countries. In the present study, ARMS-PCR has been standardized and applied for detection of β-thalassemia mutations. The common mutations that are prevalent in the Indian subcontinent have been chosen for this study.
Out of the six mutations studied, 619 bp deletion at the 3′ end of β-globin gene makes it nonfunctional. Frame shift mutations +1 codon 8/9 (+G) and −4 codon 41/42 (–TCTT) change the ribosome reading frame and causes premature termination of translation. Mutation IVS I-1 (G–T) causes change in splice junction causing ineffective RNA processing. IVS I-5 (G–C) destroys consensus sequences around splice junction which are essential for splicing. Mutation at codon 15 (G–A) causes a nonsense codon, terminating the synthesis of β-globin at a premature stage [16].
In the present study two mutations accounted for 94 subjects (74.60 %) out of total 126 subjects, these two mutations were IVS I-5 (G–C), and IVS I-1 (G–T). The other mutations namely, codon 8/9 (+G) and codon 15 (G–A) were present in eight subjects each, codon 41/42 (–TCTT) was present in five subjects and 619 bp deletion was found to be present in three subjects. There were eight (6.34 %) subjects who did not show any of the six mutations studied.
IVS I-5 (G–C) substitution which is β+ mutation was found to be the most frequent mutation in our study. This is totally in agreement with previous studies, reported from India and the Indian sub-continent. In Maharashtra, incidence of IVS I-5 mutation was reported to be 54 % [13, 17]. The incidence of this mutation reported at various regions from Indian sub-continent is, from Sindh region 12 %, Punjab 38 %, Gujarat 41 %, Bengal 60 %, and from Tamilnadu 81 %. The incidence reported from Tamilnadu is the highest reported incidence from any of the regions. Similar results were observed from Haryana (57 %), Uttar Pradesh (58 %), and eastern India (72 %) which correlate with our studies [12, 18, 19].
IVS I-1 (G–T) is one of the common mutation among Asian Indians [13] and phenotype is of β+ thalassemia. It is second common mutation which is observed in our study in 12 (9.52 %) subjects. The frequency was found to be similar to the previous studies from different geographic regions in India, Haryana (10 %), Uttar Pradesh (11 %), Eastern India (11 %), and southern India (5 %). In western India it was reported to be 20 % [20–22].
Codon 41/42 (–TCTT) mutation is one of the common mutations found in the Indian subcontinent. In our study the mutation was found in five subjects (3.96 %) out of 126 subjects. Incidence of this mutation was observed throughout India, from north-west Pakistan to Bangladesh and Bengal, and from Punjab to Tamilnadu. The incidence of this mutation was found to be 20 % in Bengal, 10 % in Tamilnadu, in Maharashtra 7.2 %, in Haryana it was 9 %, 3 % in Uttar Pradesh, 6 % in Eastern India, 5 % in Punjab, and 2 % in western India [12, 19].
Codon 8/9 (+G) mutation was present in eight (6.34 %) subjects out of total 126 subjects studied. This mutation has been reported from northern India (11 %) and from Gujarat (8.6 %) [15, 22, 23]. Varawalla et al. [12] reported incidence of this mutation from Sindh, Punjab, Gujarat, and Tamilnadu region.
In the present study, codon 15(G–A) mutation was detected in eight (6.3 %) subjects. The percent prevalence of codon 15 (G–A) was 2 % in Haryana, 11 % in Punjab, 6 % in Uttar Pradesh. In Gujarat it was 1.5 %, and in Maharashtra it was 18 % [12, 19, 20].
619 bp deletion was found only in three (2.3 %) of total subjects studied. This mutation was seen in Asian-Indians originally coming from Gujarat and Punjab [24]. Spectrum of 619 bp deletion mutation in India was found to be 16 % in Haryana, 14 % in Punjab, 5 % Uttar Pradesh, 2 % eastern India, 5 % in southern India and in western India it was found to be 8 % [20, 21]. Since this mutation originates from Punjab and Gujarat and the presence of these communities is relatively low and scattered in south-western Maharashtra region, incidence of this mutation may be low.
Since there is no proper screening program in place and usually the high risk couple (with a previous known thalassemia baby) approaches to the clinician for the sake of prenatal diagnosis at advanced stage of pregnancy, there is less time available for the mutational workup, Present study provides true picture of the spectrum of mutations in the subjects of β-thalassemia, which is vital from the view of prenatal diagnosis.
Electronic supplementary material
References
- 1.Weatherall DJ, Clegg JB. Thalassemia: a global public health problem. Nat Med. 1996;2:847–849. doi: 10.1038/nm0896-847. [DOI] [PubMed] [Google Scholar]
- 2.Christianson A, Howson CP, Modell B. March of dimes global report on birth defects. White Plains: March of Dimes Birth Defects Foundation; 2006. [Google Scholar]
- 3.WHO Guidelines for the control of hemoglobin disorders: report of the VIth annual meeting of the WHO working group on haemoglobinopathies, Cagliari, Sardinia, 8–9 April, 1989. Geneva, World Health Organization (unpublished document WHO/HDP/WG/HA/89.2).
- 4.Jain V, Sachdev A, Lokeshwar MR. Prevention of thalassemia. In: Sachdeva A, Lokeshwar MR, Shah N, Agarwal BR editors. Hemoglobinopathies. New Delhi: Jaypee Brothers Publishers; 2006. p. 94–104
- 5.Sobti PC, Gautam A. Beta- and-related thalassemias: clinical aspects. In: Sachdeva A, Lokeshwar MR, Shah N, Agarwal BR editors. Hemoglobinopathies. New Delhi: Jaypee Brothers Publishers; 2006. p. 49–59
- 6.Huisman THJ, Carver MFH, Baysal E. A syllabus of thalassemia mutations. Augusta: The Sickle Cell Anemia Foundation; 1997. [Google Scholar]
- 7.Bittles AH, Hussain R. An analysis of consanguineous marriage in the Muslim population of India at regional and state levels. Ann Hum Biol. 2000;27:163–171. doi: 10.1080/030144600282271. [DOI] [PubMed] [Google Scholar]
- 8.Steinberg MH, Forget B, Higgs DR, Nagel RL. Disorders of hemoglobin. New York: Cambridge University Press; 2001. [Google Scholar]
- 9.Weatherall DJ, Clegg JB. The thalassemia syndromes. Oxford: Oxford Blackwell Sciences; 2001. [Google Scholar]
- 10.Boehm CD, Antonarakis SE, Phillips JA, 3rd, Stetten G, Kazazian HH., Jr Prenatal diagnosis using DNA polymorphism: report on 95 pregnancies at risk for sickle-cell disease or β-thalassemia. N Engl J Med. 1983;308:1054–1058. doi: 10.1056/NEJM198305053081803. [DOI] [PubMed] [Google Scholar]
- 11.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning a laboratory manual. 2. New York: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 12.Varawalla NY, Old JM, Sarkar R, Venkatesan R, Weatherall DJ. The spectrum of beta thalassemia mutation on the Indian subcontinent: the basis of prenatal diagnosis. Brit J Haematol. 1991;78(2):242–247. doi: 10.1111/j.1365-2141.1991.tb04423.x. [DOI] [PubMed] [Google Scholar]
- 13.Varawalla NY, Old JM, Weatherall DJ. Rare β-thalassemia mutations in Asian Indians. Brit J Haematol. 1991;79:640–644. doi: 10.1111/j.1365-2141.1991.tb08094.x. [DOI] [PubMed] [Google Scholar]
- 14.Newton CR, Graham A, Heptinstall LE, Powell SJ, Summers C, Kalsheker N, et al. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS) Nucleic Acids Res. 1989;17:2503–2516. doi: 10.1093/nar/17.7.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bandyopadhyay A, Bandyopadhyay S, Chowdhury MD, Dasgupta UB. Major β-globin gene mutations in eastern India and their associated haplotypes. Hum Hered. 1999;49:232–235. doi: 10.1159/000022880. [DOI] [PubMed] [Google Scholar]
- 16.Haig HK, Jr, Corinne OB. Molecular basis and prenatal diagnosis of beta-thalassemia. Blood. 1988;72(4):1107–1116. [PubMed] [Google Scholar]
- 17.Agarwal S, Hattori Y, Agarwal SS. Rare beta thalassemia mutation in Asian Indians. Am J Hematol. 2000;65(4):322–323. doi: 10.1002/1096-8652(200012)65:4<322::AID-AJH14>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 18.Ambekar SS, Phadke MA, Balpande DN, Mokashi GD, Khedkar VA, Bankar MP, et al. The prevalence and heterogeneity of beta thalassemia mutations in the western Maharashtra population: a hospital based study. Int J Hum Genet. 2001;1(3):219–223. [Google Scholar]
- 19.Panigrahi I, Marwaha RK. Mutational spectrum of thalassemia in India. Indian J Hum Genet. 2007;13(1):36–37. doi: 10.4103/0971-6866.32034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verma IC, Saxena R, Thomas E, Jain PK. Regional distribution of thalassemia mutation in India. Hum Genet. 1997;100:109–113. doi: 10.1007/s004390050475. [DOI] [PubMed] [Google Scholar]
- 21.Vaz FE, Thakur CB, Banerjee MK, Gangal SG. Distribution of beta thalassemia mutation in the Indian population referred to diagnostic centre. Hemoglobin. 2000;24:181–194. doi: 10.3109/03630260008997526. [DOI] [PubMed] [Google Scholar]
- 22.Agarwal S, Naveed M, Gupta UR, Kishore P, Agarwal SS. Characterization of beta thalassemia mutation in 57 beta thalassemia families seen at Lucknow. Indian J Med Res. 1994;100:106–110. [PubMed] [Google Scholar]
- 23.Madan N, Sharma S, Rusia U, Sen S, Sood SK. Beta thalassemia in northern India (Delhi) Indian J Med Res. 1998;107:134–141. [PubMed] [Google Scholar]
- 24.Nigam N, Munshi N, Patel M, Soni A. Distribution of beta thalassemia mutation and its correlation with alpha thalassemia in Gujarati families. Int J Hum Genet. 2003;3(4):221–224. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.