

Abstract
The ability of a protein to carry out a given function results from fundamental physicochemical properties that include the protein’s structure, mechanism of action, and thermodynamic stability. Traditional approaches to study these properties have typically required the direct measurement of the property of interest, oftentimes a laborious undertaking. Although protein properties can be probed by mutagenesis, this approach has been limited by its low throughput. Recent technological developments have enabled the rapid quantification of a protein’s function, such as binding to a ligand, for numerous variants of that protein. Here, we measure the ability of 47,000 variants of a WW domain to bind to a peptide ligand and use these functional measurements to identify stabilizing mutations without directly assaying stability. Our approach is rooted in the well-established concept that protein function is closely related to stability. Protein function is generally reduced by destabilizing mutations, but this decrease can be rescued by stabilizing mutations. Based on this observation, we introduce partner potentiation, a metric that uses this rescue ability to identify stabilizing mutations, and identify 15 candidate stabilizing mutations in the WW domain. We tested six candidates by thermal denaturation and found two highly stabilizing mutations, one more stabilizing than any previously known mutation. Thus, physicochemical properties such as stability are latent within these large-scale protein functional data and can be revealed by systematic analysis. This approach should allow other protein properties to be discovered.
Keywords: deep mutational scanning, epistasis, high-throughput DNA sequencing
The sequence of a protein determines the protein’s physicochemical properties, which include structure, thermodynamic stability, ability to interact with other molecules, and catalytic capacity (1). These properties, in turn, determine the function of the protein. Because sequence determines function, mutagenesis has been a fundamental tool for understanding how proteins work. A mutation can impact the function of a protein when it alters one or more properties of the protein, such as its structure, catalytic activity, or stability. Understanding the mechanism by which a mutation impacts protein function has traditionally required specialized assays to measure these properties (for example, thermal denaturation has been used to measure stability).
Coupling of selection and high-throughput DNA sequencing has enabled methods to measure the function of large numbers (up to millions) of mutated versions of a protein (referred to here as variants) (2–4). These methods, known as “deep mutational scanning” (4), link the function of each variant with its abundance in a population of variants under selection for that function. Variant frequencies within the population are measured en masse by high-throughput DNA sequencing of the gene encoding the protein. The change in frequency of each variant is quantified by comparing each variant’s frequency before selection with its frequency after selection. The enrichment or depletion of each variant through selection serves as a proxy for the variant’s function; variants containing highly functional (beneficial) mutations enrich after selection, whereas variants containing poorly functional (deleterious) mutations deplete. Deep mutational scanning enables measurement of the functional consequences of large numbers of protein variants in parallel and therefore produces a large-scale set of protein functional data. Based solely on this dataset, we present an analysis to identify mutations that stabilize a protein.
Mutations that stabilize proteins are important both for understanding protein activity and for successful protein engineering. Stabilizing mutations in protein drugs, such as insulin (5) and antibodies (6), and commercial enzymes, such as subtilisin (7), can prevent proteolysis or misfolding, thereby increasing effective activity. Proteins are marginally stable and become nonfunctional if destabilized past a threshold. Thus, protein stability is linked to measures of protein function like catalytic activity or ligand binding (8–10). For example, a single mutation that decreases stability beyond the threshold can dramatically reduce protein function. However, such a destabilizing mutation can be rescued by the introduction of a second stabilizing mutation. The resulting protein is above the stability threshold and consequently, has increased function.
Most mutations are destabilizing, and thus, in a set of randomly generated double mutants, the rare stabilizing mutations will generally be paired with destabilizing mutations. Thus, we hypothesized that we could identify stabilizing mutations based on their ability to rescue many other (mostly destabilizing) mutations. This hypothesis raises the intriguing possibility that fundamental physicochemical properties of certain protein variants (e.g., those variants with the highest stability) might be inferred solely from large-scale measurements of protein function. Here, we show that a systematic analysis of these measurements for a large number of variants of a protein can be used to calculate partner potentiation, a metric that reveals stabilizing mutations.
Results
Deep Mutational Scanning of a WW Domain.
We used deep mutational scanning to measure the ability of 47,000 unique variants of the hYAP65 WW domain to bind to their polyproline peptide ligand (2, 4). WW domains mediate protein–protein interactions, have a well-defined structure, and fold through a two-state mechanism, simplifying subsequent measurements of thermodynamic stability (11–13). We displayed a library of variants of the hYAP65 WW domain on the surface of T7 bacteriophage. The library was created by doped oligonucleotide synthesis, with each library member containing, on average, two mutations in a 102-base variable region encoding 34 amino acids that span the majority of the domain. The library was subjected to three rounds of selection for binding to a biotinylated form of the GTPPPPYTVG peptide ligand, which had been immobilized on magnetic streptavidin beads. We performed high-throughput DNA sequencing of the input and libraries from rounds 1–3, acquiring at least 10 million reads for each library (Fig. S1 and Table S1).
We used this high-throughput DNA sequencing data to derive a functional score for each WW domain variant in the library based on the variant’s frequency at each round of selection. Variant frequencies were corrected for nonspecific carryover, which occurs when nonfunctional variants are carried from one round to the next because of background bead binding and incompletely effective washing (14, 15). The nonspecific carryover rate was estimated from the performance of variants containing stop codons, because these variants should be nonfunctional. From the nonspecific carryover-corrected frequencies, we made linear models of round-to-round enrichments for each of the 47,000 variants present in the input library and all three rounds of selection. For each variant, the slope of the resulting line indicates that variant’s enrichment or depletion during the assay. To calculate a functional score, we divided each variant’s slope by the wild type (WT) slope (Fig. S2). We used a goodness-of-fit cutoff (slope R2 ≥ 0.75) to eliminate variants that behaved erratically.
Interaction Between Single and Double Mutants in the WW Domain.
We hypothesized that stabilizing mutations could be found based on their ability to rescue other mutations, most of which are destabilizing. In our protein function dataset, a rescue effect would be seen when two single mutations combine in a doubly mutated variant to produce unexpected functional gains. These unexpected functional gains resulting from combinations of single mutations can be described in terms of epistasis (9, 10, 16, 17). Here, we define epistasis as occurring when two single mutations (a and b) combine to impact protein function differently than expected based on their individual functional effects and an interaction model. We used the most common model, called the product interaction model, with the epistasis score 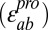 computed as (Eq. 1)
computed as (Eq. 1)
where Wab represents the functional score of the double mutant and Wa and Wb represent the functional scores of the single mutants.
Using the product model, we calculated 5,010 individual epistasis scores from the functional scores of 47,000 variants. The product model dictates that the product of two single-mutant functional scores (Wa • Wb) should equal the double-mutant functional score (Wab) if no epistasis is present. We found that single-mutant functional scores predicted double-mutant functional scores with a Pearson’s R2 of 0.67 (Fig. 1A). In a previous study examining variants of the hYAP65 WW domain that survived after six rounds of selection for peptide binding (2), we obtained a value of 0.68. Thus, despite altering our analysis to combine consecutive rounds of selection, additional sequencing, and extensive data filtering, we did not improve predictions of double-mutant functional scores (Fig. S2 and Table S1). Furthermore, we tested the logarithmic, minimal, and additive interaction models, with the epistasis scores 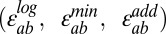 computed as
computed as
Fig. 1.

Relationship between function and epistasis in a massive collection of double mutants. (A) The functional score of 5,010 doubly mutated variants was predicted from the functional scores of the component singly mutated variants using the product model. Predicted functional score is plotted against observed functional score and the two are highly correlated (Pearson’s R2 = 0.67). For each doubly mutated variant, the linear models used to generate the functional score had an R2 ≥ 0.75. (B) Epistasis scores calculated using the product model for the 5,010 variants are plotted against the functional score of the doubly mutated variant. The distribution of epistasis scores is shown in the Inset. Dashed lines are placed at ±1 SD from the mean.
None of these commonly used models of epistasis (18) resulted in improved predictions of double-mutant functional scores (Fig. S3 A–D). We conclude that the limiting factor in predicting double-mutant functional scores is the accuracy of the model rather than the quality of the measurements of function. These results argue that epistasis is an intrinsic property of the hYAP65 WW domain rather than an artifact of data quality or model choice.
Under the product model, which predicted double-mutant functional scores most accurately, the mean epistasis score for all variants was near zero 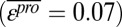 , with a SD of 0.65 and 86% of the scores within 1 SD of the mean. The magnitude of epistasis scores scales with the magnitude of variant functional scores (Fig. 1B). This scaling effect arises because raw variant functional scores are used to calculate epistasis scores. Therefore, if two single mutants and the corresponding double mutant have small functional scores, they cannot yield a large epistasis score. We observed both high magnitude-positive and -negative epistasis scores but no population mean tendency to positive or negative epistasis (Fig. 1B). The most functional double mutants have positive epistasis scores, suggesting that highly functional mutants may be difficult to predict.
, with a SD of 0.65 and 86% of the scores within 1 SD of the mean. The magnitude of epistasis scores scales with the magnitude of variant functional scores (Fig. 1B). This scaling effect arises because raw variant functional scores are used to calculate epistasis scores. Therefore, if two single mutants and the corresponding double mutant have small functional scores, they cannot yield a large epistasis score. We observed both high magnitude-positive and -negative epistasis scores but no population mean tendency to positive or negative epistasis (Fig. 1B). The most functional double mutants have positive epistasis scores, suggesting that highly functional mutants may be difficult to predict.
To gain insight into the patterns of epistasis in the WW domain, we constructed a network view, which shows that, in some regions of the WW domain, mutations that yield positive epistatic interactions occur at positions that also harbor mutations that yield negative interactions (Fig. 2A). Epistasis scores are distributed nonhomogenously, resulting in hotspots (individual positions at which some mutations have many positive epistatic interactions and other mutations have many negative epistatic interactions) (Fig. 2 A and B). These hotspots occur in regions of high epistasis that comprise both loops as well as a portion of the N terminus (Fig. S3E) (Wilcoxon rank sum test, P = 7.85 × 10−22).
Fig. 2.

Epistasis alone does not reliably identify stabilizing mutations. (A) A network view of epistatic interactions between mutations is shown. Individual mutations are presented as nodes in the graph and colored by functional scores (red corresponds to mutations with higher functional scores than WT, and blue corresponds to mutations with lower functional scores than WT). Mutations are arranged first by position and second by alphabet along the circumference of the graph in clockwise order from the 12:00 coordinate (zoom in to see individual mutations). The WT sequence is shown around the outside of the graph. Positive and negative epistatic interactions between mutations are shown as gradient red and blue edges, with width and shading proportional to the magnitude of the interaction. The position of the β-strands in the WW domain is indicated by the blue arrows. For clarity, only epistatic interactions at least 1 SD from the mean are shown. (B) For each position in the domain, the fraction of epistasis scores that are negative is plotted on the x axis, and the fraction of epistasis scores that are positive is plotted on the y axis. The fractions of positive and negative epistasis scores are correlated among positions (R = 0.60, P = 8.8 × 10−5). (C) The average epistasis score of each of the 192 single mutants found in 10 or more double mutants is plotted against the single-mutant functional score of each mutation. The known stabilizing (A20R, L30K, and D34T) and activity-enhancing (K21R and Q35R) mutations are highlighted in red and blue, respectively.
Identification of Thermodynamically Stabilizing Mutations.
Because stabilizing mutations could potentially rescue many destabilizing mutations, the simplest strategy to find them would rely on the expectation that stabilizing mutations are among the most highly represented mutations after selection. As a gold standard, we used three known stabilizing hYAP65 WW domain mutations (19, 20) present in the dataset (A20R, L30K, and D34T), which under this expectation, should become highly enriched. However, for these three mutations, postselection representation was not a useful predictor of stability (Fig. S3F). This strategy likely failed because although we measured a large number (5,010) of epistasis scores, these scores represent only a small sample of the 211,200 possible epistasis scores in the 34 positions that were varied.
Regardless of overall postselection representation, stabilizing mutations should rescue many other mostly destabilizing mutations. Each of these rescue interactions would have a positive epistasis score. Thus, we hypothesized that stabilizing mutations should be those rare mutations with a large positive average epistasis score. We calculated a mean epistasis score for each single mutation a
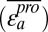 , which consists of the mean of all of the epistasis scores arising from double mutants containing a and any other single mutation b
, which consists of the mean of all of the epistasis scores arising from double mutants containing a and any other single mutation b
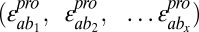 . However, the mean epistasis score was also unable to separate all three known stabilizing hYAP65 WW domain mutations present in our dataset from the bulk of the mutations as well as two known activity-enhancing mutations (19–22) (Fig. 2C). The mean epistasis score failed to correctly identify stabilizing mutations because of two inherent biases. One bias is the scaling effect, where the magnitude of an epistasis score scales with the functional scores of the participating variants. The other bias is a sampling bias inherent in deep mutational scanning caused by its reliance on selection; highly deleterious mutations are either not observed or observed only when they pair with beneficial mutations.
. However, the mean epistasis score was also unable to separate all three known stabilizing hYAP65 WW domain mutations present in our dataset from the bulk of the mutations as well as two known activity-enhancing mutations (19–22) (Fig. 2C). The mean epistasis score failed to correctly identify stabilizing mutations because of two inherent biases. One bias is the scaling effect, where the magnitude of an epistasis score scales with the functional scores of the participating variants. The other bias is a sampling bias inherent in deep mutational scanning caused by its reliance on selection; highly deleterious mutations are either not observed or observed only when they pair with beneficial mutations.
To address these biases, we derived a third strategy that employs an epistasis-based metric, which we termed partner potentiation (Fig. 3A). Partner potentiation quantifies the degree to which an individual single mutation (a) improves, or potentiates, the functional effect of its partner single mutations (b1, b2, … bx) in the collection of double mutants in which it is found (ab1, ab2, … abx). In a given double mutant (ab), a has a partner-normalized epistasis score with the other mutation b (Pa→b) calculated as (Eq. 5)
Fig. 3.

Partner potentiation reveals stabilizing mutations. (A) Partner potentiation is calculated for a query mutation a that forms doubly mutated variants with mutations b1, b2, b3 … bn. Partner-normalized epistasis scores are calculated for a by dividing the epistasis score of each double-mutant combination that a is found in by the functional score of the partner mutation (an example calculation for b1 is shown). The partner potentiation of a is calculated as the mean of the normalized epistasis scores (in this case, the scores for b1 … bn). (B) Partner potentiation is plotted for each single mutation against its functional score. The known stabilizing (A20R, L30K, and D34T) and activity-enhancing (K21R and Q35R) mutations are highlighted in red and blue, respectively. Mutations with a partner potentiation score greater than 0.4 and a functional score greater than 0.9 were considered to be stabilizing. (C) Stabilizing mutations were validated by thermal denaturation. The WT hYAP65 WW domain, the known stabilizing mutant D34T, and four candidate stabilizing mutants (L30I, I33R, Q35K, and T36R) are shown in black, purple, red, green, orange, and blue, respectively. (D) Positions in the hYAP65 NMR structure (Protein Data Bank ID code 1k9q) with stabilizing variants as judged by partner potentiation are shown, colored by the magnitude of the mean partner potentiation score using the PyMol software. Stabilizing mutations are distributed throughout the WW domain. Positions of previously known (superscript 1) and previously unknown (superscript 2) validated stabilizing mutations (20, 30, 34, 35) are highlighted.
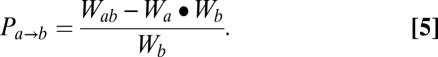 |
The partner potentiation score of a (PPa) is calculated as the mean of the partner-normalized epistasis scores (Pa→b1, Pa→b2, … Pa→bx). We calculated partner potentiation scores for mutations that occurred in at least 10 double mutants. Partner potentiation accounts for the functional effects imparted by the partner mutations, reducing the impact of both the scaling and sampling biases. Unlike change in representation or mean epistasis, partner potentiation separated all three known stabilizing mutations from the bulk of points as well as the known activity-enhancing mutations (Fig. 3B). As expected, mutations with high partner potentiation scores frequently resulted in positive epistasis (Fig. S3G).
We defined a candidate list of 15 stabilizing mutations having a partner potentiation score greater than 0.4 and a functional score greater than 0.9 (Table S2). The list harbors the three known stabilizing mutations and includes none of the known activity-enhancing mutations. We chemically synthesized six candidate-stabilizing WW domain variants (D10Q, P12H, L30I, Q35K, I33R, and T36R) as well as the known stabilizing D34T variant as a positive control (20) to characterize their stability by thermal denaturation (Fig. 3C). Far UV circular dichroism spectroscopy was used to record the denaturation curves from which ΔGfolding and ΔΔGfolding data were extracted (Table S2). The stability of the D10Q and P12H variants could not be quantified because of the absence of pretransition baselines resulting from their low stability. These strongly destabilizing mutations are located near the N terminus of the WW domain. These mutations may act to stabilize the phage capsid–WW domain interface, and therefore, they may be stabilizing the WW domain in the phage assay but not in the context of the isolated WW domain. Of the remaining five variants, the L30I, D34T, and Q35K mutations resulted in significant stabilization, I33R was a neutral mutation, and T36R was slightly destabilizing. Among these five variants, ΔΔGfolding highly correlated with partner potentiation but not with average epistasis or functional score (Spearman’s ρ = −0.81) (Fig. S4). Moreover, the identification of two stabilizing mutations in a grouping with the three known stabilizing mutations shows that thermodynamic stability, a fundamental protein property, is implicit in large-scale functional data.
To assess the false-negative rate of our approach, we compared our findings with a study of stability in the Pin1 WW domain, which shares a high degree of sequence and structural homology with the hYAP65 WW domain. The stability of 47 alanine or glycine mutants distributed throughout the Pin1 domain was assessed by thermal denaturation (23). Of these mutants, one (2.1%) mutant was significantly stabilizing, suggesting that ∼14 stabilizing mutations should exist among the 646 possible single mutations in the hYAP65 domain. Assuming that our validation rate of 33% (2/6) generalizes to all 12 candidate mutations, we would expect to find a total of four stabilizing mutations in addition to the three known stabilizing mutations. These seven mutations represent one-half of the total predicted from the Pin1 data. False negatives may be caused by the incompleteness of the data, which allowed a calculation of partner potentiation scores for 192 of 646 possible single mutations, differences in stability between Pin1 and hYAP65, or intrinsic limitations of this approach. We compared our validation rate (2/6) to the rate of random discovery of stabilizing mutations suggested by the Pin1 data (1/47) and found that our rate was significantly higher (binomial exact test, P = 0.0067). Additionally, we conducted our analysis on a restricted set of high quality data and obtained nearly identical results (Fig. S5).
The 15 candidate mutations that we identified occur at a total of just eight positions. These positions are scattered throughout the WW domain and are not confined to the loop regions (Figs. 2 and 3D). In fact, the candidate stabilizing mutations occur at positions in both loops and strands as well as ligand-contacting and -noncontacting positions. One mutation, L30I, increased the Tm by a striking 12 °C and thus, is more stabilizing than any other known stabilizing mutation in the hYAP65 WW domain. Position 30 makes contact with the peptide ligand and is the site of another known stabilizing mutation, L30K. The identification of L30I highlights the use of finding stabilizing mutations based on functional data, because these mutations will not hinder peptide binding, even if they occur at a contact position.
Finally, we used FoldX (24), a widely used computational tool for predicting the thermodynamic impact of mutations in proteins, to analyze the effects of single mutations on WW domain stability. None of the known or candidate stabilizing mutations were classified as stabilizing by FoldX (Dataset S1). This result underscores the difficulty of computational prediction of the thermodynamic impact of mutations in proteins and highlights the effectiveness of our strategy.
Candidate Stabilizing and Activating Mutations Synergize to Enhance Function.
Our data offer an opportunity to explicitly examine the behavior of candidate stabilizing mutations in a protein. We tested the theory that the candidate stabilizing mutations enable the acquisition of activity-enhancing but destabilizing mutations (10, 25, 26). We classified single mutations that had beneficial effects on function but were not classified as candidate stabilizing mutations as activating mutations. Among variants with functional scores greater than WT, those variants containing two activating mutations generally had higher functional scores than those variants with a single activating mutation (and no candidate stabilizing mutation) (Fig. 4A). Variants with candidate stabilizing mutations (and no activating mutation) had higher functional scores than variants that relied solely on activating mutations, which suggests that the WW domain is only marginally stable (Fig. 4A) (Wilcoxon rank sum test, P = 6.65 × 10−14). The largest functional score increases arose in double mutants that combine a candidate stabilizing mutation with an activating mutation (Wilcoxon rank sum test, P = 8.56 × 10−13) (Fig. 4A).
Fig. 4.

Stabilizing mutations combine with activating mutations to drive large functional gains. (A) Mutations were classified as either stabilizing (functional score > 0.9 and partner potentiation > 0.4) or activating (functional score > 1 and partner potentiation ≤ 0.4). Double mutants with functional scores greater than WT were grouped into those mutants harboring both activating and stabilizing mutations (purple; n = 170), a single stabilizing mutation but no activating mutations (red; n = 156), a single activating mutation but no stabilizing mutation (solid blue; n = 1071), or two activating mutations (dashed blue; n = 852). The functional score distributions for each class are presented. Curves for variants with greater functional scores than WT containing paired stabilizing mutations are not drawn because of their low numbers (n = 4). (B) For each stabilizing (red) and activating mutation (blue), the fraction of deleterious single mutations rescued (i.e., found in double mutants with greater functional scores than WT) was calculated. Analysis was restricted to deleterious mutations that both stabilizing and activating mutations were paired with in double mutants in our library. Experimentally validated beneficial mutations are highlighted in bold; 1 denotes stabilizing mutations validated in this study, 2 denotes previously identified stabilizing mutations, and 3 denotes previously identified activating mutations.
We compared the ability of candidate stabilizing and activating mutations to rescue deleterious mutations. Stabilizing mutations should rescue deleterious mutations more effectively, because most deleterious mutations are destabilizing, a defect directly resolved by stabilizing mutations but not activating mutations. To test this prediction, we identified rescue events, in which a deleterious single mutation paired with a candidate stabilizing or activating mutation in a double mutant with a functional score at least as good as WT. To avoid sampling biases, we restricted this analysis to the set of deleterious mutations paired with both activating and candidate stabilizing mutations in the double-mutant functional data. Within this set, the deleterious mutations that are rescued by candidate stabilizing mutations and activating mutations largely overlapped (∼70%). However, candidate stabilizing mutations rescued, on average, three times as many deleterious mutations than did activating mutations. To examine the rescue effects on a per mutation basis, we ranked candidate-stabilizing and activating mutations by the fraction of deleterious mutations that they rescued. Independently verified stabilizing and activating mutations rescued the largest fraction of deleterious mutations within their class (Fig. 4B). Thus, candidate stabilizing and activating mutations can rescue many of the same deleterious mutations, but candidate stabilizing mutations enhance tolerance to deleterious mutations to a much greater degree than activating mutations.
Stabilizing mutations rescue destabilizing mutations by buffering decreases in stability, whereas activating mutations exert their rescue effect by buffering functional costs through increased activity. The fraction of mutations rescued by a given activating or candidate stabilizing mutation was more highly correlated with functional score for activating mutations (Spearman’s ρ = 0.92) than candidate stabilizing mutations (Spearman’s ρ = 0.59; P ≤ 1 × 10−4) (Fig. S6). This finding suggests that rescue by activating mutations occurs through a cost exchange and rescue by candidate stabilizing mutations occurs through a different mechanism.
Discussion
High-throughput approaches like deep mutational scanning can measure the function of protein variants on an unprecedented scale. As a simple list, the large-scale functional data that these approaches produce identify beneficial and deleterious mutations as well as positions important for protein activity. Here, we have shown that we can use these data to identify a feature not immediately obvious from functional scores alone: the identity of stabilizing mutations. To accomplish this goal, we developed a metric, partner potentiation, that enabled us to identify stabilizing mutations without having to explicitly measure stability. We found 15 candidate stabilizing mutations and validated 2 previously unknown mutations among the ∼600 possible single mutations within the WW domain, which supports the notion that stabilizing mutations are uncommon. Three of these mutations had been previously identified by rational design efforts, but most mutations could not have been predicted. Thus, large-scale functional data can be analyzed to reveal at least one fundamental protein property.
Additionally, we characterized epistasis in a WW domain on a massive scale. Most single mutations did not show strong epistasis when combined, and there was no mean tendency to positive or negative epistasis. Recently, a large-scale analysis of epistasis in the HIV protease described a geographic enrichment of epistasis (27). In the WW data, we found similar evidence for strong epistatic interactions occurring between particular regions. In addition, our data show that the occurrences of positive and negative epistatic interactions are correlated at positions in the WW domain, highlighting a limited number of hotspot positions where epistatic interactions, both positive and negative, are most likely.
Our analysis enabled us to classify a large number of mutations as either potentially activating or stabilizing. The interplay between activating mutations and stabilizing mutations has implications for both protein evolution and protein engineering (9, 10, 16). This work offers an explicit, large-scale test of the protein evolutionary theory that predicts that stabilizing mutations permit the existence of other activating but destabilizing mutations. The finding that WW domain double mutants with one candidate stabilizing mutation and one deleterious mutation have increased function relative to those mutants with one activating mutation and one deleterious mutation supports this theory. Furthermore, we find that candidate stabilizing mutations enhance tolerance to deleterious mutations to a greater degree than activating mutations. This result illustrates the importance of stabilizing mutations in preserving diversity during protein evolution.
Computational and experimental approaches have been developed to identify stabilizing mutations. Computational methods generally rely on physicochemical models to estimate the thermodynamic impact of mutations (28, 29). Stabilizing mutations can also be identified by analyzing evolutionary conservation or proteins from hyperthermophilic organisms (30, 31). Rational design draws on protein structure as well as the knowledge of the experimenter to predict stabilizing mutations (32). Selection-based methods, including directed evolution, attempt to distinguish stabilizing mutations by selecting for activity among a library of variants of a protein under conditions that include high temperature, denaturant, or the presence of protease (31–33). The validation rate of the approach described here (33%) is broadly similar to the other approaches; however, stabilizing but deactivating mutations, which plague other strategies, are eliminated.
Systematic analyses of the kind presented here could allow us to disentangle and consequently, quantify other properties that contribute to protein function. For example, the prediction of protein structure might benefit from large-scale protein functional data that reveal amino acid preferences within particular structural elements (e.g., the paucity of proline residues in β-strands) and the functional effects of mutations that occur at spatially proximal positions. The feasibility of this approach is illustrated by existing structural prediction methods that are founded on these concepts but require extensive existing sequence alignment or structural training data (34, 35). Another example relates to the understanding of enzyme mechanism, which might be uncovered by an analysis of the pattern of mutations that increase or decrease catalytic activity in large-scale protein functional data. In particular, the study of rare strongly activating mutations represents a systematic method for exploring mechanism. Finally, protein–protein interaction interfaces could be mapped in detail by analysis of large-scale protein functional data collected in the presence and absence of an interacting protein partner. Partner-dependent changes in variant function would indicate positions important for the binding interaction, and amino acid preference at those positions could reveal the nature of the binding surface. Thus, we predict that the increasing accessibility of large-scale protein functional data will provide exciting new tools for understanding how proteins function.
Materials and Methods
We briefly discuss key methods here and refer readers to SI Text for full experimental and analytical details.
WW Domain Phage Display, Selection, and Sequencing Library Construction.
We performed the phage display and selection as described previously (2). Three rounds of selection of the WW domain library against the GTPPPPYTVG peptide bound to magnetic beads were carried out. High-throughput sequencing libraries were prepared using PCR and then sequenced on a GAIIx (Illumina).
High-Throughput Sequencing and Quality Filtration.
The 102-base variable region was sequenced using partially overlapping reads to increase quality (2). The data were analyzed using the Enrich software package (36).
Calculation of Variant Functional Scores.
To calculate variant functional scores, we used nonspecific carryover-corrected data from consecutive rounds of selection to construct a linear model for each variant. The slope of the line in this model is proportional to variant function; variants that enrich throughout the selection have positive slopes, whereas variants that deplete have negative slopes. Goodness-of-fit filters (R2 > 0.75) were employed as described in SI Text.
Supplementary Material
Acknowledgments
We thank Charlie Lee and Jay Shendure for assistance with DNA sequencing as well as Elhanan Borenstein, Christine Queitsch, David Baker, Richard McLaughlin, and Carlos Araya Rodríguez for helpful comments and discussion. S.F. is an investigator of The Howard Hughes Medical Institute. This work was supported by National Institutes of Health Grants F32GM084699 (to D.M.F), GM051105 (to J.W.K.), and P41GM103533 (to S.F.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The raw sequence data reported in this paper have been deposited in the Sequence Read Archive (accession no. SRA058752).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1209751109/-/DCSupplemental.
References
- 1.Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 2.Fowler DM, et al. High-resolution mapping of protein sequence-function relationships. Nat Methods. 2010;7:741–746. doi: 10.1038/nmeth.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hietpas RT, Jensen JD, Bolon DN. Experimental illumination of a fitness landscape. Proc Natl Acad Sci USA. 2011;108:7896–7901. doi: 10.1073/pnas.1016024108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Araya CL, Fowler DM. Deep mutational scanning: Assessing protein function on a massive scale. Trends Biotechnol. 2011;29:435–442. doi: 10.1016/j.tibtech.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brange J, et al. Monomeric insulins obtained by protein engineering and their medical implications. Nature. 1988;333:679–682. doi: 10.1038/333679a0. [DOI] [PubMed] [Google Scholar]
- 6.Ewert S, Honegger A, Plückthun A. Stability improvement of antibodies for extracellular and intracellular applications: CDR grafting to stable frameworks and structure-based framework engineering. Methods. 2004;34:184–199. doi: 10.1016/j.ymeth.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 7.Gupta R, Beg QK, Lorenz P. Bacterial alkaline proteases: Molecular approaches and industrial applications. Appl Microbiol Biotechnol. 2002;59:15–32. doi: 10.1007/s00253-002-0975-y. [DOI] [PubMed] [Google Scholar]
- 8.Taverna DM, Goldstein RA. Why are proteins marginally stable? Proteins. 2002;46:105–109. doi: 10.1002/prot.10016. [DOI] [PubMed] [Google Scholar]
- 9.Bloom JD, Arnold FH. In the light of directed evolution: Pathways of adaptive protein evolution. Proc Natl Acad Sci USA. 2009;106(Suppl 1):9995–10000. doi: 10.1073/pnas.0901522106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bershtein S, Segal M, Bekerman R, Tokuriki N, Tawfik DS. Robustness-epistasis link shapes the fitness landscape of a randomly drifting protein. Nature. 2006;444:929–932. doi: 10.1038/nature05385. [DOI] [PubMed] [Google Scholar]
- 11.Sudol M, Chen HI, Bougeret C, Einbond A, Bork P. Characterization of a novel protein-binding module—the WW domain. FEBS Lett. 1995;369:67–71. doi: 10.1016/0014-5793(95)00550-s. [DOI] [PubMed] [Google Scholar]
- 12.Koepf EK, et al. Characterization of the structure and function of W —> F WW domain variants: Identification of a natively unfolded protein that folds upon ligand binding. Biochemistry. 1999;38:14338–14351. doi: 10.1021/bi991105l. [DOI] [PubMed] [Google Scholar]
- 13.Koepf EK, Petrassi HM, Sudol M, Kelly JW. WW: An isolated three-stranded antiparallel beta-sheet domain that unfolds and refolds reversibly; evidence for a structured hydrophobic cluster in urea and GdnHCl and a disordered thermal unfolded state. Protein Sci. 1999;8:841–853. doi: 10.1110/ps.8.4.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jolma A, et al. Multiplexed massively parallel SELEX for characterization of human transcription factor binding specificities. Genome Res. 2010;20:861–873. doi: 10.1101/gr.100552.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao Y, Granas D, Stormo GD. Inferring binding energies from selected binding sites. PLoS Comput Biol. 2009;5:e1000590. doi: 10.1371/journal.pcbi.1000590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Minasov G, Shoichet BK. Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J Mol Biol. 2002;320:85–95. doi: 10.1016/S0022-2836(02)00400-X. [DOI] [PubMed] [Google Scholar]
- 17.Weinreich DM, Delaney NF, Depristo MA, Hartl DL. Darwinian evolution can follow only very few mutational paths to fitter proteins. Science. 2006;312:111–114. doi: 10.1126/science.1123539. [DOI] [PubMed] [Google Scholar]
- 18.Mani R, St Onge RP, Hartman JL, 4th, Giaever G, Roth FP. Defining genetic interaction. Proc Natl Acad Sci USA. 2008;105:3461–3466. doi: 10.1073/pnas.0712255105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pires JR, et al. Solution structures of the YAP65 WW domain and the variant L30 K in complex with the peptides GTPPPPYTVG, N-(n-octyl)-GPPPY and PLPPY and the application of peptide libraries reveal a minimal binding epitope. J Mol Biol. 2001;314:1147–1156. doi: 10.1006/jmbi.2000.5199. [DOI] [PubMed] [Google Scholar]
- 20.Jiang X, Kowalski J, Kelly JW. Increasing protein stability using a rational approach combining sequence homology and structural alignment: Stabilizing the WW domain. Protein Sci. 2001;10:1454–1465. doi: 10.1110/ps.640101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yanagida H, Matsuura T, Yomo T. Compensatory evolution of a WW domain variant lacking the strictly conserved Trp residue. J Mol Evol. 2008;66:61–71. doi: 10.1007/s00239-007-9061-5. [DOI] [PubMed] [Google Scholar]
- 22.Toepert F, Pires JR, Landgraf C, Oschkinat H, Schneider-Mergener J. Synthesis of an array comprising 837 variants of the hYAP WW protein domain. Angew Chem Int Ed Engl. 2001;40:897–900. doi: 10.1002/1521-3773(20010302)40:5<897::AID-ANIE897>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 23.Jäger M, Dendle M, Kelly JW. Sequence determinants of thermodynamic stability in a WW domain—an all-beta-sheet protein. Protein Sci. 2009;18:1806–1813. doi: 10.1002/pro.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: A study of more than 1000 mutations. J Mol Biol. 2002;320:369–387. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 25.Baroni TE, et al. A global suppressor motif for p53 cancer mutants. Proc Natl Acad Sci USA. 2004;101:4930–4935. doi: 10.1073/pnas.0401162101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bloom JD, Labthavikul ST, Otey CR, Arnold FH. Protein stability promotes evolvability. Proc Natl Acad Sci USA. 2006;103:5869–5874. doi: 10.1073/pnas.0510098103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hinkley T, et al. A systems analysis of mutational effects in HIV-1 protease and reverse transcriptase. Nat Genet. 2011;43:487–489. doi: 10.1038/ng.795. [DOI] [PubMed] [Google Scholar]
- 28.Potapov V, Cohen M, Schreiber G. Assessing computational methods for predicting protein stability upon mutation: Good on average but not in the details. Protein Eng Des Sel. 2009;22:553–560. doi: 10.1093/protein/gzp030. [DOI] [PubMed] [Google Scholar]
- 29.Dantas G, et al. High-resolution structural and thermodynamic analysis of extreme stabilization of human procarboxypeptidase by computational protein design. J Mol Biol. 2007;366:1209–1221. doi: 10.1016/j.jmb.2006.11.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bloom JD, Glassman MJ. Inferring stabilizing mutations from protein phylogenies: Application to influenza hemagglutinin. PLOS Comput Biol. 2009;5:e1000349. doi: 10.1371/journal.pcbi.1000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Magliery TJ, Lavinder JJ, Sullivan BJ. Protein stability by number: High-throughput and statistical approaches to one of protein science’s most difficult problems. Curr Opin Chem Biol. 2011;15:443–451. doi: 10.1016/j.cbpa.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bommarius AS, Broering JM, Chaparro-Riggers JF, Polizzi KM. High-throughput screening for enhanced protein stability. Curr Opin Biotechnol. 2006;17:606–610. doi: 10.1016/j.copbio.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 33.Foit L, et al. Optimizing protein stability in vivo. Mol Cell. 2009;36:861–871. doi: 10.1016/j.molcel.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marks DS, et al. Protein 3D structure computed from evolutionary sequence variation. PLoS One. 2011;6:e28766. doi: 10.1371/journal.pone.0028766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aydin Z, Singh A, Bilmes J, Noble WS. Learning sparse models for a dynamic Bayesian network classifier of protein secondary structure. BMC Bioinformatics. 2011;12:154. doi: 10.1186/1471-2105-12-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fowler DM, Araya CL, Gerard W, Fields S. Enrich: Software for analysis of protein function by enrichment and depletion of variants. Bioinformatics. 2011;27:3430–3431. doi: 10.1093/bioinformatics/btr577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.