
FIGURE 7.
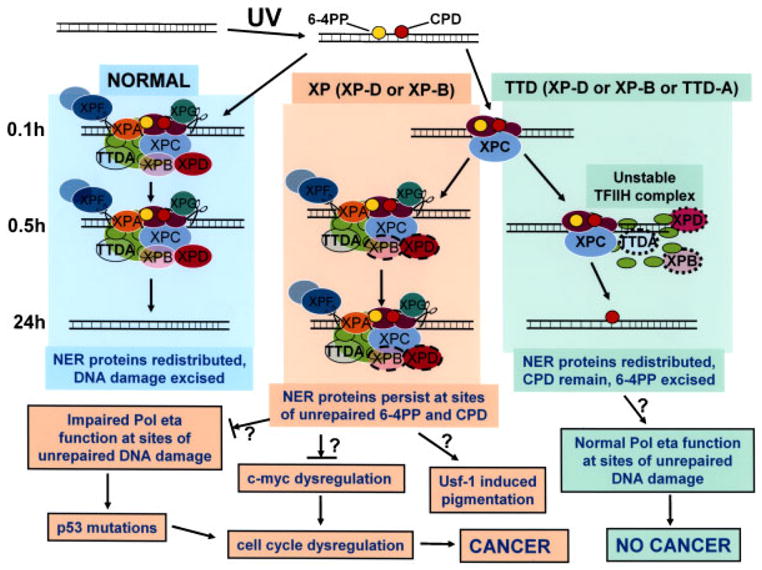
Schematic diagram of repair of UV-induced DNA damage (6-4PP and CPD) in normal, XP (XP-D or XP-B), and TTD (XP-D, XP-B, or TTD-A) cells. DNA is damaged by UV producing 6-4PP (yellow circles) and CPD (red circles). In normal cells (left column) by 0.1hr NER proteins are localized at the site of DNA damage. These proteins remain localized at 0.5 hr but by 24 hr the NER proteins are redistributed and the DNA damage has been excised in the normal cells. In contrast, in XP (XP-D or XP-B) cells (center column) by 0.1hr only XPC is visualized at the site of localized DNA damage. By 0.5 hr the NER proteins, including mutated XPD (dark pink oval with dashed border) and mutated XPB (light pink oval with dashed border), are recruited to the site of localized DNA damage. These proteins persist at 24 hr at sites of unrepaired 6-4PP and CPD. In TTD (XP-D, or XP-B or TTD-A) cells (right column) by 0.1hr only XPC is visualized at the site of localized DNA damage as in the XP cells. By 0.5 hr the NER proteins includingmutated XPD (dark pink oval with dotted border), mutated XPB (light pink oval with dotted border), and mutated TTDA (blue/green oval with dotted border) are recruited to the site of localized DNA damage in only a small proportion of the cells because of the instability of the TFIIH complex. At 24 hr the NER proteins have redistributed, CPD remain and 6-4PP are repaired. The NER proteins include DDB1-DDB2 (XPE) (purple ovals); XPC (light blue oval), which is complexed to HR23b and centrin 2; components of TFIIH: XPD (dark pink oval), XPB (light pink oval), TTDA (blue/green oval), and seven other proteins (light green ovals); and endonucleases XPF (dark gray oval) complexed to ERCC1 (light gray oval) and XPG (dark green oval). In the XP (XP-D or XP-B) cells the persistent NER proteins at the site of unrepaired 6-4PP and CPD may block progression of the translesion polymerase, pol eta, thereby creating a partial mimic of XP variant cells. This impaired pol eta function could result in increased mutagenesis of genes such as p53, which lead to cell cycle dysregulation that my eventually lead to cancer. In addition, mutations in TFIIH components have been shown to result in c-myc dysregulation with resulting cell-cycle dysregulation. The increased pigmentation in XP patients may be related to stimulation of tyrosinase activity via the UV-responsive transcription factor, Usf-1. In contrast, in TTD (XP-D, XP-D, or TTD-A) cells the unstable TFIIH complex is sufficient to repair 6-4PP but NER proteins do not persist at the site of unrepaired CPD. The translesion polymerase, pol eta can function normally to bypass the unrepaired DNA damage and thus does not induce an elevated frequency of cancer. (See discussion for more details.)