

Abstract
The treatment of patients with hematopoietic progenitor and stem cells (HPSCs) to reconstitute hematopoiesis after myeloablative therapy or to repair ischemia after myocardial infarction has significantly improved clinical outcomes. Successful blood or bone marrow transplants require a sufficient number of HPSCs capable of homing to the injured site to regenerate tissue. Granulocyte-colony stimulating factor (G-CSF) is widely used clinically for stem cell mobilization. However, in some patients the response is poor, thus a better understanding of the mechanisms underlying G-CSF-regulated stem cell mobilization is needed. The pasminogen (Plg) system is the primary fibrinolytic pathway responsible for clot dissolution after thrombosis. Recent evidence suggests that Plg plays a pivotal role in stem cell mobilization from the bone marrow to the peripheral circulation, particularly in HPSC mobilization in response to G-CSF. This paper will discuss the potential mechanisms by which the Plg system regulates stem cell mobilization, focusing on stepwise proteolysis and signal transduction during HPSC egress from their bone marrow niche. Clear elucidation of the underlying mechanisms may lead to the development of new Plg-based therapeutic strategies to improve stem cell mobilization in treating hematological and cardiovascular diseases.
1. Introduction
Recruitment of hematopoietic progenitor and stem cells (HPSCs) to the blood followed by chemotherapy or cytokine treatment is a clinical process termed stem cell mobilization. This process mimics enhancement of the physiological release of stem cells and progenitors from the bone marrow (BM) reservoir in response to stress signals during injury and inflammation [1]. Currently, stem cells mobilized to the peripheral blood after treatment are the preferable source of HPSCs harvested for stem cell transplantation because of faster engraftment and reduced procedural risks compared with the direct harvest of the BM cells [2, 3]. Successful stem cell therapy, both autologous and allogeneic, requires the infusion of a sufficient number of HPSCs capable of homing to the injured site to promote tissue repair. Granulocyte-colony stimulating factor (G-CSF) is the most commonly used mobilizing agent to recruit HPSC from the BM; however, impaired response to G-CSF is observed in 25% of patients and 10% to 20% of healthy donors [4–6]. This has led to studies to identify the mechanisms underlying cytokine-induced stem cell mobilization that could offer better strategies to enhance stem cell mobilization.
Plasmin, a serine protease, degrades fibrin and is the primary enzyme for clot lysis, a process called fibrinolysis [7]. Plg, the zymogen of plasmin, can be converted to plasmin at different locations by tissue Plg activator (tPA) or urokinase Plg activator (uPA). While tPA is distributed in the ECM of most cell types, uPA is mainly localized on the cell surface via its highly specific cell surface receptor, uPAR [8]. The activities of uPA and tPA are regulated by Plg activator inhibitor-1 (PAI-1). In addition, plasmin activity can be inhibited directly by α2-antiplasmin and α2-macroglobulin [9]. Several studies [10, 11] have reported a marked increase in Plg binding to differentiated cells compared to undifferentiated cells, suggesting an important role for Plg activation for cell differentiation that requires the release and migration of cells from microenvironments. Recent evidence reveals that the Plg system is crucial for cytokine- or chemotherapy-induced stem cell mobilization. Here, we will discuss the role of the Plg system in regulating stem cell mobilization.
2. Regulation of Stem Cell Mobilization via a Plg-Dependent Proteolytic Pathway
2.1. Stem Cell Mobilization and Plg Function
The egress of stem cells from the BM involves interfering with the physiological interaction between stromal cells and hematopoietic cells, which regulate both cell transmigration and the BM remodeling processes. The dynamic secretion and inactivation of the chemokines, stromal cell derived factor-1 (SDF-1), membrane-bound Kit ligand (KitL), and interleukin-8 (IL-8), which anchor HPSCs in the BM microenvironment, are critical for establishing the chemoattractant gradient between the BM and the peripheral blood for stem cell mobilization. Degradation and functional inactivation of the BM extracellular matrix proteins (ECM) by proteases such as elastase, cathepsin G and MMP-9, are also indicated as major players in stem cell mobilization [1, 12].
In addition to its role in fibrinolysis [13], Plg has many other functions. Interaction of Plg with its cell surface receptors and activation to plasmin, results in degradation of matrix proteins and activation of cytokines [14]. Plg directly binds to the ECM and upon its conversion to plasmin, degrades multiple ECM proteins including fibrin, laminin, and fibronectin [15–17]. Plasmin can also activate other proteases, such as MMP-3, MMP-9, MMP-12, and MMP-13 [18, 19] to degrade other matrix components such as collagens [20].
2.2. Role of Plg in Chemotherapy-Induced Stem Cell Mobilization
Many studies indicate that the Plg system facilitates HPSC mobilization through plasmin-mediated proteolytic mechanisms, by which plasmin inactivates chemotactic cytokines and degrades ECM in the BM compartment. Heissig et al. [21] have shown that deletion of Plg prevents hematopoietic stem cells from entering the cell cycle and undergoing multilineage differentiation after 5-FU treatment, causing lethality in mice. Activation of Plg by administration of tPA promoted cleavage of KitL mediated by MMP-9 secreted from stromal cells, subsequently enhancing HPSC proliferation, differentiation, and mobilization. This data suggests that Plg regulates HPSC function via MMP-9-mediated KitL release. Consistently, Tjwa et al. [22] reported that hematopoietic recovery upon delivery of 5-FU was impaired in Plg, tPA and uPA deficient mice. Moreover, depletion of fibrinogen in Plg deficient (Plg−/−) mice did not restore hematopoietic recovery, indicating that Plg-regulated stem cell mobilization is fibrinolysis-independent. Instead, Plg deficiency inhibited breakdown of the BM matrix proteins fibronectin, VCAM-1, and laminin, which are required for adhesion of HPSCs to their BM microenvironment and also in transendothelial migration of HPSCs. These studies indicate that Plg and its activators are required for hematopoietic regeneration by regulating HPSC mobilization, proliferation, differentiation through MMP-9-mediated release of KitL, and plasmin-mediated degradation of ECM in the BM.
2.3. Role of Plg in G-CSF-Induced Stem Cell Mobilization
Plg is also necessary for HPSC mobilization in response to G-CSF. Tjwa et al. [23, 24] have found that genetic loss of PAI-1 or plasmin inhibitor α2-antiplasmin, which enhances plasmin generation, increased HPSC mobilization in response to G-CSF, and thrombolytic agents such as tenecteplase and microplasmin, enhanced HPSC mobilization in mice and humans. Tenecteplase is a mutant of recombinant human tPA, which has a prolonged half-life and is used for treatment of acute cardiovascular and cerebrovascular syndromes. Microplasmin is a truncated form of plasmin that has an improved safety profile (less bleeding) and is easier to produce as a recombinant protein than is plasmin. Studies of Plg−/− mice [24, 25] validated that Plg is required for G-CSF-induced stem cell mobilization. Fewer HPSCs in the peripheral blood were detected in Plg−/− mice compared with wild-type (WT) mice after treatment with G-CSF. Similarly, WT mice treated with the Plg inhibitor tranexamic acid also impaired HPSC mobilization into the circulation. Furthermore, cleavage of the uPA receptor, (uPAR), by plasmin may account for Plg-regulated stem cell mobilization. Studies suggested that cleavage of the receptor uPAR is an important factor in regulating stem cell function [24]. First, uPAR was found to be expressed on a subpopulation of HPSCs, and HPSC mobilization was impaired in uPAR-deficient mice (uPAR−/−). Second, intact uPAR is required for adhesion of HPSCs to the BM as well as homing and engraftment of HPSCs. During stem cell mobilization, uPAR is cleaved and subsequently stem cells are released from the BM to the circulation. Cleavage of uPAR was detected only in WT mice but not in Plg−/− mice during stem cell mobilization, suggesting that plasmin regulates stem cell mobilization by inactivating uPAR via proteolytic cleavage [24]. These findings suggest that uPAR is essential for Plg-regulated stem cell mobilization. However, less inhibition in HPSC mobilization was observed in uPAR−/− mice compared to Plg−/− mice [24], suggesting that there are other mechanisms for Plg regulation of G-CSF induced HPSC mobilization besides uPAR cleavage. While kitL is important in myelosuppression-induced HPSC mobilization, it does not seem to be involved in G-CSF-induced HPSC mobilization since G-CSF does not affect its levels [25, 26].
2.4. Role of Plg in the Regulation of SDF-1/CXCR4 Pathway
Another crucial pathway controlling stem cell mobilization is the SDF-1/CXCR4 signal. Under basal conditions, SDF-1/CXCR4 anchor HPSC in the BM and keep them in a quiescent state. During stem cell mobilization, SDF-1 in the BM is downregulated and HPSCs are released and mobilized into the circulation in response to the higher SDF-1 concentration in the peripheral blood [27, 28]. Our recent study has established the interplay between Plg and SDF-1/CXCR4 signals. Our data have shown that Plg is required for G-CSF-induced HPSC egress to sinusoidal capillaries in the BM and subsequent mobilization to peripheral circulation. G-CSF induced Plg-dependent activation of MMP-9 in the BM, and MMP-9 neutralization or deficiency suppressed HPSC migration and mobilization. Reconstitution of MMP-9 activity by the BM transplantation after lentiviral overexpression rescued HPSC mobilization in Plg−/− mice, indicating that MMP-9 activation is required for Plg-mediated HPSC mobilization. Interestingly, after G-CSF simulation, Plg downregulated SDF-1 in the BM and spatiotemporally regulated the expression of CXCR4 on mobilized HPSC. Reconstitution of MMP-9 activity in Plg−/− mice reversed CXCR4 expression on HPSC in plasma and the BM, suggesting that CXCR4 serves as a newly identified downstream signal of Plg/MMP-9 in HPSC mobilization [25].
Taken together, these data indicate that the Plg system plays a crucial role in chemotherapy- or cytokine-induced stem cell mobilization. It functions through activating plasmin-mediated proteolytic activity to degrade the ECM in the BM (such as fibronectin or laminin) or by inactivating some key cytokines in the BM niches, such as KitL/c-Kit (KitL receptor), uPAR, and SDF-1/CXCR4, thus eventually leading to the release of HPSCs and the facilitation of their egress from the BM to the circulation (see proposed pathway in Figure 1).
Figure 1.
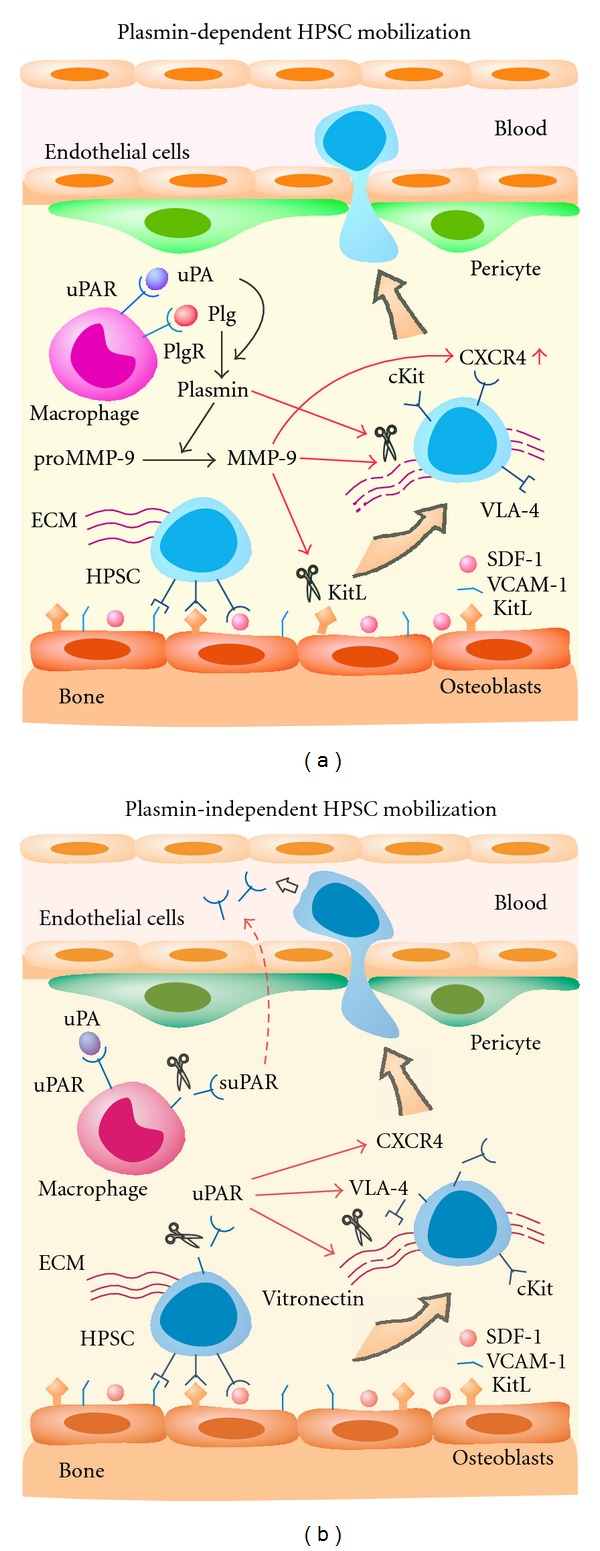
Plasmin-dependent and -independent mechanisms for HPSC mobilization from the BM to circulation. (a) Plasmin-mediated proteolysis induces HPSC mobilization. uPA converts Plg into protease-active plasmin that activates pro-MMP-9. Active MMP-9 cleaves KitL and upregulates CXCR4, and MMP-9 and plasmin degrade ECM, both of which release HPSC from the BM, leading to HPSC egress to circulation. (b) Plasmin-independent proteolysis and chemotaxis induce HPSC mobilization. uPAR is cleaved to chemotactic suPAR that drives HPSC migration to circulation. Cleavage of membrane uPAR also directly disrupts the interaction between uPAR and VLA-4, degrades vitronectin, and desensitizes the CXCR4 signal, which leads to HPSC mobilization.
3. Regulation of Stem Cell Mobilization in the Plg-Independent Signaling Pathway
3.1. Urokinase Plg Activator Receptor, uPAR
In addition to proteolytic activity, other regulatory pathways are involved in the Plg system-mediated HPSC mobilization. uPAR was originally identified as a key factor for the activation of Plg to plasmin and thereby the regulation of cell surface proteolysis in space and time [29]. The structure of uPAR consists of three homologous domains of ~90 amino acids each (D1, D2, and D3 as numbered from the NH2 terminus) and is anchored to the cell membrane through a glycosyl-phosphatidylinositol tail, attached to the C terminal D3 domain [30]. Interestingly, uPAR, independent of proteolytic activity, regulates migration and adhesion of cells through binding to integrins and G-protein-coupled receptors and initiates intercellular signaling cascades [31]. Previous reports [32–34] have shown that uPAR regulates cell adhesion and migration by activating its downstream intracellular signaling pathways in various cell types [35].
3.2. Stem Cell Mobilization and uPAR
Recently, a critical role of uPAR in stem cell mobilization has also been documented [24, 36–39]. During G-CSF-induced HPSC mobilization in humans, uPAR expression significantly increased on peripheral blood mononuclear cells (PBMNCs), in particular on CD33+ myeloid precursors and on CD14+ monocytic cells released from the BM into the circulation. By contrast, CD34+ cells and T and B lymphocytes were uPAR-negative, suggesting that uPAR may play a selective role in stem cell mobilization [36]. In uPAR−/− mice, stem cell mobilization induced by G-CSF treatment was impaired [24]. A very recent study [39] has demonstrated that uPAR is required to mobilize mesenchymal stem cells (MSC) from the BM of mice stimulated with G-CSF in vivo. Down- and up-regulation of uPAR inhibited and stimulated MSC differentiation into vascular smooth muscle cells, respectively. Consistently, infusion of MSCs isolated from uPAR−/− mice impaired its engraftment to injured femoral artery. These data indicate a role of uPAR in stem cell mobilization and engraftment. Additional evidence suggests several mechanisms by which uPAR plays a role in stem cell mobilization: chemotactic role of cleaved uPAR, regulation of integrins, and regulation of CXCR4 signaling.
3.3. The Chemotactic Role of Cleaved uPAR in Stem Cell Mobilization
Proteolytic cleavage of membrane bound uPAR in the linker region between D1 and D2 and at the juxtamembrane domain from the cell surface by plasmin or other proteases releases truncated uPAR (suPAR) into the extracellular space, where it may be proteolytically cleaved into smaller fragments (c-suPAR) [40–43]. uPAR fragments generated from uPAR cleavage are essential for uPAR-regulated stem cell mobilization. Previous studies have shown [36] that G-CSF treatment induced an increase in uPAR as well as suPAR. c-suPAR were released in vitro by the PBMNCs and were also detectable in the serum of G-CSF-treated donors. Fietz et al. [44] have confirmed that both uPAR and cleaved forms of uPAR are increased in HPSC donors following G-CSF treatment. Moreover, c-suPAR and its derived peptide (uPAR84–95) induce in vitro migration of bone marrow HPSCs towards SDF-1. Furthermore, the chemotactic human c-suPAR peptide has been shown to mobilize HPSC in mice. Similarly, administration of human uPAR84–95 peptides induced mobilization of CD34+ HPSCs into the circulation to an extent similar to that observed in G-CSF in mice [36].
In agreement with these findings, utilizing mice with deleted uPA, tPA, uPAR, and Plg genes, Tjwa, et al. [33] have found that uPAR is expressed on the BM cells that are in close contact with osteoblasts as well as a subset of HPSCs. At steady state, uPAR−/− mice are partially depleted of HPSCs in the BM with a decrease of cell cycle quiescence and chemoprotection. In addition, uPAR−/− mice are impaired in HPSC mobilization, homing, and short-term engraftment. The membrane-anchored uPAR retention signal on HPSCs is inactivated by plasmin via proteolytic cleavage to a c-uPAR truncated product, which stimulates HPSC mobilization. These studies suggest that uPAR serves as a new anchor factor, similar to KitL/c-Kit to maintain HPSC retention in the BM, while cleaved soluble uPAR is a new chemoattractant and mobilizer of stem cell egress from the BM to the circulation.
3.4. The Role of uPAR in the Regulation of Integrins
Lacking transmembrane and intracellular domains, uPAR must cooperate with transmembrane receptors to activate intracellular signaling. Extensive studies suggest that integrins, a major family of ECM receptors are signaling coreceptors of uPAR [45–49]. Besides uPA, the ECM glycoprotein vitronectin has been identified as a second ligand for uPAR [50]. It is reported that blocking uPAR-vitronectin binding impaired uPAR-regulated cell morphology, adhesion, and migration [45, 46]. Recent studies suggest that uPAR binding to vitronectin activates integrin signaling by simply increasing plasma membrane—ECM contact, facilitating integrin—ligand interactions [45]. Namely, vitronectin may act as an adaptor for the interaction of uPAR and integrins. The major integrins that uPAR interacts with are α1β1 and α4β1 (very late antigen-4, VLA-4) [51, 52]. The integrin α4β1 regulates the migration and adhesion of HPSCs to fibronectin and VCAM-1 during their homing and engraftment in the BM [53–56]. The function of α4β1 also depends on the presence of intact uPAR, as only intact uPAR interacts with the integrin [43, 56]. Removal of DI from uPAR reduces α4β1-mediated cellular adhesion in vitro [56]. Thus, when uPAR is depleted, such as in uPAR−/− mice, or inactive, such as after preincubation of WT HPSCs with anti-uPAR antibody, adhesion of α4β1 on HPSCs to the BM matrix is reduced, likely explaining why homing and engraftment of HPSCs are impaired [24]. Furthermore, anti-α4β1 antibodies could not further aggravate the adhesion defects of HPSCs when uPAR was absent or blocked. Likewise, homing and engraftment defects were similar in mice lacking functional uPAR or α4β1 [24]. These data suggest that cooperation of uPAR with α4β1 may partially contribute to the uPAR-mediated HPSC mobilization.
3.5. The Role of uPAR in the Regulation of CXCR4 Signal
Recent studies have revealed that functional interactions between the uPA-uPAR system and receptors for N-formylated peptides, such as the fMet-Leu-Phe (fMLP), are important for leukocyte chemotaxis [57]. The peptide, c-suPAR and its derived chemotactic peptide uPAR84–95, corresponding to the uPAR chemotactic region unmasked by D1-D2 cleavage, can induce monocyte chemotaxis by FPRL1 activation [58]. FPRL1 belongs to the family of fMLP receptors; the other 2 members are FPR and FPRL2 [59]. Interestingly, activation of both FPR and FPRL1 can lead to the desensitization of other chemokine receptors, such as CXCR4 [60, 61]. CXCR4 and its specific ligand SDF-1 strongly contribute to retention of HPSCs in the BM since the downregulation of the CXCR4/SDF1 signal pathway increases HPSC mobilization [27, 28, 62]. Several studies have investigated whether suPAR is also able to interfere with the CXCR4/SDF-1 axis through fMLP receptors [36, 37]. The results have shown that SDF-1-dependent BM HPSC in vitro migration was impaired by uPAR84–95 through the activation of FPR. Serum c-suPAR in vivo can also regulate CD34+ HPSC mobilization by downmodulating CXCR4 activity [37]. SDF-1 also induced chemotaxis of the BM CD34+ HPSCs isolated from 3 donors, and pretreatment with fMLP or uPAR84–95 completely abolished SDF1-dependent migration [36]. These data indicate that uPAR may regulate HPSC migration through FPR-mediated CXCR4 desensitization.
Altogether, these data suggest that uPAR regulates stem cell mobilization through several possible mechanisms. During G-CSF-induced HPSC mobilization, uPAR expression is first upregulated on CD33+ and CD14+ cells and is then cleaved, thus generating chemotactic forms of suPAR that present in the serum of G-CSF-treated donors. In the first case, cleavage of uPAR may disrupt the interaction between uPAR and α4β1 integrin to release HPSC from their osteoblast niche. In the second case, suPAR may inactivate CXCR4 by heterologous desensitization and further promote HPSC release from the BM. Most importantly, c-suPAR may act as a chemoattractant for the BM HPSCs and stimulate their mobilization from the BM to the circulation (see proposed pathway in Figure 1).
4. Conclusion
In multiple pathological settings, including stroke and myocardial infarction, HPSCs are mobilized from the BM to sites of injury to promote tissue repair and regeneration. Stem cell therapy, including direct transplantation of stem cells, stimulation of stem cell mobilization and homing by cytokines, for example G-CSF, has emerged as a promising approach to promote tissue repair and regeneration after ischemia. The studies on Plg have revealed an essential role of the Plg system in cytokine-induced stem cell mobilization and have elucidated the molecular mechanisms regulating Plg-mediated stem cell mobilization. This will potentially contribute to the development of new therapeutic strategies, for example, targeting Plg/MMP-9 for strengthening the established G-CSF treatment for ischemia disease. More importantly, the proposed experimental therapy with μPlm (a truncated form of plasmin with fewer side-effects) or chemotactic peptide (uPAR84–95) to promote HPSC recruitment to the damaged cardiac tissue, will confer clinical therapeutic potentials of plasmin in stem cell-mediated treatment, especially given the verified safety and efficiency of plasmin therapy (e.g., tPA) in MI treatment.
References
- 1.Cottler-Fox MH, Lapidot T, Petit I, et al. Stem cell mobilization. Hematology: American Society of Hematology, Education Program. 2003:419–437. doi: 10.1182/asheducation-2003.1.419. [DOI] [PubMed] [Google Scholar]
- 2.Al-Jurf M, Aranha F, Annasetti C, et al. Allogeneic peripheral blood stem-cell compared with bone marrow transplantation in the management of hematologic malignancies: an individual patient data meta-analysis of nine randomized trials. Journal of Clinical Oncology. 2005;23(22):5074–5087. doi: 10.1200/JCO.2005.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoover-Plow J, Gong Y. Challenges for heart disease stem cell therapy. Journal of Vascular Health and Risk Management. 2012;8:99–113. doi: 10.2147/VHRM.S25665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stiff P, Gingrich R, Luger S, et al. A randomized phase 2 study of PBPC mobilization by stem cell factor and filgrastim in heavily pretreated patients with Hodgkin’s disease or non-Hodgkin’s lymphoma. Bone Marrow Transplantation. 2000;26(5):471–481. doi: 10.1038/sj.bmt.1702531. [DOI] [PubMed] [Google Scholar]
- 5.Holm M. Not all healthy donors mobilize hematopoietic progenitor cells sufficiently after G-CSF administration to allow for subsequent CD34 purification of the leukapheresis product. Journal of Hematotherapy. 1998;7(2):111–113. doi: 10.1089/scd.1.1998.7.111. [DOI] [PubMed] [Google Scholar]
- 6.Anderlini P, Przepiorka D, Seong C, et al. Factors affecting mobilization of CD34+ cells in normal donors treated with filgrastim. Transfusion. 1997;37(5):507–512. doi: 10.1046/j.1537-2995.1997.37597293882.x. [DOI] [PubMed] [Google Scholar]
- 7.Castellino FJ, Ploplis VA. Structure and function of the plasminogen/plasmin system. Thrombosis and Haemostasis. 2005;93(4):647–654. doi: 10.1160/TH04-12-0842. [DOI] [PubMed] [Google Scholar]
- 8.Heissig B, Ohki-Koizumi M, Tashiro Y, Gritli I, Sato-Kusubata K, Hattori K. New functions of the fibrinolytic system in bone marrow cell-derived angiogenesis. International Journal of Hematology. 2012;95(2):131–137. doi: 10.1007/s12185-012-1016-y. [DOI] [PubMed] [Google Scholar]
- 9.Collen D. Ham-Wasserman lecture: role of the plasminogen system in fibrin-homeostasis and tissue remodeling. Hematology: American Society of Hematology, Education Program. 2001:1–9. doi: 10.1182/asheducation-2001.1.1. [DOI] [PubMed] [Google Scholar]
- 10.Andronicos NM, Chen EI, Baik N, et al. Proteomics-based discovery of a novel, structurally unique, and developmentally regulated plasminogen receptor, Plg-RKT, a major regulator of cell surface plasminogen activation. Blood. 2010;115(7):1319–1330. doi: 10.1182/blood-2008-11-188938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoover-Plow J, Yuen L. Plasminogen binding is increased with adipocyte differentiation. Biochemical and Biophysical Research Communications. 2001;284(2):389–394. doi: 10.1006/bbrc.2001.4984. [DOI] [PubMed] [Google Scholar]
- 12.Nervi B, Link DC, DiPersio JF. Cytokines and hematopoietic stem cell mobilization. Journal of Cellular Biochemistry. 2006;99(3):690–705. doi: 10.1002/jcb.21043. [DOI] [PubMed] [Google Scholar]
- 13.Lijnen HR. Plasmin and matrix metalloproteinases in vascular remodeling. Thrombosis and Haemostasis. 2001;86(1):324–333. [PubMed] [Google Scholar]
- 14.Plow EF, Herren T, Redlitz A, Miles LA, Hoover-Plow JL. The cell biology of the plasminogen system. FASEB Journal. 1995;9(10):939–945. doi: 10.1096/fasebj.9.10.7615163. [DOI] [PubMed] [Google Scholar]
- 15.Salonen EM, Zitting A, Vaheri A. Laminin interacts with plasminogen and its tissue-type activator. FEBS Letters. 1984;172(1):29–32. doi: 10.1016/0014-5793(84)80866-2. [DOI] [PubMed] [Google Scholar]
- 16.Salonen EM, Saksela O, Vartio T. Plasminogen and tissue-type plasminogen activator bind to immobilized fibronectin. The Journal of Biological Chemistry. 1985;260(22):12302–12307. [PubMed] [Google Scholar]
- 17.Silverstein RL, Leung LLK, Harpel PC, Nachman RL. Complex formation of platelet thrombospondin with plasminogen. Modulation of activation by tissue activator. Journal of Clinical Investigation. 1984;74(5):1625–1633. doi: 10.1172/JCI111578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okada Y, Gonoji Y, Naka K, et al. Matrix metalloproteinase 9 (92-kDa gelatinase/type IV collagenase) from HT 1080 human fibrosarcoma cells. Purification and activation of the precursor and enzymic properties. The Journal of Biological Chemistry. 1992;267(30):21712–21719. [PubMed] [Google Scholar]
- 19.He C, Wilhelm SM, Pentland AP, et al. Tissue cooperation in a proteolytic cascade activating human interstitial collagenase. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(8):2632–2636. doi: 10.1073/pnas.86.8.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gong Y, Hart E, Shchurin A, Hoover-Plow J. Inflammatory macrophage migration requires MMP-9 activation by plasminogen in mice. Journal of Clinical Investigation. 2008;118(9):3012–3024. doi: 10.1172/JCI32750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heissig B, Lund LR, Akiyama H, et al. The plasminogen fibrinolytic pathway is required for hematopoietic regeneration. Cell Stem Cell. 2007;1(6):658–670. doi: 10.1016/j.stem.2007.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tjwa M, Moura R, Moons L, et al. Fibrinolysis-independent role of plasmin and its activators in the haematopoietic recovery after myeloablation. Journal of Cellular and Molecular Medicine. 2009;13(11-12):4587–4595. doi: 10.1111/j.1582-4934.2008.00521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tjwa M, Janssens S, Carmeliet P. Plasmin therapy enhances mobilization of HPCs after G-CSF. Blood. 2008;112(10):4048–4050. doi: 10.1182/blood-2008-07-166587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tjwa M, Sidenius N, Moura R, et al. Membrane-anchored uPAR regulates the proliferation, marrow pool size, engraftment, and mobilization of mouse hematopoietic stem/progenitor cells. Journal of Clinical Investigation. 2009;119(4):1008–1018. doi: 10.1172/JCI36010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gong Y, Fan Y, Hoover-Plow J. Plasminogen regulates stromal cell-derived factor-1/CXCR4-Mediated hematopoietic stem cell mobilization by activation of matrix metalloproteinase-9. Arteriosclerosis, Thrombosis, and Vascular Biology. 2011;31(9):2035–2043. doi: 10.1161/ATVBAHA.111.229583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pelus LM, Bian H, King AG, Fukuda S. Neutrophil-derived MMP-9 mediates synergistic mobilization of hematopoietic stem and progenitor cells by the combination of G-CSF and the chemokines GROβ/CXCL2 and GROβT/CXCL2δ4. Blood. 2004;103(1):110–119. doi: 10.1182/blood-2003-04-1115. [DOI] [PubMed] [Google Scholar]
- 27.Petit I, Szyper-Kravitz M, Nagler A, et al. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nature Immunology. 2002;3(7):687–694. doi: 10.1038/ni813. [DOI] [PubMed] [Google Scholar]
- 28.Christopher MJ, Liu F, Hilton MJ, Long F, Link DC. Suppression of CXCL12 production by bone marrow osteoblasts is a common and critical pathway for cytokine-induced mobilization. Blood. 2009;114(7):1331–1339. doi: 10.1182/blood-2008-10-184754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ellis V, Behrendt N, Dano K. Plasminogen activation by receptor-bound urokinase: a kinetic study with both cell-associated and isolated receptor. The Journal of Biological Chemistry. 1991;266(19):12752–12758. [PubMed] [Google Scholar]
- 30.Behrendt N, Ronne E, Dano K. The structure and function of the urokinase receptor, a membrane protein governing plasminogen activation on the cell surface. Biological Chemistry Hoppe-Seyler. 1995;376(5):269–279. [PubMed] [Google Scholar]
- 31.Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nature Reviews Molecular Cell Biology. 2010;11(1):23–36. doi: 10.1038/nrm2821. [DOI] [PubMed] [Google Scholar]
- 32.Chapman HA, Wei Y. Protease crosstalk with integrins: the urokinase receptor paradigm. Thrombosis and Haemostasis. 2001;86(1):124–129. [PubMed] [Google Scholar]
- 33.Blasi F, Carmeliet P. uPAR: a versatile signalling orchestrator. Nature Reviews Molecular Cell Biology. 2002;3(12):932–943. doi: 10.1038/nrm977. [DOI] [PubMed] [Google Scholar]
- 34.Blasi F. uPA, uPAR, PAI-I: key intersection of proteolytic, adhesive and chemotactic highways? Immunology Today. 1997;18(9):415–417. doi: 10.1016/s0167-5699(97)01121-3. [DOI] [PubMed] [Google Scholar]
- 35.Carlin SM, Resink TJ, Tamm M, Roth M. Urokinase signal transduction and its role in cell migration. FASEB Journal. 2005;19(2):195–202. doi: 10.1096/fj.04-1644com. [DOI] [PubMed] [Google Scholar]
- 36.Selleri C, Montuori N, Ricci P, et al. Involvement of the urokinase-type plasminogen activator receptor in hematopoietic stem cell mobilization. Blood. 2005;105(5):2198–2205. doi: 10.1182/blood-2004-06-2424. [DOI] [PubMed] [Google Scholar]
- 37.Selleri C, Montuori N, Ricci P, et al. In vivo activity of the cleaved form of soluble urokinase receptor: a new hematopoietic stem/progenitor cell mobilizer. Cancer Research. 2006;66(22):10885–10890. doi: 10.1158/0008-5472.CAN-06-1311. [DOI] [PubMed] [Google Scholar]
- 38.Blasi F. The urokinase receptor in hematopoietic stem cells mobilization. Current Pharmaceutical Design. 2011;17(19):1911–1913. doi: 10.2174/138161211796718206. [DOI] [PubMed] [Google Scholar]
- 39.Vallabhaneni KC, Tkachuk S, Kiyan Y, et al. Urokinase receptor mediates mobilization, migration, and differentiation of mesenchymal stem cells. Cardiovascular Research. 2011;90(1):113–121. doi: 10.1093/cvr/cvq362. [DOI] [PubMed] [Google Scholar]
- 40.Andolfo A, English WR, Resnati M, Murphy G, Blasi F, Sidenius N. Metalloproteases cleave the urokinase-type plasminogen activator receptor in the D1-D2 linker region and expose epitopes not present in the intact soluble receptor. Thrombosis and Haemostasis. 2002;88(2):298–306. [PubMed] [Google Scholar]
- 41.Beaufort N, Leduc D, Rousselle JC, Namane A, Chignard M, Pidard D. Plasmin cleaves the juxtamembrane domain and releases truncated species of the urokinase receptor (CD87) from human bronchial epithelial cells. FEBS Letters. 2004;574(1–3):89–94. doi: 10.1016/j.febslet.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 42.Furlan F, Orlando S, Laudanna C, et al. The soluble D2D388-274 fragment of the urokinase receptor inhibits monocyte chemotaxis and integrin-dependent cell adhesion. Journal of Cell Science. 2004;117(14):2909–2916. doi: 10.1242/jcs.01149. [DOI] [PubMed] [Google Scholar]
- 43.Tarui T, Mazar AP, Cines DB, Takada Y. Urokinase-type plasminogen activator receptor (CD87) is a ligand for integrins and mediates cell-cell interaction. The Journal of Biological Chemistry. 2001;276(6):3983–3990. doi: 10.1074/jbc.M008220200. [DOI] [PubMed] [Google Scholar]
- 44.Fietz T, Hattori K, Thiel E, Heissig B. Increased soluble urokinase plasminogen activator receptor (suPAR) serum levels after granulocyte colony-stimulating factor treatment do not predict successful progenitor cell mobilization in vivo. Blood. 2006;107(8):3408–3409. doi: 10.1182/blood-2005-08-3176. [DOI] [PubMed] [Google Scholar]
- 45.Madsen CD, Ferraris GMS, Andolfo A, Cunningham O, Sidenius N. uPAR-induced cell adhesion and migration: vitronectin provides the key. Journal of Cell Biology. 2007;177(5):927–939. doi: 10.1083/jcb.200612058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith HW, Marra P, Marshall CJ. uPAR promotes formation of the p130Cas-Crk complex to activate Rac through DOCK180. Journal of Cell Biology. 2008;182(4):777–790. doi: 10.1083/jcb.200712050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wei Y, Lukashev M, Simon DI, et al. Regulation of integrin function by the urokinase receptor. Science. 1996;273(5281):1551–1555. doi: 10.1126/science.273.5281.1551. [DOI] [PubMed] [Google Scholar]
- 48.Chaurasia P, Aguirre-Ghiso JA, Liang OD, Gardsvoll H, Ploug M, Ossowski L. A region in urokinase plasminogen receptor domain III controlling a functional association with α5β1 integrin and tumor growth. The Journal of Biological Chemistry. 2006;281(21):14852–14863. doi: 10.1074/jbc.M512311200. [DOI] [PubMed] [Google Scholar]
- 49.Zhang F, Tom CC, Kugler MC, et al. Distinct ligand binding sites in integrin α3β1 regulate matrix adhesion and cell-cell contact. Journal of Cell Biology. 2003;163(1):177–188. doi: 10.1083/jcb.200304065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei Y, Waltz DA, Rao N, Drummond RJ, Rosenberg S, Chapman HA. Identification of the urokinase receptor as an adhesion receptor for vitronectin. The Journal of Biological Chemistry. 1994;269(51):32380–32388. [PubMed] [Google Scholar]
- 51.May AE, Kanse SM, Lund LR, Gisler RH, Imhof BA, Preissner KT. Urokinase receptor (CD87) regulates leukocyte recruitment via β2 integrins in vivo. Journal of Experimental Medicine. 1998;188(6):1029–1037. doi: 10.1084/jem.188.6.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.May AE, Neumann FJ, Schömig A, Preissner KT. VLA-4 (α4β1) engagement defines a novel activation pathway for β2 integrin-dependent leukocyte adhesion involving the urokinase receptor. Blood. 2000;96(2):506–513. [PubMed] [Google Scholar]
- 53.Scott LM, Priestley GV, Papayannopoulou T. Deletion of α4 integrins from adult hematopoietic cells reveals roles in homeostasis, regeneration, and homing. Molecular and Cellular Biology. 2003;23(24):9349–9360. doi: 10.1128/MCB.23.24.9349-9360.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hurley RW, McCarthy JB, Verfaillie CM. Direct adhesion to bone marrow stroma via fibronectin receptors inhibits hematopoietic progenitor proliferation. Journal of Clinical Investigation. 1995;96(1):511–519. doi: 10.1172/JCI118063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Prosper F, Stroncek D, McCarthy JB, Verfaillie CM. Mobolization and homing of peripheral blood progenitors is related to reversible downregulation of α4β1 integrin expression and function. Journal of Clinical Investigation. 1998;101(11):2456–2467. doi: 10.1172/JCI188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Montuori N, Carriero MV, Salzano S, Rossi G, Ragno P. The cleavage of the urokinase receptor regulates its multiple functions. The Journal of Biological Chemistry. 2002;277(49):46932–46939. doi: 10.1074/jbc.M207494200. [DOI] [PubMed] [Google Scholar]
- 57.Resnati M, Pallavicini I, Wang JM, et al. The fibrinolytic receptor for urokinase activates the G protein-coupled chemotactic receptor FPRL1/LXA4R. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(3):1359–1364. doi: 10.1073/pnas.022652999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gyetko MR, Todd RF, III, Wilkinson CC, Sitrin RG. The urokinase receptor is required for human monocyte chemotaxis in vitro. Journal of Clinical Investigation. 1994;93(4):1380–1387. doi: 10.1172/JCI117114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Le Y, Murphy PM, Wang JM. Formyl-peptide receptors revisited. Trends in Immunology. 2002;23(11):541–548. doi: 10.1016/s1471-4906(02)02316-5. [DOI] [PubMed] [Google Scholar]
- 60.Le Y, Shen W, Li B, Gong W, Dunlop NM, Wang JM. A new insight into the role of “old” chemotactic peptide receptors FPR and FPRL1: down-regulation of chemokine receptors CCR5 and CXCR4. Forum. 1999;9(4):299–314. [PubMed] [Google Scholar]
- 61.Li BQ, Wetzel MA, Mikovits JA, et al. The synthetic peptide WKYMVm attenuates the function of the chemokine receptors CCR5 and CXCR4 through activation of formyl peptide receptor-like 1. Blood. 2001;97(10):2941–2947. doi: 10.1182/blood.v97.10.2941. [DOI] [PubMed] [Google Scholar]
- 62.Lévesque JP, Hendy J, Takamatsu Y, Simmons PJ, Bendall LJ. Disruption of the CXCR4/CXCL12 chemotactic interaction during hematopoietic stem cell mobilization induced by gcsf or cyclophosphamide. Journal of Clinical Investigation. 2003;111(2):187–196. doi: 10.1172/JCI15994. [DOI] [PMC free article] [PubMed] [Google Scholar]