

Abstract

Turn up the heat: An asymmetric total synthesis of the epoxykinamycin FL-120B’ is reported. The synthesis establishes a route to epoxide-containing diazobenzofluorenes which could potentially serve as monomers to the dimeric lomaiviticins. Key steps include Sharpless asymmetric epoxidation, Stille coupling, and intramolecular Friedel-Crafts acylation of atropisomeric carboxylic acids at elevated temperatures to construct the FL-120B’ core structure.
Keywords: antitumor agents, natural products, Friedel-Crafts, atropisomerism, total synthesis
Diazobenzofluorene natural products are a family of structurally complex molecules with a tetracyclic (ABCD) framework bearing a diazo moiety - a functionality sparsely found in Nature (Figure 1).[1] Members of this family differ in levels of functionalization of the cyclohexene moiety (D ring). Kinamycin C (1),[2] biosynthetically[3] derived from the epoxide-containing ketoanhydrokinamycin (2),[4] contains a D ring with four contiguous stereocenters. Other epoxykinamycins include FL-120B (3) and the closely related FL-120B’ (4).[5] Monomeric diazobenzofluorenes have been shown to exhibit antitumor properties.[2,6] Numerous studies have suggested that these biological activities may result from damage to DNA mediated by bioreductive pathways leading to loss of the diazo functional group.[6b,7] This unique family of natural products gained significant attention upon isolation of the dimeric diazobenzofluorenes lomaiviticins A (5) and B (6) by He and coworkers in 2001.[8] Demonstrated to be DNA-damaging agents, the lomaiviticins were found to display antibiotic acitivity against Gram-positive bacteria and potent cytotoxicity in several cancer cell lines. The C2-symmetric lomaiviticins may originate from C(2)-C(2’) linkage of precursors that closely resemble the monomeric kinamycins.
Figure 1.

Representative diazobenzofluorene natural products.
Since 2006, numerous research groups have reported total syntheses of monomeric diazobenzofluorene natural products[9] as well as studies towards the dimeric lomaiviticins.[10] Recently, Herzon and coworkers have reported a remarkable 11-step synthesis of the lomaiviticin aglycon.[11] Their dimerization approach utilizes an oxidative homo-coupling of a silyl enol ether derived from a protected monomer resembling 7 (Figure 2, a). Given the abundance of diazobenzofluorene natural products bearing an epoxide or oxygenated functionality at the C(2) position, we believe that a biosynthetic precursor to the lomaiviticins may also be depicted by proposed monomer 8 (Figure 2, b). This dimerization process may result from a reductive epoxide opening[12] event leading to pivotal carbon-carbon bond formation.[13] In this regard, we report the total synthesis of FL-120B’ (4) and development of methodologies to prepare diazobenzofluorenes with intact epoxides which may allow future access to the lomaiviticins and related compounds.
Figure 2.

Biomimetic approaches to the lomaiviticins.
In our retrosynthetic analysis, we believed that diazo installation of FL-120B’ (4) could be derived from a two-step process from a diazobenzofluorenone intermediate such as 9 (Figure 3). This strategy would first involve sulfonylhydrazone installation to a ketone precursor and a subsequent oxidation with spontaneous desulfination of the hydrazone to form the requisite diazo functionality.[9c–e] The synthesis of 9 may be achieved from naphthalene fragment 10 and epoxide 11 utilizing a Stille coupling and intramolecular Friedel-Crafts acylation using conditions previously described in our group’s synthesis of kinamycin C.[9b]
Figure 3.

Retrosynthesis for FL-120B’ (4). P = protecting group, MOM = methoxymethyl. TBS = t-butyldimethylsilyl.
In our prior synthesis,[9b] asymmetric nucleophilic epoxidation (D-DIPT, Ph3COOH, and NaHMDS)[14] was utilized to access chiral, non-racemic 11. Unfortunately, high levels of enantioselectivity were not achieved on a larger scale. However, Sharpless asymmetric epoxidation (Ti(Oi-Pr)4, L-DIPT, and t-BuOOH) of quinone monoketal 12 was performed on a 4.4 g scale to provide 13 in 98% yield with moderate enantioselectivity (68% ee) (Scheme 1).[15] A single recrystallization provided 13 in high enantiomeric excess (99% ee). Epoxide 13 was further elaborated to our desired epoxyketone fragment 11 as previously reported.[9b]
Scheme 1.

Enantioselective synthesis of epoxide 13. t-BuOOH (2.0 equiv), Ti(Oi-Pr)4 (0.10 equiv), L-DIPT (0.13 equiv), 4Å MS, CH2Cl2, 0 °C, 60 h. DIPT = diisopropyl tartrate, MS = molecular sieves.
For the synthesis of the AB ring subunit, quinone 14[9b] was reduced (Na2S2O4) and subsequently methylated (MeI) to provide bromonaphthalene derivative 15 (Scheme 2). Stannylation ((SnBu3)2, Pd(PPh3)4)[16] of 15 afforded aryl stannane 10. Stille coupling (Pd2(dba)3·CHCl3, CuCl, AsPh3, i-Pr2NEt)[17] of stannane 10 and bromide 11 afforded epoxyketone 16 in excellent yield (90%) as a 1.5 : 1 mixture of atropisomers[18] as indicated by 1H NMR analysis.[19] Acetylation of 16 followed by reduction with Super-Hydride (LiHBEt3) provided allylic alcohol 17 as a single diastereomer.[19] At this stage in the synthesis, three protecting groups for the secondary alcohol of substrate 17 were explored. Alcohol 17 was masked as acetate (Ac), 4-azidobutyrate (C(O)(CH2)3N3),[20] and t-butoxycarbonyl (Boc) groups to provide protected intermediates 18a, 18b, and 18c, respectively. Desilylation (HF·pyridine)[21] of 18a–c and oxidation with Dess-Martin periodinane[22] gave aldehydes 19a–c. Oxidation with NaClO2[23] afforded carboxylic acids 20a–c for evaluation in the pivotal intramolecular Friedel-Crafts acylation to form the tetracyclic framework of FL-120B’.
Scheme 2.

Forward synthesis to carboxylic acids 20a–c. a) Na2S2O4 (6.0 equiv), Et2O, H2O, rt, 10 min; b) MeI (6.0 equiv), K2CO3 (6.5 equiv), DMF, 0 °C→rt, 18 h, 66% yield (2 steps); c) (SnBu3)2 (1.3 equiv), Pd(PPh3)4 (0.1 equiv), toluene, 110 °C, 48 h, 74%; d) 11 (1.0 equiv), 10 (1.1 equiv), Pd2(dba)3·CHCl3 (0.1 equiv), AsPh3 (0.3 equiv), CuCl (0.6 equiv), i-Pr2NEt (1.1 equiv), MeCN, 72 °C, 3 h, 90% yield; e) Ac2O (30 equiv), pyr, rt, 6 h; f) Super-Hydride (LiHBEt3) (2.1 equiv), THF; 74% (2 steps) g) 18a: Ac2O (30 equiv), pyr, rt, 3 h, 96%; 18b: ClC(O)(CH2)3N3 (1.1 equiv), CH2Cl2 / pyr, 0 °C, 1 h, 73%; 18c: Boc2O (5.0 equiv), DMAP (5.5 equiv), CH2Cl2, rt, 4 h, 93%; h) HF·pyr (excess), THF / pyr, 0 °C; 15–20 h; i) DMP (2.0 equiv), pyr (2.1 equiv), CH2Cl2, rt, 3–5 h, 73–88% (2 steps); j) NaClO2 (9 equiv), NaH2PO4 (8 equiv), 2-methyl-2-butene (excess), t-BuOH / H2O, 0 °C→rt, 2–3 h, 78–90%. Dba = dibenzylideneacetone, DMF = N,N-dimethylformamide, pyr = pyridine, Boc = t-butoxycarbonyl, DMAP = N,N-dimethyl-4-aminopyridine, DMP = Dess-Martin periodinane.
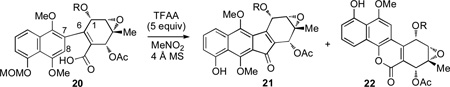
Treatment of carboxylic acids 20a–c with trifluoroacetic anhydride (TFAA) in a variety of solvents and at different temperatures gave varying ratios of the desired ketone products 21 and lactone byproducts 22 (Table 1). By adding TFAA to a preheated (80 °C) solution of 20a (R = Ac) in nitromethane as the optimal solvent, a 10 : 1 (crude 1H NMR analysis) mixture of C-acylation to O-acylation products (21a : 22a) was observed. Higher ratios (21 : 22) were observed with the use of the bulkier azidobutyrate (C(O)(CH2)3N3) and Boc groups (>20 : 1) (Entries 3–5, Table 1).[19] Interestingly, we found that higher reaction temperatures were required to achieve higher selectivities and yields for desired ketones products. For example, subjecting 20b to TFAA at rt gave a 4 : 1 (21b : 22b) selectivity and a 58% yield for ketone 21b (Entry 2, Table 1). The ratio was significantly increased (>20 : 1) when the reaction was performed at 50 °C and 80 °C affording 21b in 81% and 84% yields, respectively (Entries 3 and 4, Table 1).[19]
Table 1.
Studies on the intramolecular Friedel-Crafts acylation.
 | |||||
|---|---|---|---|---|---|
| Entry | Substrate | T [°C][a] | Selectivity [21 : 22][b] |
Yield [%][c] of 21 |
Yield [%][c] of 22 |
| 1 | 20a (R = Ac)[d] | 80 | 10 : 1 | 89 | 8 |
| 2 | 20b (R = C(O)(CH2)3N3)[d] | 23 | 4 : 1 | 58 | 15 |
| 3 | 20b [d] | 50 | >20 : 1 | 81 | - |
| 4 | 20b | 80 | >20 : 1 | 84 | - |
| 5 | 20c (R = Boc)[e] | 80 | >20 : 1 | 72[f] | - |
TFAA was added to the reaction mixture at the indicated temperature.
Selectivity was measured by crude 1H NMR analysis.
Yields are based on isolation from silica gel column chromatography.
The crude reaction mixture was further treated with TFA for complete MOM deprotection.
Treatment with pyridine[24] in EtOH was required to deprotect trifluroacetylated intermediates.
Boc group was cleaved during reactions: R = H. TFAA = trifluoroacetic anhydride, TFA = trifluoroacetic acid.
These results may be accounted for by the observation that acid substrates 20a–c were found to exist as a mixture of atropisomers (1.2–1.6 : 1 mixture by 1H NMR analysis). Variable temperature (VT) 1H NMR experiments indicated that the atropisomers equilibrate upon warming (H(8) proton shows coalescence at 80 °C, Figure 4) in which the barrier to rotation (ΔG ‡) for acid 20b was calculated to be 18 kcal/mmol.[25] This significant barrier to rotation may be directly correlated to atropisomeric acylium intermediates A1 and A2 (Scheme 3). At lower temperatures, A1 and A2 may not freely equilibrate and independently may give mixtures of intermediates B1 / C1 and B2 / C2, respectively (partial structures shown for clarity).[19,26] At higher temperatures, A1 and A2 may rapidly equilibrate leading to a thermodynamically-favored ketone intermediate. We believe that ketone intermediate B2 should be energetically preferred due to an unfavorable steric interaction between the C(16) aryl methoxy and C(1) protected alcohol in intermediate B1. In intermediate B2, the interaction between the C(16) aryl methoxy and H(1) is minimized and upon rearomatization affords the observed ketone products.
Figure 4.
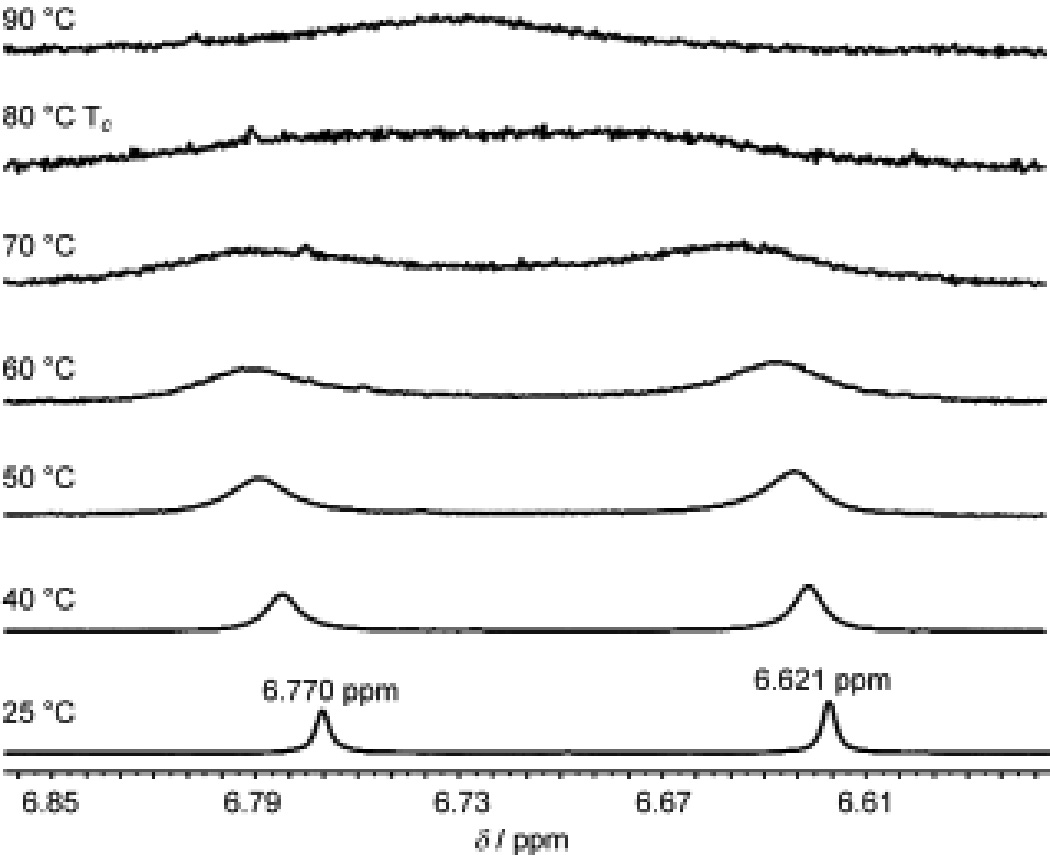
Variable temperature (VT) 1H NMR (500 MHz, CD3NO2) stackplot of acid 20b.
Scheme 3.

Proposed cyclization manifolds for intramolecular Friedel-Crafts acylation and lactone formation.
To complete the synthesis of FL-120B’, ketone 21c, derived from Boc-protected acid 20c, was protected (TBDPSCl) to give bis-silylated ketone 23 (Scheme 4). Initial attempts to form mesylhydrazone 24 failed to retain the sensitive epoxide moiety using various Lewis and Bronsted acids. Ultimately, trifluoroacetic acid (TFA) proved to be a suitable Bronsted acid with a non-nucleophilic counteranion to promote hydrazone formation to 24. Oxidation of 24 with ceric (IV) ammonium nitrate (CAN) provided the desired quinone with partial spontaneous desulfination to the desired diazo product. Treatment of the mixture with NEt3 provided full conversion to 25.[9d] In addition to 25, the parent ketone 23 was reformed (12% for three steps) through an oxidative or hydrolytic process.[27] With protected 25 in hand, desilylation with HF·pyrdine cleanly gave FL-120B’ (4). For comparison, a 4-step semi-synthesis of 4 from the closely related FL-120B (3) was also achieved.[19] Synthetic and semi-synthetic FL-120B’ gave matching 1H NMR and IR spectra as well as TLC and HPLC retention values. Furthermore, similar optical rotations for synthetic ([α]D23 = −132°) and semi-synthetic ([α]D23 = −128°) FL-120B’ allowed assignment of the absolute configuration for both FL-120B (3) and FL-120B’ (4) as depicted in Figure 1.
Scheme 4.

Completion of FL-120B’ (4). a) TBDPSCl (15 equiv), imid (20 equiv), DMAP (0.5 equiv), CH2Cl2, rt, 20 h, 84%. b) MsNHNH2 (25 equiv), TFA (8 equiv), i-PrOH / H2O, 72 h; c) CAN (3 equiv), CH3CN / pH 7 buffer, 0 °C, 1 h; d) NEt3 (10 equiv), CH2Cl2, rt, 1 h, 16% (3 steps); e) HF·pyridine (excess), THF, 0 °C→rt, 3 h, 51%. TBDPS = t-butyldiphenylsilyl, imid = imidazole, Ms = methanesulfonyl, CAN = cerium (IV) ammonium nitrate.
In summary, an asymmetric synthesis of FL-120B’ has been achieved using Sharpless asymmetric epoxidation, Stille cross coupling, and intramolecular Friedel-Crafts reactions as key steps. Notably, high reaction temperatures utilized for Friedel-Crafts acylation allowed selective formation of an intermediate that led to desired ketone products in a process likely involving atropisomeric intermediates. The completion of FL-120B’ represents the first total synthesis of an epoxide-containing, diazobenzofluorene natural product. Studies involving evaluation of reductive coupling of epoxykinamycins to access the lomaiviticins and related compounds will be reported in due course.
Acknowledgments
Financial support from the National Institutes of Health (RO1 CA137270) is gratefully acknowledged. We thank Arthur Su (Boston University) for helpful discussions, Dr. Paul Ralifo (Boston University) for NMR assistance, Dr. Norman Lee (Boston University) for HPLC assistance, and Dr. Jenn-jong Young (Institute of Preventive Medicine (Taiwan, ROC)) for generously providing a sample of FL-120B.
Footnotes
This work was presented in part at the 237th American Chemical Society National Meeting, Boston, MA, August 19–23, 2007; ORGN abstract 667.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.For a recent review on diazo natural products, see: Nawrat CC, Moody CJ. Nat. Prod. Rep. 2011;28:1426–1444. doi: 10.1039/c1np00031d.
- 2.(a) Ito S, Matsuya, T T, Omura S, Otani M, Nakagawa A. J. Antibiot. 1970;23:315–317. doi: 10.7164/antibiotics.23.315. [DOI] [PubMed] [Google Scholar]; b) Hata T, Omura S, Iwai Y, Nakagawa A, Otani M. J. Antibiot. 1971;24:353–359. doi: 10.7164/antibiotics.24.353. [DOI] [PubMed] [Google Scholar]; c) Omura S, Nakagawa A, Yamada H, Hata T, Furusaki A. Chem. Pharm. Bull. 1973;21:931–940. doi: 10.1248/cpb.21.931. [DOI] [PubMed] [Google Scholar]
- 3.For a review on the biosynthesis of diazobenzofluorene natural products, see: Gould SJ. Chem. Rev. 1997;97:2499–2509. doi: 10.1021/cr9600215.
- 4.Cone MC, Seaton PJ, Halley KA, Gould SJ. J. Antibiot. 1989;42:179–188. doi: 10.7164/antibiotics.42.179. [DOI] [PubMed] [Google Scholar]; b) Seaton PJ, Gould SJ. J. Antibiot. 1989;42:189–197. doi: 10.7164/antibiotics.42.189. [DOI] [PubMed] [Google Scholar]
- 5.Lin HC, Chang SC, Wang NL, Chang LR. J. Antibiot. 1994;47:675–680. doi: 10.7164/antibiotics.47.675. [DOI] [PubMed] [Google Scholar]; b) Young J-J, Ho SN, Ju WM, Chang LR. J. Antibiot. 1994;47:681–687. doi: 10.7164/antibiotics.47.681. [DOI] [PubMed] [Google Scholar]
- 6.(a) Hasinoff BB, Wu X, Yalowich JC, Goodfellow V, Laufer RS, Adedayo O, Dmitrienko GI. Anti-Cancer Drugs. 2006;17:825–837. doi: 10.1097/01.cad.0000224442.78211.27. [DOI] [PubMed] [Google Scholar]; (b) O’hara KA, Wu X, Patel D, Liang H, Yalowich JC, Chen N, Goodfellow V, Adedayo O, Dmitrienko GI, Hasinoff BB. Free Radical Biol. Med. 2007;43:1132–1144. doi: 10.1016/j.freeradbiomed.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Feldman KS, Eastman KJ. J. Am. Chem. Soc. 2005;127:15344–15345. doi: 10.1021/ja053880w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Feldman KS, Eastman KJ. J. Am. Chem. Soc. 2006;128:12562–12573. doi: 10.1021/ja0642616. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ballard TE, Melander C. Tetrahedron Lett. 2008;49:3157–3161. [Google Scholar]; (d) Khdour O, Skibo EB. Org. Biomol. Chem. 2009;7:2140–2154. doi: 10.1039/b903844b. [DOI] [PubMed] [Google Scholar]; (e) Heinecke CL, Melander C. Tetrahedron Lett. 2010;51:1455–1458. [Google Scholar]
- 8.He H, Ding D, Bernan VS, Richardson AD, Ireland CM, Greenstein M, Ellestad GA, Carter GT. J. Am. Chem. Soc. 2001;123:5362–5363. doi: 10.1021/ja010129o. [DOI] [PubMed] [Google Scholar]
- 9.(a) Hauser FM, Zhou M. J. Org. Chem. 1996;61:5722. [Google Scholar]; (b) Lei X, Porco JA., Jr J. Am. Chem. Soc. 2006;128:14790–14791. doi: 10.1021/ja066621v. [DOI] [PubMed] [Google Scholar]; (c) Kumamoto T, Kitani Y, Tsuchiya H, Yamaguchi K, Seki H, Ishikawa T. Tetrahedron. 2007;63:5189–5199. [Google Scholar]; (d) Birman VB, Zhao Z, Guo L. Org. Lett. 2007;9:1223–1225. doi: 10.1021/ol0629768. [DOI] [PubMed] [Google Scholar]; (e) Nicolaou KC, Li H, Nold AL, Pappo D, Lenzen A. J. Am. Chem. Soc. 2007;129:10356–10357. doi: 10.1021/ja074297d. [DOI] [PubMed] [Google Scholar]; (f) Woo CM, Lu L, Gholap SL, Smith DR, Herzon SB. J. Am. Chem. Soc. 2010;132:2540–2541. doi: 10.1021/ja910769j. [DOI] [PubMed] [Google Scholar]; (g) Kimura S, Kobayashi S, Kumamoto T, Akagi A, Sato N, Ishikawa T. Helv. Chim. Acta. 2011;94:578–591. [Google Scholar]
- 10.(a) Nicolaou KC, Denton RM, Lenzen A, Edmonds DJ, Li A, Milburn RR, Harrison ST. Angew. Chem. 2006;118:2130–2135. doi: 10.1002/anie.200504466. [DOI] [PubMed] [Google Scholar]; Nicolaou KC, Denton RM, Lenzen A, Edmonds DJ, Li A, Milburn RR, Harrison ST. Angew. Chem. Int. Ed. 2006;45:2076–2081. doi: 10.1002/anie.200504466. [DOI] [PubMed] [Google Scholar]; (b) Zhang W, Baranczak A, Sulikowski GA. Org. Lett. 2008;10:1939–1941. doi: 10.1021/ol800460a. [DOI] [PubMed] [Google Scholar]; (c) Krygowski ES, Murphy-Benenato K, Shair MD. Angew. Chem. 2008;120:1704–1708. doi: 10.1002/anie.200704830. [DOI] [PubMed] [Google Scholar]; Krygowski ES, Murphy-Benenato K, Shair MD. Angew. Chem. Int. Ed. 2008;47:1680–1684. doi: 10.1002/anie.200704830. [DOI] [PubMed] [Google Scholar]; (d) Nicolaou KC, Nold AL, Li H. Angew. Chem. 2009;121:5974–5977. [Google Scholar]; Nicolaou KC, Nold AL, Li H. Angew. Chem. Int. Ed. 2009;48:5860–5863. doi: 10.1002/anie.200902509. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Gholap SL, Woo CM, Ravikumar PC, Herzon SB. Org. Lett. 2009;11:4322–4325. doi: 10.1021/ol901710b. [DOI] [PubMed] [Google Scholar]; (f) Lee HG, Ahn JY, Lee AS, Shair MD. Chem. Eur. J. 2010;16:13058–13062. doi: 10.1002/chem.201002157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herzon SB, Lu L, Woo CM, Gholap SL. J. Am. Chem. Soc. 2011;133:7260–7263. doi: 10.1021/ja200034b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For an analogous reductive epoxide opening for dynemicin A, see: Sugiura Y, Shiraki T, Konishi M, Oki T. Proc. Natl. Acad. Sci, U.S.A. 1990;87:3831–3835. doi: 10.1073/pnas.87.10.3831.
- 13.For reductive dimerization of anthracycline antitumor drugs, see: Gaudiano G, Koch TH. Chem. Res. Toxicol. 1991;4:2–16. doi: 10.1021/tx00019a001. and references therein.
- 14.Li C, Johnson RP, Porco JA., Jr J. Am. Chem. Soc. 2003;125:5095–5106. doi: 10.1021/ja021396c. [DOI] [PubMed] [Google Scholar]
- 15.For Sharpless asymmetric epoxidation of an electron deficient quinone monoketal, see: Clark DA, Riccardis FD, Nicolaou KC. Tetrahedron. 1994;50:11391–11426.
- 16.Azizian H, Eaborn C, Pidcock A. J. Organomet. Chem. 1981;215:49–58. [Google Scholar]
- 17.Bobe F, Tunoori AR, Niestroj AJ, Gronwald O, Maier ME. Tetrahedron. 1996;52:9485–9498. [Google Scholar]
- 18.For reviews on atropisomerism, see: Bringmann G, Mortimer AJP, Keller PA, Gresser MJ, Garner J, Breuning M. Angew. Chem. Int. Ed. 2005;44:5384–5427. doi: 10.1002/anie.200462661. Clayden J, Moran WJ, Edwards PJ, LaPlante SR. Angew. Chem. Int. Ed. 2009;48:6398–6401. doi: 10.1002/anie.200901719. Bringmann G, Gulder T, Gulder TAM, Breuning M. Chem. Rev. 2011;111:563–569. doi: 10.1021/cr100155e.
- 19.See Supporting Information for complete details.
- 20.Kusumoto S, Sakai K, Shiba T. Bull. Chem. Soc. Jpn. 1986;59:1296–1298. [Google Scholar]
- 21.Carreira EM, Du Bois J. J. Am. Chem. Soc. 1995;117:8106–8125. [Google Scholar]
- 22.(a) Dess DB, Martin JC. J. Org. Chem. 1983;48:4155–4156. [Google Scholar]; (b) Ireland RE, Liu L. J. Org. Chem. 1993;58:2899. [Google Scholar]
- 23.(a) Lindgren BO, Nilsson T. Acta. Chem. Scand. 1973;27:888–890. [Google Scholar]; (b) Kraus GA, Taschner MJ. J. Org. Chem. 1980;45:1175–1176. [Google Scholar]; (c) Bal BS, Childers WE, Jr, Pinnick HW. Tetrahedron. 1981;37:2091–2096. [Google Scholar]
- 24.Takatani M, Matsuo I, Ito Y. Carbohydr. Res. 2003;338:1073–1081. doi: 10.1016/s0008-6215(03)00099-5. [DOI] [PubMed] [Google Scholar]
- 25.For a recent review on the use of dynamic NMR spectroscopy for conformational analysis, see: Casarini D, Lunazzi L, Mazzanti A. Eur. J. Org. Chem. 2010:2035–2056.
- 26.For intramolecular Friedel-Crafts acylation of separable atropisomers giving different ketone and lactone product distributions, see: Yonemoto K, Nakai Y, Yamamoto G, Oki M. Chem. Lett. 1985:1739–1742.
- 27.For CAN-mediated oxidation of hydrazones, see: Bird JW, Diaper DGM. Can. J. Chem. 1969;47:145–150.