

SYNOPSIS
In the context of a neuromuscular disease diagnostic evaluation, the clinician still must be able to obtain a relevant patient and family history and perform focused general, musculoskeletal, neurologic and functional physical examinations to direct further diagnostic evaluations. Laboratory studies for hereditary neuromuscular diseases include relevant molecular genetic studies. The EMG and nerve conduction studies remain an extension of the physical examination and help to guide further diagnostic studies such as molecular genetic studies, and muscle and nerve biopsies. All diagnostic information needs to be interpreted not in isolation, but within the context of relevant historical information, family history, physical examination findings, and laboratory data, electrophysiologic findings, pathologic findings, and molecular genetic findings if obtained.
Keywords: Neuromuscular disease, lower motor neuron, hereditary, acquired, clinical assessment, history, physical examination, diagnosis, motor neuron disease, neuropathy, neuromuscular junction, myopathy
INTRODUCTION
Progressive acquired or hereditary neuromuscular diseases (NMDs) are disorders caused by an abnormality of any component of the lower motor neuron - anterior horn cell, peripheral nerve, neuromuscular junction (pre-synaptic or post-synaptic region), or muscle. The notion that a pathologic abnormality in a neuromuscular disease may be purely isolated to one anatomic region of the lower motor neuron with primary or secondary changes isolated to muscle is only true for selected conditions. Many neuromuscular diseases are multi-system disorders affecting multiple organ systems. For example, RNA toxicity generated from expansion of trinucleotide repeat sequences in myotonic muscular dystrophy gives rise to skeletal muscle, smooth muscle, myocardial, endocrine, brain and ocular abnormalities; Duchenne muscular dystrophy gives rise to abnormalities of skeletal and cardiac muscle, the cardiac conduction system, smooth muscle and the brain; Fukuyama congenital muscular dystrophy affects skeletal muscle and brain; mitochondrial encephalomyelopathies may affect the mitochondria of multiple tissues.
The most common NMDs are acquired peripheral neuropathies. Other acquired NMDS include amyotrophic lateral sclerosis (ALS), poliomyelitis, Guillain Barre syndrome, myasthenia gravis, and polymyositis. Hereditary NMDs are also quite common and include such disorders as spinal muscular atrophy (SMA), Charcot Marie Tooth disease, congenital myasthenia, and Duchenne muscular dystrophy. Clinical NMD syndromes described over the decades in the literature have recently been redefined based molecular genetic advances and documentation of genetic heterogeneity within specific syndromes. For example, at least 70 genetically distinct subtypes of Charcot Marie Tooth have been described, some with undetermined gene loci; over 14 genetically distinct subtypes of autosomal recessive limb girdle muscular dystrophy have been identified; and 3 genetically distinct subtypes of Emery Dreifuss exist.58 In fact, the gene loci for over 500 distinct neuromuscular and mitochondrial disorders have been identified at the time this manuscript went to press. The basis for the use of molecular genetic studies for diagnosis is well-described by Arnold and Flanigan in their article “A Practical Approach to Molecular Diagnostic Testing in Neuromuscular Diseases” in this issue5.
In the context of a neuromuscular disease diagnostic evaluation, the clinician still must be able to obtain a relevant patient and family history and perform focused general, musculoskeletal, neurologic and functional physical examinations to direct further diagnostic evaluations. Laboratory studies often include relevant molecular genetic studies in certain instances, however, specific genetic entities need to be strong diagnostic considerations, as these studies may be expensive with limited sensitivity.
Electrodiagnostic studies including EMG and nerve conduction studies remain an extension of the physical examination and help to guide further diagnostic studies such as molecular genetic studies (as in the case of Charcot Marie Tooth), muscle and nerve biopsies, or even motor point biopsies applied to the evaluation of congenital myasthenic syndromes. All diagnostic information needs to be interpreted not in isolation, but within the context of relevant historical information, family history, physical examination findings, laboratory data, electrophysiologic findings and pathologic information if obtained.
A skilled synthesis of all available information may provide the patient and family with 1) a precise diagnosis or as accurate a diagnosis as is medically possible; 2) prognostic information (if available for a specific entity); 3) information as to eligibility for molecular based therapeutic agents such as antisense oligonucleotides or morpholinos for exon skipping, or stop codon read-thru agents, and 4) anticipatory guidance for the near future. Knowledge of the natural history of specific neuromuscular disease conditions helps in the ongoing rehabilitative management of progressive impairments, activity limitations, and disabilities.
This article briefly reviews the clinical approach to the diagnostic evaluation of progressive neuromuscular diseases, including relevant history, family history, clinical examination findings, laboratory studies, and where pathological studies play a role diagnostically.
NEUROMUSCULAR DISEASE HISTORY
Important Elements of the neuromuscular disease history are shown in Box 1.
Box 1. Clinical History in Neuromuscular Diseases.
-
Weakness
Anatomic distribution / pattern of weakness and focal wasting or hypertrophy of muscle groups (arms versus legs, proximal versus distal, symmetric versus asymmetric).
Myopathies have weakness that is usually proximal greater than distal with rare exceptions
-
Course of weakness
Acute onset (days to weeks)
chronic (months to years)
episodic
Is the weakness getting worse, staying the same, or getting better?
Ascertain the rate of progression (days, weeks, months, or years).
Fatigue or lack of endurance
Muscle cramps or stiffness
Lack of sensory loss
-
Gait characteristics
Toe walking, excessive lordosis, trendelenburg or gluteus maximus lurch, etc.
-
Functional difficulties
ambulatory distances
frequency of falls
transitions from the floor to standing
problems climbing stairs
problems dressing
problems reaching overhead
difficulty lifting
running ability, problems in physical education, and recreational or athletic performance.
Onset age (neonatal, childhood, teen, adult [20–60 years], or geriatric)
Identify factors which worsen or help primary symptoms
History of recent illnesses (e.g. recent viral illnesses, respiratory difficulties, pneumonia, pulmonary infections)
Pain
Feeding difficulties, dyspahgia, nutritional status, and body composition
Cardiac symptoms (dizziness, syncope, chest pain, orthopnea, cardiac complaints with exertion)
Pulmonary symptoms (breathing difficulties, sleep disturbance, morning headaches)
Anesthetic history (e.g. malignant hyperthermia)
-
History regarding the child’s acquisition of developmental milestones
Ascertain when the child was able to control his or her head, sit independently, crawl, stand with and without support, walk with and witout support, gain fine motor prehension, and acquire bimanual skills (bringing objects to midline, transfer of objects)
History regarding language acquisition, mental development and school performance
-
History regarding pregnancy and neonatal period
Quality of fetal movement, pregnancy complications, perinatal complications, evidence of fetal distress, respiratory difficulties in the recovery room, need for resuscitation or ventilation problems in early infancy, infantile hypotonia, weak cry, poor feeding
The Common Common presenting chief complaints from parents of children with suspected neuromuscular disorders may include infantile floppiness or hypotonia, delay in motor milestones, feeding and respiratory difficulties, abnormal gait characteristics, frequent falls, difficulty ascending stairs or arising from the floor, muscle cramps or stiffness. Adults often present with chief complaints of strength loss, fatigue or decreasing endurance, falls, difficulty ascending stairs, exercise intolerance, episodic weakness, muscle cramps, focal wasting of muscle groups, breathing difficulties, or bulbar symptoms relating to speech and swallowing.
Respiratory failure due to NMD has been reported in myasthenia gravis, myosin-loss myopathy, Acid Maltase disease, Amyloid; Desmin, Polymyositis (Jo-1), Congenital Myopathy (e.g. Rod; Centronuclear), Hydroxychloroquine toxicity, Neural injury (specifically phrenic lesions), ALS, DMD, and SMA. NMDs with associated cardiac disorders include DMD, BMD, LGMD 1B LGMD 1C, sargoglycanopathies, myotonic muscular dystrophy; McLeod; Emery-Dreifuss; Barth;; Desmin, Polymyositis; Nemaline rod, Acid Maltase; Debrancher, Carnitine deficiency, some mitochondrial myopathies; Amyloid, Drugs (Metronidazole, Emetine, and Chloroquine, Clofibrate, Colchicine), Cardiomyopathy + cores, and some periodic paralyses.
Information should be obtained regarding the recent course of the chief complaint, specifically whether the process is getting worse, staying the same or getting better. If strength is deteriorating, it is important to ascertain the rate of progression (i.e. is weakness increasing over days, weeks, months or years?). It is critical to determine whether the distribution of weakness is predominantly proximal, distal or generalized. It is also useful to identify factors which worsen or help the primary symptoms. A history of twitching of muscles may reflect fasciculations. Tremor or balance problems may be due to distal weakness or superimposed cerebellar involvement.
Bulbar involvement may be identified if the individual has difficulty chewing, swallowing, or with speech articulation. Visual complaints (blurriness or diplopia) may indicate presence of cataracts or possibly involvement of extraocular musculature. Distal stocking glove or focal sensory complaints may be consistent with a peripheral neuropathy or focal nerve entrapment.
A comprehensive past medical history and surgical history should be obtained. A history of recent illnesses should be carefully elucidated, including respiratory difficulties, aspiration pneumonias or recurrent pulmonary infections. In addition, cardiac symptoms, such as dizziness, syncope, chest pain, orthopnea or exertional cardiac complaints may indicate superimposed involvement of the myocardium. A pulmonary review of symptoms should be obtained. A history of weight loss may be due to recurrent illnesses, nutritional compromise, swallowing difficulty or progressive lean tissue atrophy.
For the pediatric patient, a detailed history regarding pregnancy (e.g. quality of fetal movement or pregnancy complications), and perinatal problems (evidence of fetal distress, respiratory difficulties in the delivery room, need for resuscitation or ventilation problems in early infancy, ongoing respiratory difficulties, swallowing/feeding difficulties and persistent hypotonia) should be obtained. Perinatal respiratory distress in the delivery room may be seen in acute infantile type I SMA, myotubular myopathy, congenital hypomyelinating neuropathy, congenital infantile myasthenia, transitory neonatal myasthenia, and severe neurogenic arthrogryposis.
In children, history regarding the acquisition of developmental milestones should be ascertained relating to head control, independent sitting, crawling, standing with and without support, walking with and without support, fine prehension, bimanual skill acquisition (bringing objects to midline, transfer of objects), and language acquisition. Information regarding gait characteristics (toe walking, excessive lordosis, etc.), running ability, transitions from floor to standing, stair climbing, falls, recreational / athletic performance, pain or muscle cramps and easy fatigue or lack of endurance may be important clues to the presence of a neuromuscular disorder. History regarding mental development, type of school, and school performance may be important indicators of superimposed CNS involvement. For the adult, detailed history regarding the age of onset of symptoms, age bracing was provided to maintain ambulation, age to wheelchair (if applicable), pattern of progression, distribution of weakness, presence of muscle cramps, fatigue, episodic weakness, presence of atrophy or fasciculations, performance in physical education, military or vocational performance and pursuits, current and past ambulatory distances, ability to transition from floor to standing, problems climbing stairs, and problems reaching overhead or dressing may all be important functional information.
Potential causes of muscle cramps are shown in Box 2. Muscle cramps in the setting of an elevated creatine kinase value and no skeletal muscle weakness has been reported and a pedigree with mild Becker muscular dystrophy.26 The etiology of myalgias can be quite varied and a definitive etiology is found in only one-fourth of those patients presenting with muscle pain as a chief complaint.52 Patterns of weakness in myopathies, neuromuscular junction disorders, anterior horn cell disorders, and diagnostic considerations are outlined in Box 3, while selective anatomical distribution of peripheral neuropathies and neuronopathies are listed in Box 4.
Box 2. Causes of Muscle Cramps.
-
Cramps at rest (usually not a neuromuscular disorder)
Benign nocturnal leg cramps.
Diurnal cramps related to exercise.
-
Cramps occurring with exertion, relieved by rest (may be associated with myoglobinuria)
Muscular dystrophy, Duchenne, Becker, limb girdle muscular dystrophy.
-
Myopathy: Rippling Muscle Syndromes
RMD1 : Chromosome 1q41; Dominant
RMD2 : Caveolin-3; Chromosome 3p25.3; Dominant
-
Metabolic disorders
-
Glycogenoses
Myophosphorylase deficiency (type V; McArdle’s disease)
Phosphofructokinase deficiency (type VII)
Phosphorylase b kinase deficiency (type VIII)
Phosphoglycerate kinase deficiency (type IX)
Phosphoglycerate mutase deficiency (type X)
Lactate dehydrogenase deficiency (type XI)
Myoadenylate deaminase deficiency
-
Lipid metabolism disorders
Carnitine palmityl transferase deficiency
uremia
electrolyte abnormality: hyponatremia, hypocalcemia, hypomagnesemia, hypoglycemia
hypothyroidism
hypoadrenalism
paroxysmal myoglobinuria
idiopathic rhabdomyolysis
-
-
Toxic myoglobinuria
alcohol
barbiturates
heroin
carbon monoxide
amphotericin B
toxic venoms
-
Inflammatory myositis
acute dermatomyositis, polymyositis
viral myositis (coxsackie etc.)
bacterial myositis (staphylococcus, clostridia)
-
Acute extracellular volume depletion
perspiration
diarrhea, vomiting
diuretic therapy
hemodialysis
-
Other lower motor neuron disorders
amyotrophic lateral sclerosis, old polio, other motor neuron disorders
radiculopathy and neuropathy
Box 3. Patterns of Weakness in Myopathies, NMJ pathology and Motor Neuron Disorders.
Extraocular muscles weak
Myasthenia Gravis (MG)
Thyroid;
Botulism
Mitochondrial: KS; PEO; MNGIE
Myopathy: Centronuclear; Multicore
Oculopharyngeal MD;
IBM + Contracture;
Oculopharyngodistal myopathy;
Congenital ophthalmoplegias
Periocular without EOM Weakness
Dystrophies: Myotonic; FSH; Oculopharyngeal
NMJ: Myasthenia Gravis (MG)
Congenital Myasthenic Syndromes
Congenital Myopathies
Inflammatory myopathy: Polymyositis
Rule out: VII nerve lesion
Bulbar dysfunction
NMJ: MG; Congenital myasthenic syndromes
Thyroid
Cranial nerve Δ
Oculopharyngeal MD
Distal myopathy (MPD2)
Polymyositis: IBM; Scleroderma
Motor neuron Δ: ALS; BSMA
Pseudobulbar; Fazio-Londe;
Brown-Vialetto-van Laere
Posterior neck weak
Common: MG; PM; ALS
Focal myopathy: Neck; Paraspinous
Rare: FSH dystrophy; LMN synd; IBM;
Rod; PROMM; Acid maltase; hypo K+;
Carnitine; Endocrine; Desmin
Proximal arms weak
Dystrophy: Scapuloperoneal; FSH
Inflammatory : Brachio-Cervial Inflammatory Myositis (BCIM)
Absent muscles; Shoulder joint Δ
NMJ: MG;
Neuropathic: ALS; P-LMN;
Brachial plexopathy
Distal & Proximal weakness
Dystrophy: Myotonic; FSH, Scapuloperoneal
Myopathy: Congenital; Distal
Glygogenoses: Debrancher,
Phosphorylase b kinase
Neuropathy + Myopathy: Paraneoplastic;
Sarcoid; Mitochondria; HIV;
Drugs (Amiodarone; Doxorubicin Colchicine; Chloroquine
Acute weakness
NMJ: Myasthenia gravis
Myoglobinuria
Myosin loss myopathy
Carnitine deficiency
Periodic paralysis: X-Episodic Xp22
Hypo K+: CACNA1S; SCN4A; KCNE3
Hyper K+: SCN4A; KCNE3
Andersen: KCNJ2
Electrolyte disorders: K+ ↑ or ↓;
Mg ↑; PO4 ↓;
Barium
Rule out: Neuropathy (AIDP, CIDP); Spinal cord
Wasting > Weakness
Pathology: Type II atrophy
Cachexia: Wt loss > 15%, Aging / sarcopenia
Disuse
Steroid myopathy
Paraneoplastic
Weakness > Wasting
Polymyositis;
Myoglobinuria;
Periodic Paralysis;
NMJ: Myasthenia gravis; Congenital myasthenia
Neuropathy + conduction block
Quadriceps weak
LGMD: 1B; 2B; 2H; Ring fib
Becker
Myositis: IBM; Mitochon; Focal
Nerve: Femoral; LS plexopathy;
Diabetic amyotrophy; L3-L4 root
Adapted from Alan Pestronk, Neuromuscular Disease Center Website, Washington University, St. Louis, MO USA, 6/20/2011; http://neuromuscular.wustl.edu
Box 4. Selective Anatomical Distribution of Peripheral Neuropathies and Neuronopathies.
(Most peripheral neuropathies are symmetric and maximal distally in the lower extremities)
Extraocular muscle
Botulism
Diabetes
Miller-Fisher
Diphtheria
Rule out:
MG; Myopathy
Proximal Motor
Immune Demyelinating:
GBS; CIDP
SMA; Porphyria
Plexopathy:
Brachial; Lumbar
Rule out: Joint pain;
Myopathy
Proximal Sensory
Hereditary: Porphyria; Tangier
Neuronopathy: Hu; Sjögren’s
Thoracic neuropathy
Rule out: Myelopathy
Skin temperature-related
Leprosy
Upper extremity
Immune: MMN; Vasculitis
CIDP variant
Amyloid: Carpal tunnel
Entrapment: HNPP; Other
Toxic: Lead; Vincristine
ALS; LMN
Rule out: Spinal; CNS
Asymmetric
Mononeuritis multiplex
Neuronopathy: ALS; Sensory
Entrapments
Plexopathies
Toxic
Mononeuritis Multiplex
Vasculopathy;
Amyloid;
Leprosy;
Diabetes;
CMV
Waldenström;
Perineuritis
Demyelinating: HNPP; Multifocal CIDP; MMN
Compression: Multiple
Lymphoma: Intraneural
Wartenberg
CNS
Spinal: Organophosphate; Hexacarbon; AMN;
MLD; Lymphoma; Cuban; Vernant’s
Optic : Disulfiram; CS2; Hg; Drugs; NARP;
CMT6; Post col & RP; Cuban; Vernant’s
Hearing loss: HMSN X, 1A, 1B, 4D, 6; Mitichondrial; Sarcoid
Cerebellum: FA; AT; MLD; Refsum; A-β-lipoproteinemia; SCA 2, 3, 4; IOSCA; Hu & CV-2
Supratentorial: Mitochondrial; Thyroid; Hu; B12; Vasculitis; Neoplastic; Sarcoid
Infection: Lyme; HIV; Rabies; Syphilis; West Nile
Hereditary: Polyglucosan; Fabry; HexA; Porphyria; Prion; ALS; Cowchock; NAD; Krabbe; MLD
Face
Bell’s Palsy
Melkersson; Tangier
Polyradiculopathies:
Sarcoid; Lyme; GBS
Motor neuron disorders:
ALS; Kennedy’s; Möbius
Rule out: MG; Myopathy
Adapted from Alan Pestronk, Neuromuscular Disease Center Website, Washington University, St. Louis, MO USA, 6/20/2011; http://neuromuscular.wustl.edu
A history should be obtained regarding dark colored urine or hematuria as a clue regarding rhabdomyolysis. Myoglobinura may be associated with Glycogenolysis; CPT II; LPIN1, malignant hyperthermia; central core, King-Denborough; DMD (Some), hypokalemia, Licorice; Li; Thiazide; Amphotericin; Laxatives, Infections; mitochondrial myopathy; muscle trauma,; muscle: Ischemia; overactivity; Polymyositis, neuroleptic malignant syndrome, drugs (Heroin; Phencylidine; Epsilon-ACA, Clofibrate + Renal failure; Cyclosporine A + Lovastatin, Toxins (e.g. venoms; IV drugs, Oral drugs (Haff); mushrooms; and EtOH.
A thorough anesthetic history should be obtained as malignant hyperthermia is associated with one of the many subtypes of primary familial malignant hyperthermia (Hypokalemic periodic paralysis, or one of the MHS loci including MHS1: Ryanodine Receptor; 19q13 (allelic with Central Core congenital myopathy), MHS2: Na+ channel (SCNA4); 17q11, MHS3: Ca++ channel (CACNL2A); 7q21, MHS4: 3q13, MHS5: Ca++ channel (CACNA1S); 1q32, MHS6: 5p, CPT2: 1p32, and King-Denborough syndrome. Other NMD conditions occasionally reported to be associated with malignant hyperthermia include: Duchenne muscular dystrophy (DMD), Becker muscular dytsrophyFukuyama congenital muscular dystrophy, limb girdle muscular dystrophy (LGMD), fascioscapulohumeral muscular dystrophy (FSH), periodic paralysis, myotonia congenita, mitochondrial myopathy, minimal change myopathy, myoadenylate deaminase deficiency, and the Schwartz-Jampel syndrome.
FAMILY HISTORY
Whenever a neuromuscular disorder is suspected with a potential genetic etiology, a detailed family history and pedigree chart is absolutely essential. Autosomal dominant conditions may have pedigrees with multiple generations affected with equal predilection to males and females. Typically one-half of offspring within a pedigree are affected. In autosomal recessive conditions, only one generation may be affected with equal proportions of males and females. Proportionally, one-fourth of offspring are clinically affected. Parents in earlier generations may be unaffected and the parents of affected children are presumptive heterozygote carriers of the condition. In many instances of autosomal recessive inheritance, no other family members within the nuclear family unit are affected making the confirmation of inheritance pattern difficult without a molecular genetic marker present or protein abnormality confirmed by immunohistochemistry techniques. In X-linked recessive conditions, males on the maternal side of the family are affected in approximately 50% of instances and females are carriers in 50% of instances.
Often, it is valuable to examine affected relatives who may be either earlier or later in the course of their neuromuscular disease relative to the patient. In addition, medical records and diagnostic evaluations of affected family members should be reviewed and the diagnosis confirmed if possible.
In some instances, the examination of a parent can help establish the diagnosis in an affected infant or child, as is frequently the case in myotonic muscular dystrophy (MMD). In this disorder, genetic anticipation with abnormal CTG trinucleotide expansion of unstable DNA results in progressively earlier onset of the disease in successive generations with increasing severity, as described elsewhere in this issue.5
In the case of dystrophic myopathies, a definitive molecular genetic or pathologic diagnosis established in a sibling or close relative may allow the clinician to establish the diagnosis in a child or adult based on clinical examination and laboratory data such as creatine kinase or molecular genetic testing, thus allowing the avoidance of further invasive testing such as a muscle biopsy.
PHYSICAL EXAMINATION
Inspection At Rest
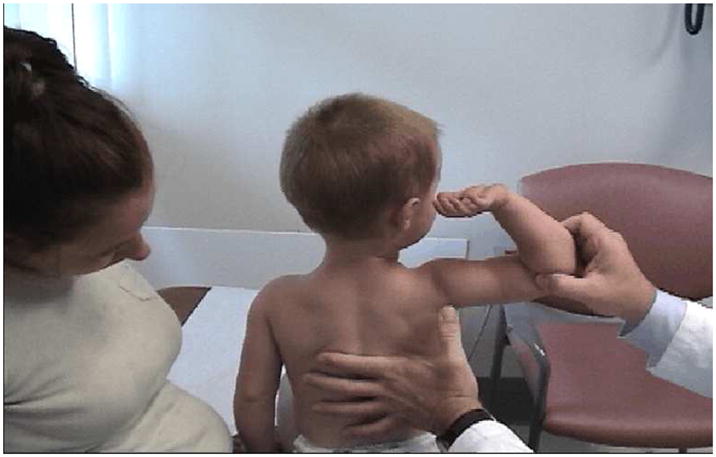
Simple inspection allows the observation of focal or diffuse muscle wasting, or focal enlargement of muscles as with the “pseudohypertrophy” seen in dystrophic myopathies such as Duchenne and Becker muscular dystrophy (see figure 1) LGMD, and lipodystrophy. Cros and colleagues16 have demonstrated that the increase in calf circumference in DMD is caused by an increase in fat and connective tissue and not secondary to true muscle fiber hypertrophy in the gastrocnemius. In contrast, the reduced bulk of the quadriceps in DMD was caused by more severe fiber loss in a more “active” dystrophic process affecting the knee extensors. In DMD, pseudohypertrophy may be present in other muscle groups such as the deltoid (figure 2).
Figure 1.

Child with Duchenne muscular dystrophy; note the calf hypertrophy, mild equinus posturing at the ankles, shoulder retraction, and mild scapular winging.
Figure 2.
Pseudohypertrophy of the posterior deltoid muscle and posterior axillary depression sign in Duchenne muscular dystrophy.
Other neuromuscular disorders may show calf pseudohypertrophy.71 Calf hypertrophy is particularly prominent in childhood type of acid maltase deficiency. In spinal muscular atrophy type III (Kugelberg-Welander syndrome), calf enlargement has been occasionally noted but wasting of affected musculature is typically more prominent. Other NMDs with enlarged muscles include myotonia conditions with overusage; hypothyroidism, acromegaly, infection with cysticercosis, Trichinosis, and Schistosomiasis, anabolic drugs (e.g. β2 adrenergic; Androgen), glycocgen storage diseases, amyloidosis, accumulation of Gangliosides, and Schwartz-Jampel Syndrome.
Children ages 6–11 with Duchenne muscular dystrophy have been noted to exhibit an unusual clinical examination sign due to selective hypertrophy and wasting in different muscles of the same region.70 When viewing these patients posteriorly with their arms abducted to 90° and elbows flexed to 90°, the DMD patients demonstrated a linear or oval depression (due to wasting) of the posterior axillary fold with hypertrophied or preserved muscles on its two borders (i.e., infraspinatus inferomedially and deltoid superolaterally), as if there were a valley between the two mounts as seen in Figure 2.
There are several characteristic facial features of myotonic muscular dystrophy which may be noted on inspection (see Figure 3). The adult with long-standing myotonic muscular dystrophy often has facial features so characteristic that it is often easy to make a tentative diagnosis from across the room. The long thin face with temporal and masseter wasting is drawn and described by some as “lugubrious”. Adult males often exhibit frontal balding. Infants and young children with a variety of myopathies may exhibit a tent-shaped mouth (Figure 3).
Figure 3.

a) adult with characteristic facial characteristics associated with myotonic muscular dystrophy (DM1). Note the long drawn face, temporal wasting, and male pattern baldness. B) Four year old child with congenital myotonic muscular dystrophy (DM1). Note the triangular or “tent-shaped” mouth and slight temporal wasting.
Focal atrophy of particular muscle groups may provide diagnostic clues to specific neuromuscular disorders. SMA gives diffuse muscle atrophy or focal atrophy in more slowly progressive subtypes. Emery-Dreifuss may present with striking wasting of the biceps, accentuated by sparing of the deltoids and forearm muscles. There may also be wasting of the calf muscles in this condition. Quadriceps selective weakness and atrophy may be a presenting sign in a variety of myopathies such as Becker muscular dystrophy, LGMD: 1B; 2B; 2H; 2L (11p13 LGMD 2L: recessive, Anoctamin 5 (ANO5, MEM16E, GDD1) ; Chromosome 11p14.3; Recessive, Emery-Dreifuss: Lamin A/C hereditary IBM3, inflammatory myopathies, sporadic inclusion body myositis, polymyositis with mitochondrial pathology, focal myositis, myopathy with ringed fibers, Spinal muscular atrophy types III and IV.
5q: Type III & IV, femoral neuropathy, and diabetic amyotrophy, and L3-L4 radiculopathy.
Patient’s with focal shoulder girdle weakness, as in Facioscapulohumeral muscular dystrophy (FSHD) and limb girdle muscular dystrophy, may show characteristic patterns of muscle atrophy and scapular displacement. In FSHD, involvement of the latissimus dorsi, lower trapezius, rhomboids and serratus anterior results in a characteristic appearance of the shoulders with the scapula positioned more laterally and superiorly, giving the shoulders a forward-sloped appearance. The upper border of the scapula rises into the trapezius, giving it a hypertrophied appearance falsely. From the posterior view, the medial border of the scapula may exhibit profound posterior and lateral winging (Figure 4). The involvement of shoulder girdle musculature in FSHD may also be quite asymmetric.
Figure 4.

Young adult with Facioscapulohumeral Muscular Dystrophy (FSHD). Note the posterior and lateral scapular winging, the high riding appearance of the scapula, and the asymmetry of winging in the photo on the right.
Most weakness in neuromuscular disorders is associated with focal atrophy. Those with CMT, particularly those with the type II axonal forms demonstrate distal atrophy or “stork leg appearance” relatively early in the disease course. Those with primarily demyelinating type I forms of CMT may show distal wasting later in the disease course.
Muscle fasciculations may be seen as nonspecific findings of a variety of lower motor neuron disorders. Fasciculations are particularly common in motor neuron disorders, such as ALS and SMA. Distal essential tremor may be seen in a large proportion of CMT patients, (30–50%),34 and other patients with weakness such as SMA. “Polyminimyoclonus”, another variant of muscle fasciculations, characterized by a fine tremor of the fingers and hands, may be evident in SMA I and II.
Palpable nerves in the cubital tunnel, posterior auricular region or around the fibular head may be indicative of onion bulbs seen in CMT I subtypes, or Dejerine Sottas Disease (CMT III).
General Examination
Important aspects of the cardiac and pulmonary assessment pertaining to NMD conditions are described in the next issue of the Physical Medicine and Rehabilitation Clinics of North America. Hepatomegaly may be seen in metabolic myopathies such as acid maltase deficiency (type 2 glycogenosis) and types 3 and 4 glycogenosis. Characteristic skin rashes and nail bed capillary changes may be present in dermatomyositis. Ullrich’s congenital muscular dystrophy patients with a collagen VI abnormality often show hyperkeratosis pilaris in the extensor surfaces of the upper arms (Figure 5). Craniofacial changes and dental malocclusion are commonly seen in congenital myotonic muscular dystrophy, congenital myopathies, congenital muscular dystrophy, and type II SMA.
Figure 5.

Hyperkeratosis pilaris (is a fine erythematous popular rash on the back and extensor surface of the upper arm) on the left (A) and distal joint hyperlaxity on the right (B) in a patient with Ullrich congenital muscular dystrophy
Cognitive Assessment
Some neuromuscular disorders such as congenital and non-congenital myotonic muscular dystrophy (DM1), PROMM (DM2), Fukuyama congenital muscular dystrophy, selected cases with mitochondrial encephalomyelopathies and a small proportion of Duchenne muscular dystrophy cases may have significant intellectual impairment. In addition other NMDs with significant cognitive involvement include hereditary IBM (9pp13), selected mitochondrial encephalomyopathies, congenital MD: (Santavuori, POMGnT1 1p32; Merosin 6q22; Fukuyama Fukutin 9q31; and Integrin-α7 12q13), and Phosphoglycerate Kinase deficiency. In these instances referral for neuropsychological testing, a neurodevelopmental evaluation, and/or a pyschoeducational evaluation may be helpful..78
Cranial Nerve Examination
Neuromuscular disorders tend not to have optic nerve involvement, however, an evaluation of vision and a funduscopic examination can be exceedingly important. For example, myotonic muscular dystrophy patients (DM1) may have cataracts giving significant visual impairment. These cataracts may have multi-colored subcapsular opacities noted on a careful slit-lamp examination. In addition to the lens opacities, retinal degeneration characterized by peripheral pigmentary changes in the macula may be present in MMD. Other ocular abnormalities, including low intraocular pressure, enophthalmos, blepharitis, and corneal lesions have been described in this disorder as well. All myotonic muscular dystrophy patients should have regular ophthalmologic evaluations.
Ptosis is a finding described in myasthenia gravis, congenital myasthenic syndromes, transient auto-immune neonatal myasthenia, oculopharyngeal muscular dystrophy, and occasionally myotonic muscular dystrophy.
Ophthalmoparesis may be a finding seen in myasthenia gravis, congenital myasthenic syndromes and oculopharyngeal muscular dystrophy. In addition, extraocular muscle involvement may occur in some of the congenital myopathies, particularly myotubular myopathy, and some of the mitochondrial myopathies. For example, progressive external ophthalmoplegia (PEO) is a mitochondrial disorder which may present with bilateral ophthalmoplegia with or without limb weakness. Congenital fibrosis of the extraocular muscles or “congenital familial external ophthalmoplegia” is an autosomal dominant, congenital, nonprogressive disorder of the ocular muscles with primary findings of bilateral ptosis and external ophthalmoplegia. Affected individuals with PEO often have associated facial weakness. Gaze limited in all directions, eye movement speed is slow, it is associated with ptosis, and it is slowly progressive.
Facial weakness is an important clinical feature of facioscapulohumeral muscular dystrophy (FSHD). The initial weakness affects the facial muscles, especially the orbicularis oculi, zygomaticus, and orbicularis oris. These patient’s often have difficulty with eye closure but not ptosis (Figure 6). The individual may assume an expressionless appearance and exhibit difficulty whistling, pursing their lips or drinking through a straw, or smiling. Even in the very early stages, forced closure of the eyelids can be easily overcome by the examiner. Masseter, temporalis, extraocular and pharyngeal muscles are characteristically spared in FSHD.
Figure 6.
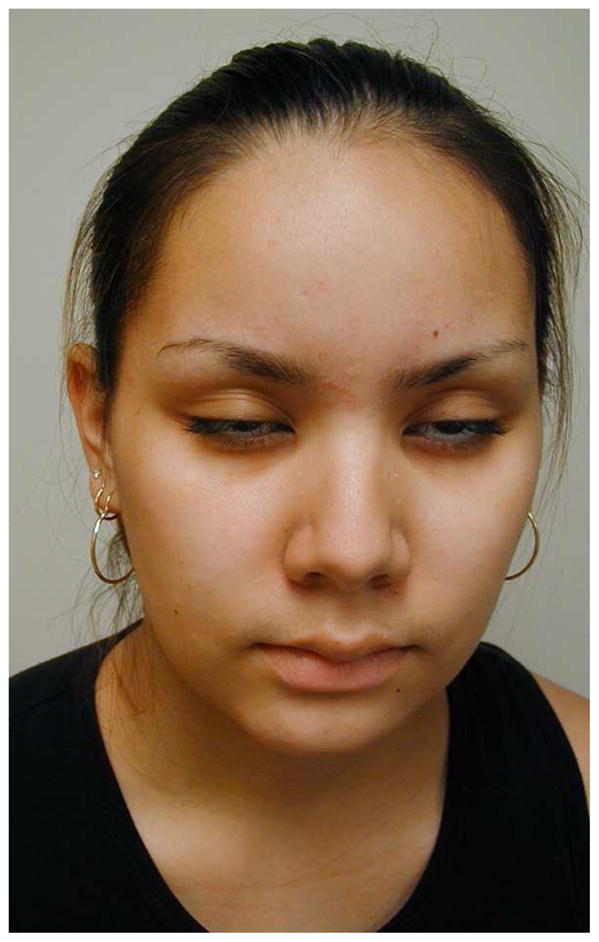
Facial weakness of orbicularis oculi in Facioscapulohumeral Muscular Dystrophy (FSHD). Eye closure is weak and weakness of orbicularis oris produces difficulty smiling, puffing out the cheeks, and pursing the lips.
Facial weakness may also be observed in oculopharyngeal muscular dystrophy, myasthenia gravis, congenital myasthenic syndromes, Moebius syndrome, congenital myopathies, and myotubular myopathy. Rare cases with FSHD have been described, secondary to a hereditary neuropathy with weakness in predominantly a scapuloperoneal distribution with involvement of the facial muscles and other limb muscles such as the shoulder girdle, ankle dorsiflexors and ankle everters.
A sensorineural hearing deficit was originally observed in “Coates syndrome” (early onset FSHD). These individuals have a myopathy presenting in infancy. The disease progression is fairly rapid with most individuals becoming wheelchair reliant by the late second or third decade. These individuals also have a progressive exudative telangiectasia of the retina. Early recognition and photocoagulation of the abnormal retinal vessels may prevent visual loss. Several studies of later onset FSHD using audiometry have demonstrated hearing deficits in many later onset FSHD patients in addition to those with “Coates” syndrome, suggesting that impaired hearing function is more common than expected in FSH muscular dystrophy.51,62,93 Thus, all patients with FSHD should have screening audiometry and ophthalmologic evaluation.
Involvement of palatal, pharyngeal, and laryngeal muscles may produce dysarthria and dysphagia. Patients at particular risk include those with ALS, SMA, myasthenia gravis, congenital myasthenic syndromes, congenital myopathies such as myotubular myopathy, oculopharyngeal muscular dystrophy, late-stage Duchenne muscular dystrophy and late-stage LGMD with autosomal recessive inheritance. The function of the swallowing mechanism is best evaluated with a fluoroscopic video dynamic swallowing evaluation.
Vocal cord paralysis is a relatively uncommon finding in hereditary neuromuscular disorders, however, distal infantile spinal muscular atrophy with diaphragm paralysis (DSMA1; SMARD1; HMN 6) linked to Chromosome 11q13.3.27,98 Vocal cord paralysis has also been described as a complication of dermatomyositis.
Examination of the tongue for muscle bulk and presence of fasciculations should be performed. Tongue fasciculations are a common finding in ALS and SMA types I, II and III. However, tongue fasciculations are not an absolute finding in ALS or SMA. For example, 56–61% of SMA I patients, 30–70% of SMA II patients and roughly half of SMA III patients late in the disease course show tongue fasciculations.33,54,57,63 Thus, absence of tongue fasciculations does not necessarily exclude these motor neuron disorders. The bulk of the tongue may be increased in some metabolic diseases such as acid maltase deficiency and often in later stages of Duchenne muscular dystrophy.
Tone
Hypotonia (see Figure 7) is an important clinical examination finding in children with neuromuscular disorders. The most common etiology for infantile hypotonia is central, accounting for approximately 80% of cases. Hypotonia remains the most common reason for referral to the pediatric electrodiagnostic laboratory. A differential diagnosis of infantile hypotonia is shown in Box 5.
Figure 7.
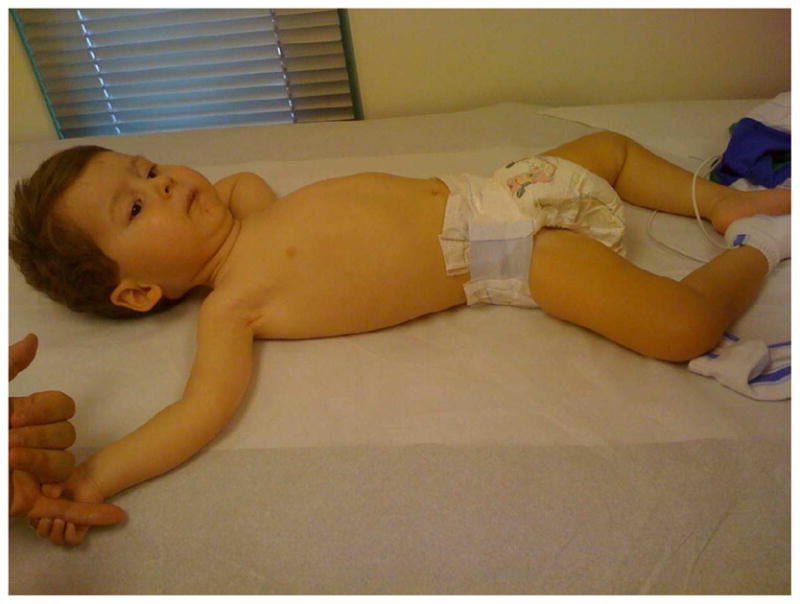
Child with severe SMA II, with hypotonia and chest wall wasting creating a bell-shaped chest.
Box 5. Differential Diagnosis of Infantile Hypotonia.
-
Cerebral hypotonia
-
Chromosome disorders
Trisomy
Prader-Willi syndrome
-
Static encephalopathy
cerebral malformation
perinatal CNS insult
post-natal CNS insult
-
Peroxisomal disorders
cerebrohepatorenal syndrome (Zellweger)
neonatal adrenoleukodystrophy
-
Inborn errors of metabolism
glycogen storage disease type II (Pompe disease)
infantile GM1, gangliosidosis
Tay-Sachs infantile GM2 gangliosidosis)
-
vitamin dependency disorders (many)
etc.
-
Amino acid and organic acid disorders
maple syrup disease
hyperlysinemia
nonketotic hyperglycinemia
-
propionyl-CoA carboxylase deficiency
etc.
-
Other genetic disorders
familial dysautonomia
Cohen syndrome
oculocerebrorenal syndrome (Lowe)
Benign congenital hypotonia
-
-
Spinal cord
-
Trauma (obstetrical; post-natal)
hypotonia early with acute paraplegia
hypertonia
-
Tumor or AVM
hypertonia may occur later or with slow growing tumor
-
Anterior horn cell
spinal muscular atrophy type I (Werdnig-Hoffman)
spinal muscular atrophy type II
poliomyelitis
neurogenic arthrogryposis
-
-
Polyneuropathies
Congenital hypomyelinating neuropathy
Chronic inflammatory demyelinating polyneuropathy
Acute inflammatory demyelinating polyradiculoneuropathy (Guillain-Barre)
Hereditary motor-sensory neuropathies (e.g. I, III,)
Toxic polyneuropathy
Leukodystrophies (Krabbe’s; Nieman-Pick)
Leigh’s syndrome
Giant axonal neuropathy
Dysmaturation neuropathy
-
Neuromuscular junction
-
Presynaptic
infantile botulism
hypermagnesemia - eclampsia
aminoglycoside antibiotics
congenital myasthenia
acetylcholine vesicle paucity
decreased quantal release
-
Postsynaptic
neonatal (autoimmune)
congenital myasthenia
acetylcholinesterase deficiency
slow changes
acetylcholine receptor deficiency
-
-
Myopathies
-
Congenital myopathies
Nemaline rod
central core
myotubular (centronuclear)
congenital fiber type disproportion
Congenital myotonic dystrophy
-
Congenital muscular dystrophy
Fukuyama type (CNS involvement)
Merosin deficiency (with or without CNS involvement)
atonic-sclerotic type (Ulrich’s disease)
undifferentiated
-
Inflammatory myopathies
infantile polymyositis
-
Metabolic myopathies
acid maltase deficiency (type II)
muscle phosphorylase deficiency (type V)
phosphofructokinase deficiency (type VII)
cytochrome c oxidase
carnitine deficiency
-
Endocrine myopathies
hypothyroidism
hypoparathyroidism
-
Strength Assessment
The distribution of weakness is often a critical piece of information allowing the clinician to categorize a patient into a specific neuromuscular diagnostic syndrome. The distribution of weakness should be noted (predominantly proximal versus distal; lower extremity versus upper extremity; focal versus generalized; isolated peripheral nerve distribution versus multiple peripheral nerves; or single versus multiple roots/myotomes). It should be noted whether extraocular, facial and bulbar muscles are involved or spared. In addition to appendicular (limb) strength, the strength of axial musculature should also be noted.
A common finding in myopathies, particularly dystrophic myopathies, is the early and selective weakness of the neck flexors as opposed to the neck extensors. For example, the neck flexors are the earliest muscle group to show weakness in Duchenne muscular dystrophy.7,38 Clinical examination of a child or adult with a suspected dystrophic myopathy should always include an evaluation of neck flexor strength (see Figure 8). Quantitative isometric strength measurements of neck strength in normal subjects with grade 5 neck flexors and extensors on manual muscle testing show the neck extensors to be stronger than the neck flexors. Absolute muscle strength is directly proportional to the physiological cross-sectional area of muscle fiber.4,95 The cross-sectional area of the neck extensors is much greater than the cross-sectional area of the neck flexors. Seventeen muscle groups act bilaterally as neck extensors, whereas only six muscle groups act bilaterally as neck flexors. Thus, with dystrophic myopathies, the progressive loss of muscle fiber over time results in significant clinically detectable weakness of the neck flexors earlier than the neck extensors. This is often accentuated in children by the large proportional size of the head relative to the rest of their body.
Figure 8.
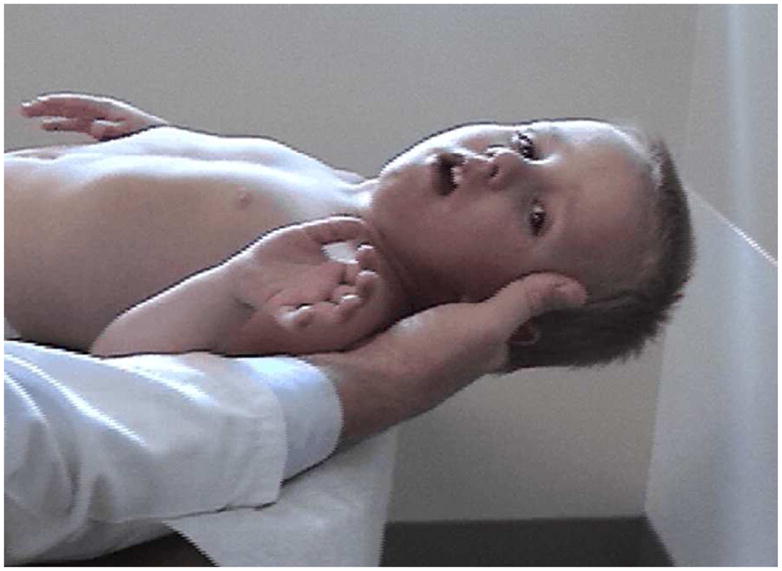
Examination for neck flexor weakness in Duchenne muscular dystrophy.
Predominantly distal lower extremity weakness is highly suggestive of an acquired or inherited peripheral neuropathy, the differential for which is quite broad. There are a number of other inherited neuromuscular disorders which can present with distal lower extremity weakness. Anterior horn cell disorders include distal chronic spinal muscular atrophy. Myopathies include inflammatory myopathies, such as inclusion body myositis, scapuloperoneal syndromes including scapuloperoneal muscular dystrophy, late adult onset autosomal dominant distal myopathy, Finnish tibial muscular dystrophy, early adult onset autosomal recessive distal myopathy (types I and II) and occasionally metabolic myopathies. Distal upper extremity weakness may be seen initially in Asian variant distal spinal muscular atrophy, and Welander type late adult onset autosomal dominant distal myopathy.
The differential diagnosis of the limb girdle syndromes presenting in childhood and adulthood and characterized by predominantly proximal weakness of shoulder and pelvic girdle muscles remains quite large and may include LGMD subtypes, polymyositis, dermatomyositis, congenital myasthenic syndromes, inclusion body myositis, type III spinal muscular atrophy, manifesting carrier of DMD, BMD, FSH, scapuloperoneal myopathy, Emery-Dreifuss muscular dystrophy, congenital myopathies occasionally presenting later in childhood or adulthood (i.e. adult onset Nemaline rod disease, central core disease, centronuclear myopathy, fiber type disproportion, multicore disease, sarcotubular myopathy, fingerprint myopathy, reducing body myopathy), mitochondrial myopathies with limb girdle weakness, other metabolic myopathies which may present in adulthood (i.e., adult onset acid maltase deficiency, debrancher enzyme deficiency, McArdle’s disease, carnitine deficiency), myopathy with tubular aggregates, and myopathy with cytoplasmic bodies.
Quantitative Strength Testing
Strength is difficult to objectively evaluate in children with motor impairments. McDonald and colleagues40 have demonstrated strength measurement to be more stable and reproducible in children older than age five. Quantitative strength measurements have been demonstrated to be far more sensitive than clinical strength testing for detecting weakness in children and adults with motor impairments.1,46 The author and his colleagues at the University of California, Davis Research and Training Center in Neuromuscular Disease have published a number of studies utilizing isometric and isokinetic quantitative strength testing as a measure of impaired strength in patients with neuromuscular disorders,1,2,12,24,25,35,39,40,46,47,48 and we have shown quantitative strength testing to be a more sensitive measure of weakness than clinical examination, particularly when strength is grade 4–5 on manual muscle testing. At age six, the reduction in tension developed by the knee extensors of DMD subjects was approximately 50% of control values for knee extension, while knee extension was between grade 4–5 on same day clinical manual muscle testing. Thus, by the time patients have progressed to grade 4 strength by manual muscle testing, substantial weakness is present.
Repetitive Strength Testing
When suspecting episodic weakness with a fatigue component, the examiner may have the patient repetitively contract a muscle against resistance for 10–15 contractions through a functional range of motion. This often brings about obvious fatigue and progressive weakness after a number of contractions in myasthenic syndromes, such as myasthenia gravis or congenital myasthenia. This can also be accomplished more quantitatively with isokinetic dynamometry, comparing peak torque with initial contractions versus later contractions (e.g., the fifth contraction or tenth contraction).
Sensory Examination
A stocking glove loss of sensation or vague distal dysesthesias may be present in a peripheral neuropathy. Focal sensory changes in one or more peripheral nerve distributions can be indicative of focal entrapments which are commonly seen in hereditary neuropathy with predisposition to pressure palsy (HNPP), which is one of the CMT subtypes.
Cerebellar Examination
The presence of tremor, dysdiadachokinesia (problems with rapid alternating movements), or axial and appendicular ataxia/balance problems can be important findings in syndromes such as ataxia telangiectasia, autosomal dominant spinocerebellar degeneration syndromes, and Friedreich’s ataxia.
Deep Tendon Reflexes
While deep tendon reflexes are generally depressed or absent in many neuromuscular diseases, they may be brisk in syndromes with superimposed upper motor neuron involvement such as ALS or some spinocerebellar degeneration syndromes. It is important to remember that the presence of deep tendon reflexes (DTRs) does not necessarily exclude the presence of a neuromuscular disease. For example, in one series,33 DTRs were absent in all four extremities in 74% of SMA I cases, but present and depressed in 26% of cases. In SMA II and III, DTRs are invariably depressed and usually become absent over time.
Myotonia
The clinical finding common to all myotonic disorders is myotonia, which is a state of delayed relaxation or sustained contraction of skeletal muscle. Grip myotonia may be demonstrated by delayed opening of the hand with difficult extension of the fingers following tight grip. Paradoxical myotonia is the situation where myotonia becomes worse with successive movements instead of improving with activity. Percussion myotonia may be elicited by percussion of the thenar eminence with a reflex hammer giving an adduction and flexion of the thumb with slow return (Figure 9). Other sites which may give a local contraction with percussion include the deltoid, brachioradialis and gluteal muscles. Occasionally, myotonia of the tongue draped over a tongue blade may be elicited with a midline tap of the finger, giving a bilateral contraction notch along the lateral portion of the tongue bilaterally with slow relaxation. Myotonic syndromes include myotonic muscular dystrophy (Steinert’s disease), myotonia congenita (Thomsen’s disease), Becker type myotonia congenita, paramyotonia congenita (Eulenburg’s disease), and Schwartz-Jampel syndrome (chondrodystrophic myotonia).
Figure 9.

Percussion myotonia in myotonic muscular dystrophy (DM1).
Schwartz-Jampel syndrome is usually distinguished by typical facial characteristics, blepharospasm, dwarfism and other skeletal abnormalities, and the presence of hypertrophic and clinically stiff muscles. Muscle hypertrophy may also be seen in myotonia congenita and paramyotonia congenita.
Myotonia may be aggravated by cold in myotonia congenita, the dominant form of Becker type myotonia congenita, and paramyotonia congenita. The myotonia seen in myotonic muscular dystrophy is not typically exacerbated by cold.
Limb Contractures
A comprehensive description of the specific contractures most often present among the more common NMD conditions is presented elsewhere in this issue.81 The presence of specific contractures can be helpful diagnostically, as in the clinical distinction between congenital muscular dystrophy which often presents with contractures versus other congenital structural myopathies which frequently present with hypotonia but no contractures. The presence of isolated elbow flexion contractures can be a diagnostic clue to Emery-Dreifuss muscular dystrophy. In general, dystrophic myopathies have a greater predilection towards the development of contractures than other myopathies and neurogenic conditions.
Spinal Deformity
A discussion of the prevalence, natural history and management of spinal deformity is discussed in the next issue of the Physical Medicine and Rehabilitation Clinics of North America. NMD populations at risk for scoliosis include DMD, SCARMD, congenital muscular dystrophy, FSH, congenital myotonic muscular dystrophy, spinal muscular atrophy, and Friedreich’s ataxia.
Functional Examination
A thorough functional examination is essential in the diagnostic evaluation of a patient suspected of a neuromuscular disease. This includes an evaluation of head control, bed/mat mobility, transitions from supine-to-sit, sit-to-stand, sitting ability without hand support, standing balance, gait, stair climbing and overhead reach.
An evaluation of overhead reach examining the patient from the front and from behind is helpful in evaluation of shoulder girdle weakness. Careful assessment of scapular winging, scapular stabilization and scapular rotation is very helpful in the assessment of patients with FSHD and LGMD. The scapulae is stabilized for overhead abduction by the trapezius, rhomboids, and serratus anterior. Abduction to 180° requires strong supraspinatus and deltoid in addition to strong scapular stabilizers.
Patients with proximal weakness involving the pelvic girdle muscles may rise off the floor using the classic “Gower’s” sign where the patient usually assumes a four point stance on knees and hands, brings the knees into extension while leaning forward on the upper extremities, substitutes for hip extension weakness by pushing off the knees with the upper extremities and sequentially moves the upper extremities up the thigh until they have achieved an upright stance with full hip extension (Figure 10). A Gower’s sign is not specific to any neuromuscular condition but may be seen in a variety of neuromuscular diseases including DMD, BMD, LGMD1, LGMD2, SMA type III, congenital muscular dystrophy, congenital myopathy, myasthenic syndromes, severe forms of CMT (e.g., CMT III and CMT IV), and other neuromuscular disease conditions producing proximal weakness.
Figure 10.

Gower’s sign in a seven-year-old boy with Duchenne muscular dystrophy
Patients with proximal lower extremity weakness often exhibit a classic myopathic gait pattern (Figure 11a). Initially, weakness of the hip extensors produces anterior pelvic tilt and a tendency for the trunk to be positioned anterior to the hip joint. Patients compensate for this by maintaining lumbar lordosis which positions their center of gravity/weight line posterior to the hip joints, thus stabilizing the hip in extension on the anterior capsule of the hip joint. Subsequently, weakness of the knee extensors produces a tendency for patients to experience knee instability and knee buckling with falls. Patients compensate for this by decreasing stance phase knee flexion and posturing the ankle increasingly over time into plantar flexion. This produces a knee extension moment at foot contact, and the plantar flexion of the ankle during mid to late stance phase of gait helps position the weight line/center of gravity anterior to the knee joint (thus producing a stabilizing knee extension moment). Patients with Duchenne muscular dystrophy will progressively demonstrate initial foot contact with the floor increasingly forward onto the mid foot and finally the forefoot as they reach the transitional phase of ambulation before wheelchair reliance. Finally, weakness of the hip abductors produces a tendency towards lateral pelvic tilt and pelvic drop of the swing phase side. Patients with proximal weakness compensate for this by bending or lurching their trunk laterally over the stance phase hip joint (Figure 11b). This produces the “so-called gluteus medius lurch” or Trendelenburg gait pattern”.
Figure 11.

Myopathic gait pattern in Duchenne muscular dystrophy due to pelvic girdle and knee extension weakness; a) lumbar lordosis to keep center of mass posterior to hip joint; anterior pelvic tilt due to hip extensor weakness; weight line/center of mass maintained anterior to an extended knee; and forefoot ground contact with stance phase plantar flexion (toe walking) to maintain a knee extension moment and knee stability; b) trendelenberg or “gluteus medius gait” with lateral lean over the stance side due to hip abductor weakness; ankle dorsiflexion weakness necessitates swing phase circumduction for clearance.
Patients with this classic gait pattern, secondary to proximal pelvic girdle weakness, often exhibit toe walking. The clinician may mistakenly provide an AFO with the ankle positioned at 90 degrees with the thought that the patient needs orthotic management of foot drop. This can produce a precipitous increase in falls because the orthotic blocks the ankle at 90 degrees, thus compromising the patient’s ability to stabilize the knee into extension with equinus posturing of the gastrocnemius-soleus complex.
Patients with distal weakness affecting the ankle dorsiflexors and ankle everters and less severe proximal weakness (e.g., CMT, Emery-Dreifuss muscular dystrophy, myotonic muscular dystrophy, FSHD and other conditions) often exhibit a foot slap at floor contact and a steppage gait pattern to facilitate swing phase clearance of the plantar-flexed ankle. Alternatively, these patients may clear the plantar-flexed ankle using some degree of circumduction at the hip or vaulting on the stance phase side. These patients often benefit from the provision of an AFO with either a solid ankle or articulated ankle with a plantar flexion stop at neutral. More mild distal lower extremity weakness may become clinically evident by testing heel walking and toe walking.
LABORATORY EVALUATIONS
Serum Laboratory Studies
A variety of neuromuscular diseases, particularly those characterized by sarcolemmal muscle membrane injury, show significant elevations in transaminases, aldolase, and creatine kinase (CK). The CK enzyme catalyzes the release of high energy phosphates from creatine phosphate. It occurs mainly in muscle and leaks into the serum in large amounts in any disorder involving muscle fiber injury. The MM fraction is specific to skeletal muscle. The CK value may be significantly elevated in the early stages of DMD and BMD with values up to 50–100 times normal. A normal CK value may help exclude DMD and BMD. Overlap in CK values occurs between DMD and BMD. Other forms of muscular dystrophy such as Emery-Dreifuss muscular dystrophy, limb girdle muscular dystrophy, FSHD, and congenital muscular dystrophy may show moderate elevations in CK. However, in congenital muscular dystrophy, the CK value may be extremely variable, ranging from normal values to a fairly marked elevation. There is no close association between disease severity and CK values. In all dystrophic myopathies, the CK values tend to decrease over time with increasing severity of the disease due to progressive loss of muscle fiber and irreversible cell death. Thus, a three year old with DMD may have a CK value of 25,000 while a ten year old with DMD may show a CK value of 2,000. Other dystrophic myopathies iwith elevated CK values (> 1,000) values include LGMD 2A-2I, LGMD 1C, distal myopathy of the Miyoshi type, immune polymyopathy with SRP & HMGCoAR Ab; Paraneoplastic syndromes, Acid Maltase deficiency, acute damage: from injection, rhabdomyolysis; and muscle trauma. In addition myopathy from hypothyroidism may be associated with high CK values. Other conditions with significant elevations in CK may include polymyositis, dermatomyositis, acute rhabdomyolysis, and malignant hyperthermia. In many of the congenital structural myopathies, such as central core disease, nemaline rod myopathy, and fiber-type disproportion syndrome, a serum CK is likely to be normal or only mildly elevated.
CK levels have been found to be normal to elevated two to four times in SMA I and II.20 SMA III patients have also been found to have normal to slightly elevated CK values with elevations generally in the range of two to five times normal. A serum CK level greater than ten times the upper limit of normal generally is an exclusionary criteria for SMA54,55,56 and, in this setting, workup for other disorders such as inflammatory or dystrophic myopathies should be pursued. Functional status and disease progression did not correlate with initial CK determination in one series of SMA III cases.18
Thus, in a child with muscle weakness, a normal CK does not exclude a myopathy or other NMD condition, a severely elevated CK is suggestive of but not diagnostic of a dystrophic myopathy, and a very high CK is no reflection of disease severity in both inflammatory and dystrophic myopathies. Normal CK values may be seen in the acute active phase of childhood dermatomyositis, even in the presence of severe weakness.
Serial CK measurement in the morning after several days of sedentary activity is still useful in the evaluation of potential female DMD carriers who do not have a detectable gene deletion on molecular genetic studies. Three normal CK values in a female is approximately 90% specific for ruling out carrier status. Even one abnormally elevated CK makes carrier status a possibility.
Lactate and pyruvate levels are useful in the setting of a possible metabolic myopathy. Presence of a lactic acidosis may be seen in mitochondrial encephalomyelopathies such as Kearns-Sayre syndrome, MERRF (myoclonus epilepsy and ragged-red fibers) and MELAS (mitochondrial encephalomyelopathy with lactic acidosis and stroke-like episodes). Whenever clinical evidence suggests a disorder of oxidative metabolism, blood lactate and pyruvate values should be obtained. Arterial lactate values are more reliable. Lactate elevations under ischemic or exercise stress suggest mitochondrial dysfunction. In a setting of lactic acidemia, the lactate/pyruvate ratio may aid in the differential diagnosis. Children with suspected encephalomyelopathy should be evaluated with CSF lactate levels, as these values are less subject to flux than either venous or arterial values.
The ischemic forearm test, initially utilized by McArdle, is the most widely used means of assessing muscle anaerobic metabolism,15,55,79 The hallmark of defects in muscle glycogenolysis is failure of the normal rise in lactate in venous blood flowing from ischemically exercised muscles. The increase in blood pyruvate is also attenuated or absent, as is the lactate/pyruvate (L/P) ratio which normally rises roughly fivefold. A virtually unchanged L/P ratio with exercise typifies myophosphorylase and muscle PFK deficiency. In muscle LDH deficiency, ischemic exercise causes a disproportionate increase in pyruvate relative to lactate. This is secondary to increasing levels of pyruvate behind the metabolic block. An exaggerated increase in ammonia and purine metabolites with heavy exercise typifies glycolytic defects. A normal lactate response but impaired ammonia production is characteristic of myoadenylate deaminase deficiency.
Laboratory Evaluation of Neuromuscular Junction (NMJ) Disorders
Patients suspected of Lambert Eaton syndrome, infantile or late acquired botulism, myasthenia gravis, or presynaptic and postsynaptic congenital myasthenic syndromes may be evaluated electrodiagnostically with repetitive stimulation studies as described in this issue.43
Those suspected of infantile botulism should have stool or a rectal irrigation sample sent for botulinum toxin. The stool studies are often helpful in establishing an early diagnosis as the electrodiagnostic studies may have less sensitivity within the first few days of presentation.
Infants suspected of transient neonatal myasthenia or congenital myasthenic syndromes may show a clinical response to intravenous edrophonium (Tensilon) testing or neostigmine testing. The author prefers comparing the degree of decrement on repetitive stimulation studies before and serially (every two minutes) after neostigmine administration in infants with suspected myasthenic syndromes as clinical response to neostigmine or edrophonium may be exceedingly difficult to judge in the intubated neonate. Decremental responses at slow rates of stimulation (2–3 Hz) are not specific to postsynaptic defects in infants with congenital myasthenic syndromes and may be seen in presynaptic or postsynaptic subtypes. Motor point biopsy is discussed in the electrodiagnstic article in this issue.
Children and adults with suspected myasthenia gravis should have acetylcholine receptor antibodies sent (binding or blocking, modulating, and striated muscle Ach antibodies). Negative antibody studies do not rule out autoimmune myasthenia gravis as some patients have antibodies of a different nature that cannot be measured with current laboratory techniques. Patients with myasthenia gravis should have a chest x-ray and/or chest CT to rule out thymoma.
Muscle Imaging
Ultrasound imaging has been used as a screening tool with muscle for pathologic change.23,28–30,99 More recently, magnetic resonance imaging has been used to evaluate the extent and distribution of involvement in neuromuscular disorders as well as disease progression.22,32,44,88 MR imaging has also been used to help differentiate between dystrophic myopathy and neurogenic atrophy due to spinal muscular atrophy.58 A recent review describes the use of skeletal muscle MRI in diagnosis and monitoring disease progression.22
While muscle imaging is generally not used to delineate a specific diagnosis, these techniques are useful for the identification of appropriate muscle biopsy sites, determining distribution and extent of involvement, and for monitoring disease progression.22,23
Body Composition by Dual-Energy X-ray Absorptiometry (DEXA)
Both lean and fat tissue mass can be accurately and reliably estimated over wide age ranges, using DEXA. Subjects with myogenic atrophy have significantly elevated fat/muscle ratio. Both functional activity scales and strength correlates with percentage of lean body mass measured by DEXA. Diffuse neurogenic atrophy is associated with decrease in the mass of all three compartments (lean mass, fat mass and bone mineral content) but relatively normal fat/muscle ratios standardize to body mass index. Regional body composition by DEXA has been proposed as a monitor of disease progression in such entities as muscular dystrophy or progressive denervating diseases such as spinal muscular atrophy and peripheral neuropathies. Skalsky and colleagues80 have recently extensively reviewed the use of regional and whole body dual-energy x-ray absorptiometry to guide treatment and monitor disease progression in NMD.
Electrodiagnostic Studies
Nerve conduction and electromyography are an extension of the clinician’s physical examination and a powerful tool for the localization of pathology within the lower motor neuron. In addition, EMG and nerve conduction studies help to guide further studies such as molecular genetic studies (e.g. to improve cost-effectiveness of molecular genetic panels in CMT by determining the nature of a peripheral neuropathy as demyelinating or axonal), and to help guide muscle biopsies by providing information regarding the most appropriate muscle site for biopsy. Lipa and Han provide a comprehensive review of the role of electrodiagnostic studies vis a vis the diagnostic evaluation of NMDs in this issue.43
Molecular Genetic Studies
The application of molecular genetic techniques has resulted in enormous gains in our understanding of the molecular and pathophysiologic basis of hereditary neuromuscular diseases. In addition, molecular genetic studies now aid in the diagnostic evaluation of many NMD conditions as described in this issue by Arnold and Flanigan.5 In MMD, the CTG repeat size of the DMPK gene inversely correlates with age of onset of DM1 (Figure 12). In addition complete sequencing of genes is critical for the determination of the potential value of genetic based therapeutic agents for particular patients.
Figure 12.
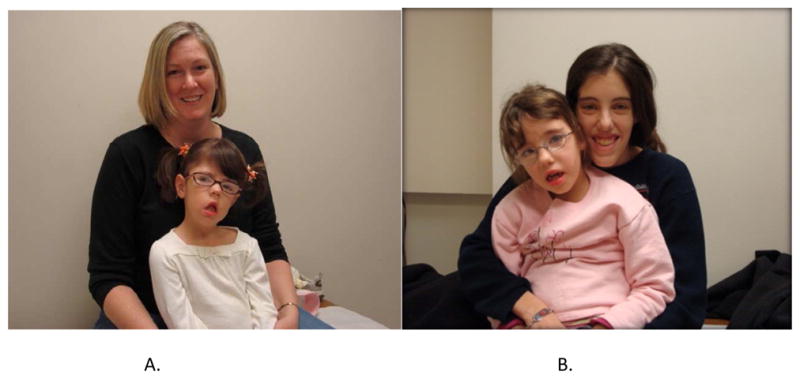
Four individuals with myotonic muscular dystrophy. The mother on the left (a) has 75 CTG repeats in the DM protein kinase (DMPK) gene loci on chromosome 19q13.3 and her daughter has 2538 CTG repeats. The mother on the right (a) is more symptomatic and has 450 CTG repeats and her daughter has 1650 repeats. This is an example of genetic anticipation with greater severity occurring in successive generations.
Muscle and Nerve Biopsy Evaluation
While molecular genetic testing has reduced the need for muscle biopsy, the appropriate acquisition of muscle biopsies is still valuable in the diagnostic evaluation of hereditary and acquired NMDs and the topic is reviewed by Joyce and colleagues in this issue.37 Nerve biopsies are somewhat useful in the characterization of more severe hereditary motor and sensory neuropathies, congenital hypomyelinating neuropathy, and neuroaxonal dystrophy. In addition, perineural immune complex deposition seen in some autoimmune neuropathies, or changes consistent with vasculitis may also be useful diagnostically. Otherwise, nerve biopsies rarely add useful specific information to the diagnostic evaluation of the NMD patient, beyond that information obtained from nerve conduction studies and EMG.
CLINICAL CLUES TO HELP WITH THE EARLY DIAGNOSIS OF COMMON NMD CONDITIONS: IMPLICATIONS FOR TREATMENT
Most neuromuscular disease syndromes give very specific patterns of presentation and strength loss. For the most common NMD conditions the patterns of strength impairment and natural history profiles for change in strength over time has been described in detail by the author and colleagues at the University of California, Davis Research and Training Center in Neuromuscular Disease.12,13,24,35,39,47,48,50 Early diagnosis is facilitated by knowledge of the common initial clinical presentations of specific NMDs. This allows for streamlined and cost effective laboratory evaluations, electrodiagnostic studies, molecular genetic diagnostic evaluations, imaging and determination if muscle biopsies are necessary. Early diagnosis is facilitated by knowledge of the common initial clinical presentations of specific NMDs, and in many cases the early diagnosis has potential implications for treatment and prevention of secondary conditions.
MOTOR NEURON DISEASES
Amyotrophic Lateral Sclerosis
Sporadic Amyotrophic Lateral Sclerosis
ALS can be defined as a rapidly progressive neurodegenerative disease characterized by weakness, spasticity, and muscular atrophy with subsequent respiratory compromise leading to premature death. It is caused by the destruction of motor neurons in the primary motor cortex, brainstem, and spinal cord. Onset of ALS is insidious and most commonly presents with painless asymmetric limb weakness. ALS most often afflicts people between 40 and 60 years of age with a mean age of onset of 58 yrs.59,69,72 Five percent of cases have onset prior to age 30.Wijesekera Men are affected more commonly than women with a ratio of 1.5:1.0. Patients with LMN pathology usually present complaining of muscle weakness. In addition, they may note muscle atrophy, fasciculations, and muscle cramping. Cramping may occur anywhere in the body, including the thighs, arms, and abdomen. Cramping of abdominal or other trunk muscles raises a red flag urging the clinician to consider a diagnosis of ALS. Patients with UMN pathology often complain of loss of dexterity or a feeling of stiffness in the limbs. They may note weakness which is caused by spasticity resulting from disinhibition of brainstem control of the vestibulospinal and reticulospinal tracts. Signs and symptoms suggesting bulbar muscle weakness include dysarthria, dysphagia, drooling, and aspiration. These signs and symptoms may be caused by UMN and/or LMN dysfunction involving the bulbar muscles. Signs of spastic dysarthria, indicating UMN pathology, include a strained and strangled quality of speech, reduced rate, low pitch, imprecise consonant pronunciation, vowel distortion, and breaks in pitch. LMN dysfunction creates flaccid dysarthria in which speech has a nasal and/or wet quality, pitch and intensity are monotone, phrases abnormally short, and inspiration audible. Patients may complain of intermittent gagging sensations due to muscle weakness with drooping of the soft palate. Complaints of difficulty chewing and swallowing, nasal regurgitation, or coughing when drinking liquids, may all indicate dysphagia.
Other signs and symptoms frequently associated with ALS are cachexia, fatigue and musculoskeletal complaints. The term “ALS cachexia” refers to a phenomenon experienced by some patients in which weight loss occurs in excess of that caused by muscle atrophy and reduced caloric intake. Both subcutaneous fat and peritoneal fat are lost, presumably because of acceleration of the basal metabolic rate.77 In patients with ALS cachexia, greater than 20% of body weight is typically lost over a 6 month period.
Familial ALS
The vast majority of ALS cases are presumably acquired and occur sporadically. However, approximately 5–10% of all ALS cases are familial (FALS) and most commonly have an autosomal dominant inheritance pattern, though, autosomal recessive, X-linked and mitochondrial inheritance patterns have been reported.65,76,83 The age of onset of FALS occurs a decade earlier then sporadic cases and progression of the disease is more rapid. Males and females are equally affected. About 20% of FALS cases result from a Copper-Zinc superoxie dismutase (SOD1) gene defect.3,74,96 Other disease causing genetic mutations have more recently been identified. These mutations have been found in genes encoding for angiogenin, chromatin modifying protein, dynactin, vesicle associated membrane protein, and TAR-DNA binding protein.96
Predominantly Proximal Spinal Muscular Atrophy
In type I SMA, onset is generally ≤ six months, the patients never sit without support, and survival is usually less than two years. In SMA type II, onset is generally ≤ 18 months, patients sit independently but never stand or walk without aids and survival is usually greater than two years and often to young adulthood. In SMA type III, onset is after 18 months, patients achieve standing or walking without support but may lose this milestone at a later age, and survival is essentially normal. In all SMA types, proximal muscles are weaker than distal muscles. Patients have symmetric weakness involving the lower extremities earlier and to a greater extent than the upper extremities.13 The diaphragm is usually relatively preserved, relative to intercostal and abdominal musculature. In SMA I, this results in a diaphragmatic breathing pattern during respiration with abdominal protrusion, paradoxical thoracic depression and intercostal retraction (see Figure 6). Patients with SMA may have both neck flexor and neck extensor weakness. Clinical features of SMA I, II, and III are shown in Table 1.
Table 1.
Childhood Onset Proximal Spinal Muscular Atrophy
| SMA I (Wernig Hoffman) | SMA II (Intermediate SMA) | SMA III (Kugelberg Welander) | |
|---|---|---|---|
| Onset | < 6 months | 6–18 months | >18 months IIIa < 3 years IIIb > 3 years |
| Genetics | SMN1: AR homozygous SMN2: ≤2 copies |
SMN1: AR homozygous SMN2: three copies |
SMN1: AR homozygous SMN2: 4–8 copies |
| Phenotype | Severe hypotonia, weak suck, weak cry, proximal weakness, absent refexes, respiratory failure common | Hypotonia, proximal weakness, muscle wasting, contractures, scoliosis, absent reflexes, tongue fasiculations | Proximal symmetric weakness, lordotic gait, Gowers’s sign, decreased reflexes, tremor, tongue fasiculations |
| Milestones | Poor head control; Never sit independently |
Sit with head control; never stand unassisted; may require ventilatory support |
Stand & walk unassisted; may lose standing or continue to walk IIIa: onset 18 mo to < 3 years (80% not walking at age 40) IIIb: onset > 3 years (40% not walking at age 40) |
| Life Expectancy | 1–2 years 10% living at age 20 |
Most live to 3rd decade; many live to 4th to 5th decade | Normal life expectancy |
SMA I (Werdnig Hoffman Disease)
The majority of cases of SMA I present within the first two months with generalized hypotonia and symmetrical weakness. The age of onset of symptoms is less than four months in the vast majority of cases. Weak sucking, dysphagia, labored breathing during feeding, frequent aspiration of food or secretions, and weak cry are frequently noted by history.
Examination shows generalized hypotonia and symmetric weakness involving the lower extremities earlier and to a greater extent in the upper extremities. Proximal muscles are weaker than distal extremities. In the supine position, the lower extremities may be abducted and externally rotated in a "frog-leg" position (Figure 6). The upper extremities tend to be adducted and externally rotated at the shoulders with a semi-flexed elbow. Volitional movements of fingers and hands persist well past the time when the shoulders and elbows cannot be flexed against gravity. The thorax is flattened anteroposteriorly and bell-shaped as a result of intercostal weakness. Pectus excavatum may be variably present. The diaphragm is usually preserved, relative to the intercostal and abdominal musculature. This results in a diaphragmatic breathing pattern during respiration with abdominal protrusion, paradoxical thoracic depression and intercostal retraction. Neck flexor weakness may result in persistent posterior head lag when the trunk is lifted forward from the supine position. Neck extensor weakness may result in forward head lag when the infant is positioned in the horizontal prone position. With advanced disease, the mouth may remain open as a result of masticatory muscle weakness. Facial weakness may be noted in up to half of patients. The diagnostic criteria for SMA outlined by the International SMA Consortium54 lists marked facial weakness as an exclusionary criterion for SMA, but this is not an absolute criterion. Tongue fasciculations have been reported in 56%-61% of patients,20 so the absence of this finding does not necessarily exclude the disease. In one series,20 deep tendon reflexes (DTRs) were absent in all four extremities in 74% of cases. Thus, the preservation of DTRs does not exclude the diagnosis of SMA. Appendicular muscle fasciculations and distal tremor are also associated examination findings. Extraocular muscles are spared, as are the myocardium. Mild to moderate hip flexion, knee flexion, and elbow flexion contractures may be observed in some patients along with wrist contractures and ulnar drift of the fingers. Severe arthrogryposis is not typically observed.
Spinal muscular atrophy II
disease onset is usually more insidious than that of SMA I. The findings of generalized hypotonia, symmetrical weakness and delayed motor milestones are hallmarks of SMA II. Weakness also involves proximal muscles more than distal muscles, and lower extremity more than upper extremity. A fine tremor of the fingers and hands occurs in a minority of patients. This “polyminimyoclonus” may be attributed to spontaneous, repetitive rhythmical discharges by the motor neurons that innervate a large territory of muscle. Wasting tends to be more conspicuous in SMA II versus SMA I. DTRs are depressed and usually absent in the lower extremities. Appendicular or thoracic muscle wall fasciculations may be observed. Tongue fasciculations have been observed in 30–70% of SMA II patients.20,54,63 Progressive kyphoscoliosis and neuromuscular restrictive lung disease is almost invariably seen in the late first decade. Contractures of the hip flexors, tensor fasciae latae, hamstrings, triceps surae, and elbow and finger flexors are quite common. Hypotonic hip dislocations have been noted commonly in SMA II patients. Sensory examination is completely normal and extraocular muscles and the myocardium are spared. In a large series from Germany,99 of 104 cases classified as SMA II (sits alone, never walks), 98% survived to the age of ten and 77% to the age of 20. Thus, a longer life span is possible with adequate supportive care.
Spinal Muscular Atrophy III (Kugelberg-Weilander Disease)
In more chronic SMA III, also referred to as Kugelberg- Welander Syndrome, weakness usually initially occurs between the ages of 18 months and late teens. Motor milestones may be delayed in infancy. Proximal weakness is observed with the pelvic girdle being more affected than the shoulder girdle.13 There is an exaggerated lumbar lordosis and anterior pelvic tilt owing to hip extensor weakness. There is also a waddling gait pattern with pelvic drop and lateral trunk lean over the stance phase side, secondary to hip abductor weakness. If ankle plantar flexion strength is sufficient, the patients may show primarily forefoot or toe contact and no heel strike similar to patients with Duchenne dystrophy. This is a compensatory measure for knee extensor weakness to maintain a stabilizing knee extension moment at the knee. The patient may exhibit a Gower’s sign when arising from the floor; stair climbing is difficult due to hip flexor weakness. Facial weakness is sometimes noted. Fasciculations are noted in about half of the patients54 and are more common later in the disease course. Fasciculations in the limb muscles and thoracic wall muscles are common. Calf pseudohypertrophy has been occasionally noted, but wasting of affected musculature is more prominent. Deep tendon reflexes are diminished and often become absent over time. Contractures are generally mild as long as patients remain ambulatory. Scoliosis may be observed in SMA III, but it occurs less frequently and is less severe than scoliosis and SMA II. While no survival data exists for patients with SMA III, cases have been followed into the eighth decade without mechanical ventilation.13,100 Ventilatory failure due to neuromuscular restrictive lung disease is a rare event in SMA III, occurring only in adulthood.7,13 Zerres and Rudnik-Schoneborn100 have proposed further subtypes, including SMA IIIa (walks without support; age of onset less than three years) and SMA IIIb (walks without support; age of onset 3–30 years). In their series, only 44% of SMA “IIIa” patients remained ambulatory 20 years after onset of weakness, whereas 89% of “IIIb” patients remained ambulatory after a similar 20 year duration. Diagnosis of SMA is confirmed by a consideration of clinical findings, molecular genetic studies and, occasionally, electrodiagnostic studies. Muscle biopsy is generally not required to confirm the diagnosis. Genetic studies have now established that SMA is caused by mutations in the telomeric SMN1 gene, with all patients having at least one copy of the centromeric SMN2 gene. At least one copy of the SMN2 must be present in the setting of homozygous SMN1 mutations; otherwise, embryonic lethality occurs. The copy number of SMN2 varies in the population, and this variation appears to have some important modifying effects on SMA disease severity.64,85,101 All SMA patients have > 2 SMN2 genes. It appears that a higher number of SMN2 copies in the setting of SMN1 mutations is associated with a less severe clinical SMA phenotype: SMA I (Severe): 2 or 3 gene copies of SMN2; SMA II: 3 copies of SMN2; SMA III: 4 to 8 copies of SMN2. However, substantial variations in SMA phenotype and disease severity can exist with a given SMN2 copy number, so it is not recommended that disease severity be predicted based soley on SMN2 copy numbers. Therapeutic interventions for SMA are reviewed in this issue.82
HEREDITARY ATAXIAS
Friedreich’s ataxia
Friedreich’s ataxia is a spinocerebellar degeneration syndrome with the onset of symptoms before age 20 years. This autosomal recessive condition has been linked in one subtype to chromosome 9q13-21.1 (FRDA) with the protein implicated being termed “frataxin”. A second subtype referred to as FRDA2 is linked to chromosome 9p23-p11.
The incidence of Friedreich’s ataxia is 1 in 25,000 to 50,000. Carrier frequency is 1 in 60 to 110. Age of onset is usually: < 20 years, typically around puberty with a range from 2 to 25 years. Obligate signs and symptoms include progressive ataxic gait, cerebellar dysfunction with tremor and dysmetria, dysarthria, decreased proprioception or vibratory sense (or both), muscle weakness, and absent deep tendon reflexes. Other common signs include cavus foot deformity, cardiomyopathy, scoliosis, and upper motor neuron signs, such as a Babinski sign and spasticity. Weakness is progressive, affects lower extremities and small muscles in hands & feet. Sensory loss is typical and especially affects vibration & joint position sensation. Tendon reflexes are often absent. An occasional patient may have chorea without ataxia. With electrodiagnostic studies, sensory nerve potentials may be absent or reduced. Progression is slow with mean time to wheel chair is 15 years of age and death from cardiomyopathy ranges from the third to seventh decade. The prevalence of scoliosis approaches 100% but some cases have more severe progressive spinal deformity than others. Those Friedreich’s ataxia cases with onset of disease before the age of ten years generally have more severe progressive scoliosis. Those with the onset of disease during or after puberty have later onset spinal deformity which may not require surgical intervention.
Frataxin is a mitochondrial protein located on the inner mitochondrial membrane. It is likely required for maintenance of mitochondrial genome and it is involved in iron homeostasis and iron transport into mitochondria. Idebenone is a power antioxidant and a synthetic analogue of coenzyme Q. It may improve iron homeostasis and mitochondrial function in Friedreich’s ataxia. In andomized clinical trials, longer-term idebenone treatment has been shown to prevent progression of cardiomyopathy and cardiac hypertrophy in both pediatric and adult patients with Friedreich’s ataxia. Its stabilizing effect on neurological dysfunction has been shown to be present only in the pediatric population, mainly before puberty. This suggests that the age at which idebenone treatment is initiated may be an important factor in the effectiveness of the therapy.67
Spinicerebellar ataxias
The hereditary ataxias are a group of genetic disorders characterized by slowly progressive incoordination of gait and often associated with poor coordination of hands, speech, and eye movements. Frequently, atrophy of the cerebellum occurs. A peripheral neuropathy can occur in many of the subtypes. The hereditary ataxias are categorized by mode of inheritance and causative gene or chromosomal locus. The genetic forms of ataxia are diagnosed by family history, physical examination, and neuroimaging. Molecular genetic tests are available for the diagnosis of many but not all spinocerebellar ataxias (SCA). Over 60 genetically distinct autosomal dominant and autosomal recessive hereditary ataxias and SCA subtypes have been identified.
PERIPHERAL NERVE DISORDERS
Acute Inflammatory Demyelinating Polyradiculoneuropathy (AIDP- Guillain-Barré Syndrome)
Acute inflammatory demyelinating polyradiculoneuropathy (AIDP) is a primarily demyelinating neuropathy with autoimmune etiology. Motor axons are affected more than sensory axons. Incidence in children is similar to that seen in adults. Children often have a prodromal respiratory or gastrointestinal infection occurring within one month of onset. Common precipitating infections include Mycoplasma, cytomegalovirus, Epstein-Barr virus, Campylobacter jejuni, and various vaccinations. Weakness generally begins distally in the lower extremity with a progressive ascending paralysis ultimately involving the upper limbs. Pain and sensory symptoms are not uncommon. The most common cranial nerve abnormality is an ipsilateral or bilateral lower motor neuron facial paralysis. Objective sensory loss has been documented in the minority of children.36 In one series, only 15% required mechanical ventilation. 61 The maximal degree of weakness generally reaches a peak within two weeks of onset and time to maximum recovery was 7 +/- five months in one series.10 Complete recovery occurs in most children. Classic criteria for poor recovery in adults (low median CMAPs and fibrillation potentials) may not apply to children.10 Disturbances of the autonomic nervous system are common in children, including transient disturbances of bowel and bladder, excessive sweating or vasoconstriction, mild hypertension or hypotension and occasionally cardiac arrhythmias.
The acute motor axonal neuropathy (AMAN) involves predominantly motor nerve fibers with a physiologic pattern suggesting axonal damage, whereas the acute inflammatory demyelinating polyneuropathy (AIDP) involves both motor and sensory nerve fibers with a physiologic pattern suggesting demyelination. Another clinical variant is the Miller- Fisher syndrome characterized by acute onset ataxia, ophthalmoparesis, and areflexia.
Diagnosis is generally confirmed by electrodiagnostic studies43, and the CSF protein is characteristically elevated in a majority of children. Serum autoantibodies which may be elevated include IgM and IgG versus Beta-Tubulin and Heparin sulfate. AMAN patients may show increased IgG antibodies to GM1 ganglioside. The Miller Fisher syndrome is associated with a high frequency of the IgG GQ1b antibodies. The major considerations in differential diagnosis of AIDP or AMAN include transverse myelitis, toxic neuropathies, tick paralysis, infantile botulism, myasthenia gravis, and dermatomyositis.
Treatment has typically included corticosteroids, plasma exchange, or more recently, intravenous immune globulin.21, 41,42,75 AIDP patients respond to both plasma exchange and intravenous immunoglobulin (IVIG). Patients with Acute Motor Axonal Neuropathy (AMAN) respond preferentially to IVIG over plasma exchange. Recovery is often quite good in children without treatment. After standard intravenous immunoglobulin therapy, children with axonal forms of GBS recover more slowly than those with the demyelinating form, but outcome at 12 months appears to be equally favorable in two groups.87
Chronic Inflammatory Demyelinating Polyradiculoneuropathy (CIDP)
Children or adults with chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) often have a presentation similar to AIDP, however, the disorder continues with a chronic or relapsing course. The disorder may begin as early as infancy, but is seen in children and adults. Electrophysiologic studies show focal conduction block, temporal dispersion of CMAPs, prolongation of distal motor latencies, markedly slow conduction velocities, and absent or prolonged H-wave and F-wave latencies. CIDP cases often demonstrate axonal loss on EMG. The CSF protein is elevated in most cases. The differential diagnosis usually includes CMT types I and III. The presence of acute relapsing episodes point towards CIDP. Due to the more severe involvement of proximal nerves and nerve roots, a distal sural nerve biopsy may not always show inflammatory changes and demyelination. Treatment may include corticosteroids (Prednisone), and IVIG as first line approaches and subsequently plasma exchange.
Charcot Marie Tooth
Charcot-Marie-Tooth (CMT) neuropathy (also called hereditary motor sensory neuropathy-HMSN) is a heterogenous group of inherited disease of peripheral nerve that affects both children and adults and causes significant progressive neuromuscular impairment.8,14 It has been estimated that 1 per 2,500 to 3,000 persons has a form of CMT. CMT 1 denotes individuals with a hypertrophic demyelinating neuropathy ("onion bulbs") and reduced nerve conduction velocities, whereas CMT 2 refers to individuals with an axonal neuropathy and normal or slightly reduced nerve conduction velocities. Individuals with CMT 3 (Dejerine-Sotttas disease) have a primarily demyelinating peripheral neuropathy with a more severe phenotype presenting in infancy. Historically types 1, 2, and 3 were felt to be autosomal dominant conditions with type 3 CMT patients exhibiting point mutations with frame shift and either dominant or recessive inheritance. CMT 4 refers to autosomal recessive CMT. However, recently axonal forms of CMT have been identified with autosomal recessive inheritance (deemed AR-CMT 2A, 2B, etc.)
In general, in most CMT subtypes onset is usually during the first or second decade of life. Both motor and sensory nerve function are affected. The clinical features include distal muscle weakness, impaired sensation and absent or diminished deep tendon reflexes. Weakness usually is greatest initially present in the foot and hand inrinsics and distal lower extremities and subsequently in the distal upper extremities. Slow progressive weakness, more proximally in the knees, elbows and pelvic and shoulder girdles may occur over decades. 12 There is variable penetrance in most subtypes. Weakness is usually initially greatest in the distal lower extremities and subsequently in the distal upper extremities. Slow progressive weakness more proximally in the knees, elbows and pelvic and shoulder girdles may occur over decades.13 The various gene locations and known protein abnormalities associated with various forms of CMT (HMSN) and the clinical subtypes are described in Table 2.
Table 2.
CMT subtypes: Comparison of clinical features
| Disorder | Gene | Location | Usual onset | Early or distinct symptoms | Tendon reflexes | Average NCVs |
|---|---|---|---|---|---|---|
| CMT1: Dominant; Demyelinating | ||||||
| CMT 1A | PMP-22 | 17p11 | 1st decade | Distal weakness | Absent | 15 to 20 M/s |
| CMT 1B | P0 | 1q22 | 1st decade | Distal weakness | Absent | <20 M/s |
| CMT 1C | LITAF | 16p13 | 2nd decade | Distal weakness | Reduced | 16 to 25 M/s |
| CMT 1D | EGR2 | 10q21 | 2nd decade | Distal weakness | Absent | 26 to 42 M/s |
| CMT 1E (Deafness) | 8p21.2 | R159C point mutation89 Inheritance: Dominant Late onset: 5th decade | Sensory loss: Distal Weakness: Distal Vocal cord dysfunction in some patients | Reduced | NCV: Axon loss | |
| CMT 1F | Neurofilament light chain (NEFL) | 8p21.2 | Less than 13 years | Weakness Legs & Arms Distal > Proximal May be severe Early: Delayed motor milestones or gait disorder Dominant | Absent | Motor NCV: 15 to 38 M/s SNAPs: Often absent F-waves: Normal or Slow |
| CMT X (S-D*) | Connexin-32 | Xq13 | 2nd decade | Distal weakness | Absent distal | 25 to 40 M/s |
| HNPP | PMP-22 | 17p11 | 3rd decade | Focal episodic weakness | Normal | Entrapments |
| Dejerine-Sottas (HMSN 3) |
PMP-22 8q23 EGR2 |
17p11 8q23 10q21 |
2 years | Severe weakness | Absent | <10 m/s |
| CMT Intermediate NCV |
DNM2 10q24 1p34 P0 CMT-X |
19p12 10q24 1p34 1q22 Xq13 |
1st or 2nd decade | Distal weakness | 25 to 50 M/s | |
| CMT2: Dominant; Axonal | ||||||
| CMT 2A | KIF1Bβ | 1p36 | 10 yrs | Distal weakness | Absent distal | > 38 M/s |
| Mitofusin 2 | 1 | |||||
| CMT 2B | RAB7 | 3q13 | 2nd decade | Distal weakness Sensory loss Acromutilation | Absent distal | Axon loss |
| CMT 2C | TRPV4 | 12q24 | 1st decade | Vocal cord & Distal weakness | Absent | > 50 M/s |
| CMT 2D | GARS | 7p15 | 16 to 30 yrs | Distal weakness Arms > Legs | Reduced | Axon loss |
| CMT 2E | NF-68 | 8p21 | 1 to 40 yrs | Distal weakness | Reduced | Axon loss |
| CMT 2F/ Distal HMN | HSPB1 (HSP 27) | 7q11 | 6 to 54 years | Difficulty walking | Reduced ankle | Axon loss |
| CMT 2G | 12q12 | 15 to 25 years | Distal weakness | Reduced | CMAPs, SNAPs Small in legs 42 to 58 M/s | |
| CMT 2I | Po | 1q22 | Late onset | Distal weakness Sensory loss (90% to 100%) Severe Panmodal Distal > Proximal Weakness (80% to 100%) Legs (80%) > Arms (35%) Distal Mild to Severe | Rduced | Velocity: Usually > 20 m/s to Normal Often not clearly demyelinating CMAPs & SNAPs: Reduced amplitude or Absent |
| CMT 2J | P0 | 1q22 | Age: Adult; Usually after 30 years Legs Paresthesias, Hypoesthesia (85%) | CMT with hearing loss & pupillary abnormalities | Predominantly axonal neuropathy (Adult onset) | |
| CMT 2K | Ganglioside-induced differentiation-associated protein 1 GDAP1 | 8q21 | Weakness (100%) Distal Severe Feet > Hands: Hand onset later in 1st decade Proximal: Moderate; Legs > Arms | Vocal cord paralysis Onset: 2nd decade Hoarseness (80%) Not present in some families: Gait disorder: | NCV: Axon loss; Velocities preserved | |
| CMT 2L | HSPB8 | 12q24 | 15 to 33 years | Distal weakness | Reduced | Axon loss |
| CMT 2M | DNM2 | 19p13 | Onset Age: Congenital to 4th decade Cataracts: Early Neuropathy: Childhood | Legs > Arms Sensory: Pan modal loss; Sensory ataxia; Paresthesias Weakness: Distal; Legs > Arms Progression: Mild | Reduced | NCV: Axonal loss |
| CMT 2N | AARS; | 16q22 | Onset Age: Mean 28 yrs; Range 6 to 54 yrs | Leg weakness Occasional asymptomatic patient | Tendon reflexes Knees: Reduced Ankles: Absent Reduced | : Axonal to Intermediate NCV: 32 to 50 M/s; CMAP amplitudes reduced SNAP amplitudes: Small |
| CMT 2O | DYNC1H1; | 14q32 | Onset: Early childhood Motor milestones: | Delayed Weakness: Distal; Legs > Arms Sensory loss: Distal; Pan-modal; Normal in some patients Pes cavus: Some patients have CNS: Learning difficulties | Tendon reflexes: Normal or Reduced | |
| CMT 2P | LRSAM1 | 9q33 | Onset age: 27 to 40 years | Clinical Weakness & Wasting Distal Legs > Arms | Tendon reflexes: Reduced | NCV: SNAP & CMAP amplitudes reduced |
| HMSN-P | 3q13 | 17 to 50 yrs | Proximal weakness Cramps | Absent | Axon loss | |
| HSMN + Ataxia | 7q22 | 13 to 27 yrs | Gait ataxia | Absent | Axon loss | |
| CMT 2 P0 | P0 | 1q22 | 37 to 61 years | Leg weakness Pupil or Hearing | Reduced | < 38 M/s to Normal |
| AR-CMT2: Recessive; Axonal | ||||||
| AR-CMT2A | Lamin A/C | 1q21 | 2nd decade | Distal weakness | Reduced | Axon loss |
| AR-CMT2B | MED25 | 19q13.3 | 3rd & 4th decade | Distal weakness | Absent distal | Axon loss |
| AR-CMT2 Ouvrier | Autosomal | Onset: Early childhood; 1st decade | Weakness: Distal; Symmetric; Legs before arms Sensory loss: Mild Progression: Slow; Severe distal weakness by 20 years | Reduced | Axon loss | |
| HMSN 3: Infantile | ||||||
| Dejerine-Sottas (HMSN 3) |
P0 PMP-22 Periaxin |
Autosomal Dominant / Recessive | 2 years | Severe weakness | Absent | <10 m/s |
| Congenital Hypomyelinating Neuropathy | P0 EGR2 PMP-22 |
Autosomal Recessive | Birth | Severe weakness | Absent | <10 m/s |
| CMT4: Recessive; Demyelinating | ||||||
| CMT 4A | GDAP1 | 8q13 | Childhood | Distal weakness | Reduced | Slow |
| CMT 4B | MTMR2 | 11q22 | 2 to 4 yrs | Distal & Proximal weakness | Absent | Slow |
| CMT 4B2 | SBF2 | 11p15 | 1st 2 decades | Distal weakness Sensory loss | Absent | 15–30 m/s |
| CMT 4C | KIAA1985 | 5q23 | 5 to 15 yrs | Delayed walking | Reduced | 14 to 32 M/s |
| CMT 4D (Lom) | NDRG1 | 8q24 | 1 to 10 yrs | Gait disorder | Absent | 10 to 20 M/s |
| CMT 4E | EGR2 | 10q21 | Birth | Infant hypotonia | Absent | 9 to 20 M/s |
| CMT 4F | Periaxin | 19q13 | 1 to 3 yrs | Motor delay | Absent | Absent |
| CMT 4H | FGD4 | 12q12 | 10 to 24 mo | Walking delay | Absent | < 15 M/s |
| CCFDN | CTDP1 | 18q23 | 1st or 2nd decade | Distal leg weakness | Reduced | 20 to 34 m/s |
CMT 1
The majority of CMT 1 pedigrees (70%), demonstrate linkage to chromosome 17p11.2-12 and are designated CMT 1A.36 CMT 1A duplication results in increased expression of peripheral myelin protein-22 (PMP-22). Conduction velocities are uniformly slow in all nerves with a mean of 17 to 20 M/s and a range 5 to 34 M/s. Onset is typically in the first decade with leg arreflexia, gait disorder (toe walking or steppage gait), foot muscle atrophy or pes cavus, occasionally short achilles tendons, and enlarged nerves owing to onion bulb formation in half of patients. Distal weakness develops initially in intrinsic muscles of the feet and hands. Ankle dorsiflexion, ankle eversion, and extensor hallucis longus weakness develops with more normal strength proximally. Progress cavus foot deformities with clawing of the toes often develops. Orthopedic procedures are limited to soft tissue procedures and correcting wedge osteotomies and joint fusion should be avoided if possible to avoid late pain. Late in the disease diaphragm or bulbar weakness may develop in rare cases. Progression is slow over many decades. Defects in the human myelin zero gene (P0) on chromosome 1q22-q23 leads to CMT 1B. P0 is the major protein structural component of peripheral nervous system myelin. The clinical presentation is similar to CMT1A however onset may lag into the second to third decade in a minority of patients and there is more variability in severity. Nerve conduction velocities are usually less than 20 m/s. P0 mutations may lead to other clinical variants referred to as CMT 1E (demyelinating CMT with deafness), and predominantly axonal neuropathy with late adult onset (e.g. CMT 2I, and CMT 2J with hearing loss & pupillary abnormalities).
CMT 2
CMT 2 is a less common disorder than CMT 1. Generally, CMT 2 patients demonstrate later age of onset, less involvement of the small muscles of the hands, and no palpably enlarged nerves. Wasting in the calf and anterior compartment of the leg may give rise to an "inverted champagne bottle" or "stork-leg" appearance. Conductions velocities are mildly reduced and CMAP amplitudes and SNAP amplitudes are usually reduced. CMT 2A2 with mitofusin abnormality accounts for approximately 20% of CMT 2 probands. CMT 2C linked to chromosome 12q23-q24 has interesting features of early onset in the first decade, and diaphragm & Intercostal weakness producing shortness of breath. Vocal cord paralysis may alter the voice of these patients. The disease may progress to proximal & face muscles. Arthrogryposis is present in some patients. Phrenic nerve CMAPs are often reduced. CMT 2E with abnormality in neurofilament light chain (NFL) linked to chromosome 8p21 may have associated hearing loss in 30% of cases. While most axonal CMT is autosomal dominant, emerging pedigrees are being identified with recessive inheritance.
CMT 3
Dejerine-Sottas disease (CMT 3) is a severe hypertrophic demyelinating polyneuropathy with onset in infancy or early childhood. Most achieve ambulation but some may subsequently progress to wheelchair reliance. Nerve conduction velocities are greatly slowed (often below 10 m/s), and elevations in cerebrospinal fluid protein may be present. Dejerine-Sottas disease may be associated with point mutations in either the PMP-22, P0 or EGR2 genes.36 While this disorder was previously felt to be autosomal recessive, many cases are due to denovo point mutations and actually have dominant inheritance.
Congenital Hypomyelinating Neuropathy
This is a severe and often fatal newborn disorder often presents with respiratory distress in the delivery room. These infants often have severe generalized hypotonia and associated arthrogryposis. Diagnostically, these infants have absent sensory nerve action potentials (SNAPS) or low amplitude SNAPS with prolonged distal latencies. Compound muscle action potentials are either absent or low amplitude with motor conduction velocities ranging from 3–10 meters per second. The disorder has been linked to PMP-22, P0 and EGR2 genes. Sural nerve biopsy may be useful. Inheritance is usually autosomal recessive with some dominant inheritance linked to EGR2.
CMT 4
Autosomal recessive CMT 4 is relatively rare. Most are demelinating with more severe phenotypes and onset is often in childhood. CMT 4C linked to 5q23 is a relatively more common form of CMT 4.
Toxic Neuropathies
Toxic polyneuropathies are rare occurrences in children in North America. Toxic exposure to heavy metals and environmental toxins may be more common in other regions of the world. Expeditious diagnosis is critical to identify and remove the source of the toxicity and to establish treatment with agents such as penicillamine.
Arsenic polyneuropathy
Arsenic toxicity produces is a sensorimotor neuropathy that may be axonal or, at times, predominantly demyelinating, simulating Guillain-Barré syndrome or CIDP. GI symptoms are common as well as tachycardia and hypotension. Mee’s lines may be seen in nails along with other skin changes and allopecia. The diagnosis is established by obtaining levels of arsenic in blood, urine, hair and nail samples.
Lead polyneuropathy
Lead toxicity is most commonly observed in children who have ingested old lead-based paint. Acute exposures cause lead encephalopathy more commonly. Clinical findings may include anorexia, nausea and vomiting, gastrointestinal disturbance, fatigue, clumsiness and ataxia, and occasionally cognitive impairment, seizures, mental status changes, papilledema, and coma. The weakness is predominantly in the lower limbs, but the upper limbs may be involved. Electrophysiologic studies show a primarily axonal degeneration affecting motor greater than sensory axons. A microcytic hypochromic anemia with basophilic stippling of red blood cells establishes the diagnosis. Lead lines may be evident in long bone films. Lead levels may or may not be elevated in urine and blood but levels of delta aminolevulinic acid are usually elevated in the urine.
Mercury poisoning
Mercury poisening may occur from the ingestion of mercuric salts, exposure to mercury vapor or use of topical ammonia mercury ointments. Patients present with a generalized encephalopathy, fatigue, and occasionally a skin rash. A predominantly distal motor axonal neuropathy occurs. Deep tendon reflexes may be absent and the gait is often ataxic. Sensory examination is often normal, although patients may complain of distal paresthesias. Electrophysiologic studies show motor axonal degeneration with normal sensory conduction studies.
Organophosphate poisoning
This entity is caused by to exposure to insecticides or high-temperature lubricants or softeners used in the plastic industry. Patients present with an encephalopathy manifested by confusion and coma. In acute exposure cholinergic crisis, manifested by sweating, abdominal cramps, diarrhea, and constricted pupils, may be present. A predominantly motor polyneuropathy is a late effect. However, the disorder may present as a rapidly progressive polyneuropathy mimicking Guillain-Barré syndrome. Severe paralysis with respiratory failure requiring ventilatory support may occur and in this situation there may be a superimposed post-synaptic defect in neuromuscular transmission.
Glue-sniffing (N-Hexane)
Glue sniffing neuropathy may be seen in teenage recreational glue sniffers. Repeated use may cause symptoms and signs of a predominantly distal motor and sensory polyneuropathy which is predominantly demyelinating. Motor and sensory nerve conduction studies demonstrate moderate slowing.
Chemotherapeutic agents
Vincristine, in particular, often produce a relatively pure motor axonal polyneuropathy. Severity is dose-dependent. Clinical findings include distal weakness, absent deep tendon reflexes, and at times foot drop. The disorder is often readily apparent by clinical examination and electrophysiologic studies or nerve biopsy is usually not necessary. The neuropathy usually improves with discontinuation of the medication, although significant electrophysiologic abnormalities (reduced CMAP) amplitudes and neuropathic recruitment) may persist. Vincristine may beparticularly troublesome for children with hereditary motor sensory neuropathy.
Metabolic Neuropathies
Uremic neuropathy
Uremic polyneuropathy often occurs in children and adults with end-stage renal disease. If clinical manifestations are present, they consist of a predominantly distal motor and sensory polyneuropathy with glove and stocking loss of sensation, loss of vibratory sense and distal weakness, particularly involving peroneal innervated musculature. With successful renal transplantation, clinical findings and electrophysiologic abnormalities normalize.
Diabetic polyneuropathy
Diabetes produces is a mixed motor and sensory polyneuropathy with both axonal changes and mild demyelination. The polyneuropathy is less common in children with diabetes mellitus, as compared with adults. The severity of the neuropathy may be related to the degree of glucose control.31
Alcoholic polyneuropathy
Chronic ethanol ingestion produces a polyneuropathy. Studies show that 9% of alcoholics with clinically manifest polyneuropathy with females showing more severe neuropathy. Alcohol abuse is generally severe over years with intakes of > 100 grams of alcohol per day. Nutritional deficiency and skipped meals exacerbates the neuropathy. Those with ethanol neuropathy show weight loss of 30 to 40 lbs in 50% of cases. The majority show clinical signs of polyneuropathy but approximately 40% are asymptomatic. Muscles are thin and tender, distal tendon reflexes reduced, there is variable loss of distal pain & temperature sensation. Patients complain of pain consisting of a dull ache & burning in feet and legs. They occasionally complain of lancinating pains. The distribution of signs is distal and symmetric. There is commonly hyperesthesia. Weakness involves the legs greater than hands. Tendon reflexes are reduced at the ankle in 80%. As far as autonomic findings patients frequently exhibit hyperhidrosis in the feet & hands. Electrodiagnostic studies show Nerve conduction studies show distal axonal loss in sensory and motor nerves, small sensory potentials, and mildly slowed conduction velocities. Findings are more severe in lower extremities. Nerve biopsy shows distal axonal loss. Disease course of the neuropathy shows slow improvement with reduced alcohol intake.
NEUROMUSCULAR JUNCTION TRANSMISSION DISORDERS
Autoimmune Myasthenia Gravis
This disorder is similar to the autoimmune myasthenia gravis observed in adults. The onset is often insidious, but at times patients may present with acute respiratory difficulties. Patients usually present with variable degrees of ophthalmoparesis and ptosis. In addition, patients may exhibit facial weakness, swallowing difficulties, speech problems and weakness of the neck, trunk and limbs. Proximal muscles are more affected than distal, and the upper limits are more affected than the lower. Fluctuation in the disease course with relapse and remission is common. Patients often complain of fatigue and diplopia, as well as progressive difficulty with chewing or swallowing. Patients are often worse with fatigue towards the end of the day. Thymoma, which occurs in about 10% of adult cases, is not a feature of the childhood onset disease.
Serum AChR antibodies are an important diagnostic screening tool. Anti-AChR antibodies can be detected in the serum in about 85–90% of patients with generalized myasthenia gravis and greater than 50% of those with ocular myasthenia. The most common antibodies detected are AChR binding, followed by AChR modulating and then Striational AChR antibodies. MUSK antibodies are an additional marker present in some seronegative patients and many patients with ocular myasthenia.
Diagnosis may also be confirmed by clinical response to an anticholinesterase drug such as Edrophonium (Tensilon®) Alternatively, neostigmine, a longer acting agent, can be used. Repetitive nerve stimulation studies show a characteristic decrement in the compound muscle action potential with slow stimulation rates (2–5 Hz) over a train of 4–5 stimuli. A decrement greater than 12–15% is often noted.
Congenital myasthenia syndromes (CMS)
CMS is a term used for a heterogenous group of disorders which are genetically determined rather than autoimmune mediated. Patients may present in the neonatal period, later in childhood, or even in adult life. Patients often exhibit ptosis, external ophthalmoparesis, facial weakness, general hypotonia, proximal greater than distal muscle weakness and variable degrees of functional impairment. Patients show absence of anti-AChR antibodies. Over 20 subtypes have been described and congenital myasthenia may be classified according to the following: 1) presynaptic defects (e.g. CHATCholine Acetyltransferese deficiency causing CMS with epidodic apnea; paucity of synaptic vesicles & reduced quantal release; or Congenital Lambert-Eaton-like syndrome), 2) Synaptic basal lamina defects (e.g. Endplate Acetylcholinesterase (AChE) deficiency at NMJs), and 3) Postsynaptic defects: (e.g. AChR disorders involving α, β, δ, e subunits; kinetic abnormalities in AChR function caused by AChR deficiency; Slow AChR channel syndromes; Fast-channel syndromes; Endplate rapsyn deficiency, etc.).
Several congenital myasthenic syndromes have been associated with Arthrogryposis syndromes. For example “multiple pterygium syndrome” (Escobar Syndrome) has been associated with AChR gamma, alpha 1, and delta subunit mutations.
For diagnostic workup, standard EMG with repetitive nerve stimulation is utilized initially and subsequently stimulated single fiber EMG may be useful. Ultrastructural evaluation of the neuromuscular junction with electron microscopy usually is performed on a biopsy of the deltoid or biceps, including the muscle region containing the neuromuscular junction (NMJ) or the "motor point". For in vitro electrophysiologic and immunocytochemical studies of the neuromuscular junction, a short muscle usually is removed from origin to insertion along with its motor branch and NMJ (a “motor point biopsy”). Muscles obtained have included the anconeus muscle near the elbow, the external intercostal muscle in the fifth or sixth intercostal space near the anterior axillary line or the peroneus tertius muscle in the lower extremity. Such in- vitro electrophysiologic studies allow specific delineation of the congenital myasthenic syndrome into one of the numerous specific subtypes. More recently the diagnostic evaluation of CMS has increasingly relied upon molecular genetic studies.
For treatment of a CMS subtype a definitive diagnosis is important because some CMS syndromes deteriorate with empiric treatment with AChE inhibitors such as pyridostigmine (Mestinon®). For example, slow channel syndromes may deteriorate on pyridostigmine and endplate acetylcholinesterase deficiency may deteriorate or show no response. Some pre-synaptic syndromes may show response to 3,4 diaminopyridine, which increases release of acetylcholine at the presynaptic terminal. This drug has been used in Lambert Eaton syndrome and in pre-synaptic CMS on a compassionate use basis.
Infantile Botulism
Infants with botulism usually present between ten days to six months of age with an acute onset of hypotonia, dysphagia, constipation, weak cry and respiratory insufficiency. The neurologic examination shows diffuse hypotonia and weakness, ptosis, ophthalmoplegia with pupillary dilation, reduced gag reflex and relative preservation of deep tendon reflexes. The diagnosis may be made by electrodiagnostic studies42 or by measurement Clostridium Botulinum Toxin in a rectal aspirate containing stool.
Non-Infantile Acquired Botulism
Older children and adults acquire botulism through poorly cooked, contaminated food with the toxin or through a cutaneous wound that becomes contaminated with soil-containing Clostridium Botulinum. The toxin can often be identified in the serum and the food source. Clinical findings include acute onset of constipation, ptosis, diplopia, bulbar weakness, respiratory difficulties, ophthalmoparesis, pupillary dilation, and diminished deep tendon reflexes. Recovery may take months. The diagnosis is generally made from electrodiagnostic studies.
MYOPATHIES
Dystrophinopathies
Duchenne Muscular Dystrophy (DMD)
DMD is an X-linked disorder caused by a gene abnormality at the Xp21 gene loci. The gene codes for dystrophin, which is a protein localized to the intracellular side of the plasma membrane of all myogenic cells, certain types of neurons, and in small amounts of other cell types. Dystrophin deficiency at the plasma membrane of muscle fibers disrupts the membrane cytoskeleton and leads to the secondary loss of other components of the muscle cytoskeleton. The primary consequence of the cytoskeleton abnormalities is membrane instability, leading to membrane injury from mechanical stresses, transient breaches of the membrane, and membrane leakage. Chronic dystrophic myopathy is characterized by aggressive fibrotic replacement of the muscle and eventual failure of regeneration with muscle fiber death and fiber loss. Generally loss of the reading frame causes complete absence of dystrophin (<5% by Western blot) and a Duchenne phenotype.
While the history of hypotonia and delayed motor milestones are often reported in retrospect, the parents are often unaware of any abnormality until the child starts walking. There has been variability reported in the age of onset.48 In 74% to 80% of instances, the onset has been noted before the age of four years.47,48 The most frequent presenting symptoms have been abnormal gait, frequent falls, and difficulty climbing steps. In DMD) the earliest weakness is seen in the neck flexors during preschool years. Parents frequently note the toe walking, which is a compensatory adaptation to knee extensor weakness and a lordotic posture to the lumbar spine, which is a compensatory change due to hip extensor weakness (figure 10). The vast majority of cases are identified by 5–6 years of age. Occasionally, DMD is identified pre-symptomatically in situations where a CK value is obtained with a markedly elevated value, malignant hyperthermia occurs during general anesthesia for an unrelated surgical indication, or a diagnosis is pursued in a male with an affected older sibling. Difficulty negotiating steps is an early feature as is a tendency to fall due to the child tripping or stumbling on a plantar-flexed ankle or the knee buckling or giving way due to knee extensor weakness. There is progressive difficulty getting up from the floor with presence of a Gower's sign (Figure 9).
Pain in the muscles, especially the calves, is a common symptom. Enlargement of muscles, particularly the calves (Figure 1), is commonly noted. The deltoid may also be hypertrophied. The tongue is also frequently enlarged. There is also commonly an associated wide-arch to the mandible and maxilla with separation of the teeth, presumably secondary to the macroglossia.
Weakness in DMD is generalized but predominantly proximal early in the disease course. Pelvic girdle weakness predates shoulder girdle weakness by several years. Ankle dorsiflexors are weaker than ankle plantar flexors, ankle everters are weaker than ankle inverters, knee extensors are weaker than knee flexors, hip extensors are weaker than hip flexors, and hip abductors are weaker than hip adductors.48 Molecular genetic studies to confirm DMD are summarized by Arnold and Flanigan5 in this issue, and muscle biopsy immunohistochemistry is summarized by Joyce and collegues37 in this issue.
Glucocorticoid therapy had become the standard of care in DMD throughout the lifespan. The past several years have seen a markedy increased interest by pharmaceutical companies in conducting ground-breaking research and development into effective treatment agents for DMD. Therapeutic approaches under development for clinical trials in DMD include antisense oligonucleotide (AON) exon skipping therapies, gene therapy strategies, stem cell therapies, as well as a host of small-molecule therapies (e.g. compounds that induce read-through of premature stop-codon mutations, promotion of muscle growth via myostatin inhibition, utrophin upregulation, and steroid analogs with improved side effect profiles). Diagnostic and clinical features of DMD are shown in Table 3.
Table 3.
Characteristics of Dystrophinopathies (DMD and BMD)
| DMD | BMD | |
|---|---|---|
| U.S. Prevalence (est.) | 15,000 | 3,700–8,300 |
| Incidence rate | 1/3500 male births | unknown |
| Inheritance | X-linked | X-linked |
| Gene Location | Xp21 (reading frame shifted) | Xp21 (reading frame maintained) |
| Protein | Dystrophin | Dystrophin |
| Onset | 2 to 6 yrs. | 4–12 years (severe BMD) Late teenage to adulthood (mild BMD) |
| Severity & Course |
|
|
| Ambulation Status |
|
|
| Weakness | Proximal > Distal
|
Proximal > Distal
|
| Cardiac | Dilated cardiomyopathy 1st to second decade; Onset of signs second decade. |
Cardiomyopathy (may occur before weakness); third to 4th decade frequent |
| Respiratory |
|
|
| Muscle size |
|
|
| Musculoskeletal |
|
|
| CNS |
|
|
| Muscle pathology |
|
|
| Blood chemistry & hematology |
|
|
Becker muscular dystrophy
In Becker muscular dystrophy, patient’s have similar distribution of weakness to those with DMD, however, onset may be delayed to the late first decade, second decade, or in mild BMD, the third or fourth decade in some instances. Some patients with BMD may present initially in the late second or third decade with signs of cardiomyopathy with clinically normal strength or mild strength loss. Diagnostic and clinical features of BMD are shown in Table 3. In severe BMD, there can be overlap in the age at diagnosis with DMD.47,48 For cases with a deletion mutation, the “reading frame” hypothesis predicts that BMD patients with in-frame deletions produce a semifunctional, internally deleted dystrophin protein. Thus DMD patients with frameshift point mutations or “out of frame deletions,” on the other hand, produce a severely truncated protein that is unstable.
Facioscapulohumeral muscular dystrophy
FSHD is a slowly progressive dystrophic myopathy with predominant involvement of facial and shoulder girdle musculature. The condition has autosomal dominant inheritance with linkage to the chromosome 4q35 locus. It is the second most common inherited muscular dystrophy in the adult population, with a prevalence estimate of 1 to 5/100,000.19 Overall, FSHD is the third most common of the dystrophies, behind DMD and myotonic muscular dystrophy. Presentation ranges from congenital to late in life but typical age of presentation is generally before age 20. Initially, 85% of patients show predominant involvement of facial and shoulder girdle musculature with facial weakness commonly being the initial manifestation. Facial weakness (Figure 6) typically involves the orbicularis oris, zygomaticus, and orbicularis oculi. Patients often have an expressionless face. Even in the very early stages, forced closure of the eyelids can be easily overcome by the examiner. The patient will typically have difficulty burying the lashes and pursing the lips, smiling, drinking through a straw, or whistling. The Face is spared in 5%-15% of patients who usually exhibit later onset of facial weakness (in the 4th or 5th decade), and often have a smaller deletion. By age 30, 95% show facial weakness. The facial weakness predominates in approximately 7% of patients. Masseter, temporalis, extraocular, and pharyngeal muscles are characteristically spared in FSHD. Scapular stabilizers, shoulder abductors, and shoulder external rotators may be significantly affected but at times the deltoids are surprisingly sparred if tested with the scapulae stabilized. Posterior and lateral scapular winging is common and scapulae are high riding (Figure 4). The biceps and triceps may both be more affected than the deltoids.39 Over time, ankle dorsiflexion weakness often becomes significant in addition to pelvic girdle weakness and some patients (approximately 13%) exhibit ankle dorsiflexion weakness very early in the disease course. Bilateral proximal lower extremity weakness occurs with disease progression with female gender and larger deletions being risk factors. Late in the disease course of early onset FSH, patients may show marked wrist extension weakness. Some authors have found asymmetric weakness in the dominant upper extremity.39 Lower abdominal weakness leads to a positive Beevor’s sign in many. Muscles usually spared include bulbar, extraocular, deltoid & respiratory. Severe respiratory weakness does occur, but fewer than 5% will require ventilatory assistance. Pectus excavatum and progressive thoracolumbar hyperlordosis may occur. Wheelchair reliance occurs in 20% of patients. Prognosis is worse with younger onset. There is linear decline in strength between 20 to 50 years of age, but report some question that strength decline stabilizes in later life. Life expectancy in FSHD is often normal. Clinical Features of FSHD are shown in Table 4. Functional consequences for facial weakness includes Sleeping with eyes open, bulbar dysfunction using straws, blowing up balloons, dysarthria (especially labial consonants), transverse smile, and misinterpretation of patients having a dour or flat affect.
Table 4.
FSHD Clinical Characteristics
| FSHD | |
|---|---|
| U.S. Prevalence (est.) | 15,000 |
| Prevalence rate | 1/20,000 |
| Inheritance | 70 – 90% AD; 10–30% sporadic |
| Gene Location | 4q35 FSHD1: deletion in units of the D4Z4 DNA repeat sequence (a D4Z4 contraction) FSHD2: |
| Protein | N/A |
| Onset |
|
| Severity & Course | Variable progression
|
| Ambulation Status |
|
| Weakness |
|
| Cardiac | Some conduction defects |
| Respiratory |
|
| Muscle size |
|
| Quality of Life |
|
| Musculoskeletal |
|
| CNS |
|
| Muscle pathology |
|
| Blood chemistry & hematology | CK: Normal to 5 times upper limit of normal range |
In FSH disease with locus at 4q35 (95% of all FSHD), there are 2 different abnormalities in the D4Z4 DNA fragment. In 90% of FSHD patients termed FSHD1, there is deletion in units of the D4Z4 DNA repeat sequence (a D4Z4 contraction) resulting in a reduced D4Z4 fragment size. This allows increased expression of the DUX4 gene in the distal repeat. The D4Z4 contraction produces permissive sequences in the 4qA region distal to the repeats which allows polyadenylation & stabilization of the distal DUX4 transcript. In some 5% of patients with facioscapulohumeral muscular dystrophy (FSHD), no D4Z4 repeat contraction on chromosome 4q35 is observed. Such patients, termed FSHD2, show loss of DNA methylation and heterochromatin markers at the D4Z4 repeat that are similar to patients with D4Z4 contractions (FSHD1).18 Thus, the D4Z4 DNA methylation in both FSHD1 & FSHD2 patients is reduced resulting in an open chromatin structure that polyadenylates and upregulates DUX4 transcriptional activity in the distal repeat. This commonality suggests that a change in D4Z4 chromatin structure and polydenylated and upregulated DUX4 expression in myoblasts unifies FSHD1 and FSHD2. DUX4 is localized to the nucleus and is toxic by being proapoptotic, is involved in transcriptional regulation, creates sensitivity to oxidative stress, represses MyoD and its target genes diminishing myogenic differentiation, and interferes with Pax7 in satellite cells to inappropriately regulate Pax targets during muscle regeneration.
FSHD2 is identical to FSHD1 in its clinical presentation and clinical features. Notable differences include a higher incidence (67%) of sporadic cases in FSHD2, the absence of gender differences in disease severity in FSHD2, and possibly later symptom onset in FSHD2. Overall, average disease severity in FSHD2 was similar to that reported in FSHD1 and was not influenced by D4Z4 repeat size.18 However, in FSHD2, a small effect of the degree of hypomethylation on disease severity was observed.18 In approximately 5% of FSH-like families, there is no linkage to 4q35.
Limb girdle muscular dystrophy (LGMD)
Before the advent of genetic testing, a group of patients commonly sharing a progressive pattern of greater proximal than distal muscular weakness with either autosomal dominant (LGMD1) or autosomal recessive (LGMD2) inheritance were said to have limb – girdle muscular dystrophies. Recent advances in molecular and genetic analyses have now identified a number of distinct genetic mutations in these patients. In the various subtypes of limb girdle muscular dystrophy, those patients with autosomal recessive inheritance (LGMD2) generally have earlier age of onset and are weaker than those with autosomal dominant inheritance and LGMD1. The lower extremities tend to be more affected than upper extremities. In autosomal dominant late-onset limb girdle muscular dystrophy, distal upper extremity muscles tend to show little progressive weakness over the years.50 In LGMD2, the distribution and pattern of weakness tends to be similar to DMD, however, the rate of progression tends to be slower than that observed in DMD.48,50 In one series,50 several differences between DMD and LGMD2 (SCARMD) were noted. The limb extensors were not weaker than limb flexors. In particular, ankle dorsiflexors were similar in strength to ankle plantar flexors, knee extensors showed similar strength compared to knee flexors, and hip extensors and hip flexors showed similar strength values. Clinical features of the more common Limb Girdle Muscular Dystrophies are shown in Tables 5 and 6.
Table 5.
Characteristics of Common Autosomal Dominant Limb Girdle Muscular Dystrophies (AD-LGMD)
| LGMD 1A | LGMD 1B | LGMD 1C | |
|---|---|---|---|
| U.S. Prevalence | 4,200 | 2,850 | 675 |
| Inheritance | AD | AD | AD |
| Gene Location | 5q31 | 1q11-q21 | 3p25 |
| Protein | Myotilin | Lamin A/C | Caveolin-3 |
| Onset |
|
|
|
| Severity & Course |
|
|
|
| Weakness |
|
|
|
| Ambulation Status |
|
|
|
| Cardiac |
|
|
No Cardiomyopathy |
| Respiratory |
|
|
|
| Muscle size | – | – |
|
| Musculo-skeletal |
|
|
|
| CNS |
|
|
|
| Muscle pathology |
|
|
|
| Blood chemistry |
|
|
|
Table 6.
Characteristics of Common Autosomal Recessive Limb Girdle Muscular Dystrophies (AR-LGMD)
| LGMD 2A | LGMD 2B | LGMD 2C | LGMD 2D | LGMD 2E | LGMD 2F | LGMD 2G | LGMD 2I | |
|---|---|---|---|---|---|---|---|---|
| U.S. Prevalence | 4,200 | 2,850 | 675 | 1,260 | 675 | 105 | 450 | |
| Inheritance | AR | AR | AR | AR | AR | AR | AR | AR |
| Gene Location | 4p21 | 2p12-14 | 13q12 | 17q21 | 4q12 | 5q33 | 17q12 | 19q13.3 |
| Protein | Calpain-3 | Dysferlin | Gamma-Sarcoglycan | Alpha Sarcoglycan (Adhalin) | Beta Sarcoglycan | Delta Sarcoglycan | Telethonin | Fukutin-related protein |
| Onset |
|
|
|
|
|
|
Mean 12.5 years Range 9–15 years |
|
| Severity & Course |
|
Slow Progression Mild weakness |
Variable progression (some like DMD; others like BMD)
|
Variable
|
Moderate progression & severity | Rapid progression
|
Slow progression Mild weakness |
Variable
|
| Ambulation Status |
|
|
|
|
|
|
40% non-ambulatory in third to 4th decade |
|
| Weakness |
|
|
|
|
|
|
Arms: proximal Legs: proximal and distal (foot drop) |
|
| Cardiac |
|
|
|
|
|
|
Cardiac involvement in 55% of patients |
|
| Respiratory |
|
|
|
|
|
|
|
|
| Muscle size |
|
|
|
|
|
|
Calf hypertrophy 50% Calf atrophy 50% |
|
| Musculo-skeletal |
|
|
|
|
|
|
|
|
| CNS |
|
|
|
|
|
|
No intellectual defect reported |
|
| Muscle pathology |
|
|
|
|
|
|
Myopathic Fiber degeneration Fiber regeneration Rimmed vacuoles Telethonin absent from muscle |
|
| Blood chemistry | CK: 7 to 80 times normal | CK: 10 to 72 times normal | CK: Very high | CK: Very high (often > 5,000) | CK: Very high (often > 5,000) | CK: 10 to 50 times normal | CK 3–30 times normal | CK : Very high 1,000 – 8,000) |
Emery Dreifuss Muscular Dystrophy
EMD refers to a group of muscular dystrophies with weakness, contractures, and cardiac conduction abnormalities. Inheritance pattern is variable among subtypes.
Emery-Dreifuss Muscular Dystrophy 1 (EMD1)
EMD1 is an X-linked recessive progressive dystrophic myopathy caused by an abnormality of the protein emerin with a gene locus identified at Xq28.53,94 Patients usually present in the teenage years, but age of presentation can vary from the neonatal period with hypotonia to the third decade. Early elbow flexion contractures are a hallmark of the disease.94
Severe contractures, including elbow flexion, ankle equinus, rigid spine, and neck extension contractures, are often more limiting than weakness, which begins in a scapulohumeral peroneal distribution. The biceps and triceps show wasting and weakness, and the deltoids and forearms are more spared. The calf frequently shows wasting. Ankle dorsiflexors often are weaker than ankle plantar flexors, leading to the equinus contractures.94 Scapular winging is frequent. Tightness of the cervical and lumbar spinal extensor muscles, resulting in limitation of neck and trunk flexion, with inability to flex the chin to the sternum and to touch the toes, also has been reported in EMD. The face is either spared or affected late. Functional difficulties are experienced walking or climbing stairs. Progression is slow, and loss of ambulation is rare. Some cases with EMD1 can have nocturnal hypoventilation as a result of restrictive expansion of the chest in association with the rigid spine, and partly because of involvement of the diaphragm.
Progressive cardiac disease is almost invariably present with onset in the early second decade to the 40s. Arrhythmia can lead to emboli or sudden death in early adult life. The cardiomyopathy can progress to left ventricular myocardial dysfunction or four-chamber dilated cardiomyopathy resulting from fibrosis with complete heart block and ventricular arrhythmias.53,94 Atrial arrhythmia usually appears before complete heart block. Frank syncope can develop in the late second and early third decades, and patients often require a cardiac pacemaker by age 30 (with an indication being bradycardia with heart rate <50). ECG changes include slow heart rate, absent or small P waves, atrioventricular block, and atrial fibrillation/flutter. 53,94 Evidence of cardiac arrhythmia often requires 24-hour Holter monitoring. A significant percentage of female carriers have conduction defects and arrhythmias; therefore they warrant monitoring with annual ECGs.
Laboratory evaluation is usually done with molecular genetic studies and/or muscle biopsy. Serum CK is mildly elevated to less than 10 times normal, and levels decrease with age. Muscle biopsy reveals emerin loss by immunohistochemistry in more than 95% of patients.
Emery-Dreifuss Muscular Dystrophy 2
EMD2 is caused by a lamin A/C protein abnormality and has been linked to chromosome 1q21.2. Inheritance can be dominant or recessive, and lamin A/C mutations can be either frameshift or missense.53 Those with missense mutations have childhood onset with a mean age of onset of 2.4 years. Weakness is in a scapuloperoneal distribution. Patients demonstrate paravertebral weakness or rigidity, and tendon contractures are common. Those with frameshift mutations producing a truncated protein have adult onset with mean age of 30.5 years, and cardiomyopathy is more frequent than weakness.53 Contractures are rare, and weakness is in a limb – girdle distribution. The disorder is allelic with autosomal dominant LGMD1B.
Congenital Muscular Dystrophy
The term congenital muscular dystrophy has been widely used for a group of infants presenting with hypotonia, muscle weakness at birth or within the first few months of life, congenital contractures, and immunohistochemical findings of dystrophic changes on muscle biopsy: muscle fiber necrosis and regeneration, increased endomysial connective tissue, and replacement of muscle with fat tissue. The early contractures might include equinovarus deformities, knee flexion contractures, hip flexion contractures, and tightness of the wrist flexors and long f nger flexors. The contractures can become more severe over time with prolonged static positioning and lack of adequate passive range of motion (ROM) and splinting/positioning. Classical CMDs are clinically confined to the musculoskeletal system, but other CMDs, including muscle – eye – brain disease and Walker-Warburg syndrome, are characterized by significant cerebral neuronal migration defects and eye abnormalities. Classical CMDs are further subdivided according to the presence or absence of merosin (laminin 2).57 An additional subgroup with collagen VI abnormalities has been identified and referred to as Ullrich congenital muscular dystrophy (Figure 12).
Congenital Myopathies
The term congenital myopathy is used to describe a group of heterogenous disorders usually presenting with infantile hypotonia as a result of genetic defects causing primary myopathies.17 There is an absence of any structural abnormality of the central nervous system or peripheral nerves. A specific diagnosis of each entity is made based on specific histologic and electron microscopic changes found on muscle biopsy. Molecular genetic studies are increasingly being used to confirm subtypes diagnostically.17 Although patients can be hypotonic during early infancy, they later develop muscle weakness that is generally nonprogressive and static. The weakness is predominantly proximal, symmetric, and in a limb – girdle distribution. The serum CK values are frequently normal, and the EMG can be normal or might show mild, nonspecific changes, usually of a myopathic character (small amplitude polyphasic potentials). The only congenital myopathy consistently associated with spontaneous activity is myotubular (centronuclear) myopathy. In this disorder, the EMG reveals myopathic motor unit action potentials with frequent complex repetitive discharges and diffuse fibrillation potentials. These myopathies can be considered primarily structural in nature, and patients do not actively lose muscle fibers, as is the case in dystrophic myopathies. Examples include Central Core Myopathy, Nemaline myopathy, Centronuclear (myotubular) myopathy (non X-linked), Severe X-Linked Centronuclear (Myotubular) Myopathy, and Congenital Fiber-Type Size Disproportion.
Myotonic Disorders
Myotonic muscular dystrophy type 1 (DM1)
DM1 is an autosomal dominant multisystem muscular dystrophy with an incidence of 1/8000.19 It represents the most common inherited neuromuscular disease of adults. The disorder affects skeletal muscle, smooth muscle, myocardium, brain, and ocular structures. Associated findings include baldness and gonadal atrophy (in males), cataracts, and cardiac dysrhythmias. Insulin insensitivity can be present. The gene has been localized to the region of the myotonin – protein kinase (DMPK) gene at 19q13.3. Patients demonstrate expansion of an unstable CTG trinucleotide repeat within the region. Molecular genetic testing is available for diagnosis. Normal individuals generally have fewer than 37 repeats, which are transmitted from generation to generation. DM1 patients can have 50 to several thousand CTG repeats with remarkable instability. The age of onset is inversely correlated to the number of repeat links. 61 Mild, late-onset DM1 usually is associated with 50 to 150 repeats; classic adolescent or young adult-onset DM1 shows 100 to 1000 repeats; and congenital DM1 patients show more than 1000 repeats (Figure 11).
The expanded CTG repeat further expands as it is transmitted to successive generations, providing a molecular basis for genetic anticipation. Several characteristic facial features of DM1 can be noted on inspection. The adult with long-standing DM1 often has characteristic facial features. The long thin face shows temporal and masseter wasting. Adult males often exhibit frontal balding. Myotonia, which is a state of delayed relaxation or sustained contraction of skeletal muscle, is easily identified in school-aged children, adolescents, and adults with DM1. Grip myotonia can be demonstrated by delayed opening of the hand with difficult extension of the fingers after tight grip. Percussion myotonia can be elicited by percussion of the thenar eminence with a reflex hammer, giving an adduction and flexion of the thumb with slow return (Figure 8). Symptomatic myotonia can be treated with agents such as mexiletine or membrane stabilizers such as carbamazepine or phenytoin sodium, which have been shown to affect the symptoms. The treated patients, however, have shown little functional gain. 91
DM1 is one of the few dystrophic myopathies with greater distal weakness than proximal weakness, and weakness initially is often most predominant in the ankle dorsiflexors, ankle everters and inverters, and hand muscles. Neck flexors, shoulder girdle musculature, and pelvic girdle musculature can become significantly involved over decades. As with other dystrophic myopathies, significant muscle wasting can occur over time. In DM1 patients with infantile onset, a congenital club foot or talipes equinovarus is a fairly common deformity. Many novel pharmacologic agents are on the horizon (e.g. antisense oligonucleotides) to decrease organ system effects from the trinucleotide repeat expansion and resultant RNA toxicity.
Proximal Myotonic Myopathy (DM2)
Proximal myotonic myopathy, also referred to as myotonic muscular dystrophy 2 (DM2), is a disorder with clinical similarities to DM1.90 The abnormal protein in this autosomal dominant disorder is the zinc finger protein 9 with genetic loci at chromosome 3q21. Clinical severity is unrelated to variable size CCTG repeats. The prognosis is more benign than DM1, and there is not a severe congenital onset form. Onset is 8 to 60 years of age, and there is intrafamilial variability. Patients present with muscle stiffness and pain. Weakness involves the proximal legs (hip flexors and extensors) more than the proximal arms. The distal arms (particularly the thumb and finger flexors) can also show involvement early in the course of the disease. Facial weakness is seen in a minority of patients. Respiratory muscles and distal legs are not clinically affected. A hallmark is the enlargement of calf muscles. Muscle pain is present in proximal muscle groups, is induced by palpation, occurs with exercise or at rest, and is unrelated to the myotonia. The myotonia is induced with grip or percussion in distal upper extremities, and is often asymmetric. The myotonia in DM2 increases with warmth and decreases with cold. Cataracts are noted in all patients over 20 years with slit lamp examinations. Cardiac conduction defects are present in 20%, diabetes mellitus in 20%, and hearing loss in 20%. MRI shows white matter hyperintensity on T2-weighted images. CK is normal to less than 10 times elevated. EMG shows profound myotonia, and CMAP amplitudes increment by 60% with exercise and reduce by 40% with rest. No decrement is noted on short bouts of exercise or slow or rapid repetitive stimulation. Myopathic motor units are seen proximally. MRI shows selective muscle involvement of the erector spinae and gluteus maximus. Diagnosis is confirmed by molecular genetic studies. A comparison of DM1 with the less common DM2 subtype is shown in Table 7.
Table 7.
Comparison of Myotonic Muscular Mystrophy types 1 and 2
| Feature | DM 1 | DM 2 |
|---|---|---|
| GENERAL | ||
| Epidemiology | Widespread | European |
| Onset Age | 0 to Adult | 8 to 60 years |
| Anticipation | + | Mild |
| Cogenital form | + | Rare |
| MUSCLE | ||
| Weakness | ||
| Face | + | Mild |
| Ptosis | + | Mild |
| Sternomastoid | + | Variable |
| Proximal legs | Late | Early |
| Distal | + | Hands |
| Any location | + | + |
| Muscle pain | ± | + |
| Myotonia | + | + |
| Calf hypertrophy | - | + |
| SYSTEMIC | ||
| Cataracts | + | + |
| Balding | + | + |
| Cardiac arrhythmias | + | Variable |
| Gonadal failure | + | 20% |
| Hypersomnia | + | Variable |
| Hyperhidrosis | Variable | + |
| Cognitive disorder | Mild to Severe | Mild |
| LABORATORY | ||
| Hyperglycemia | + | 20% |
| EMG: Myotonia | + | + |
| Muscle | Varied | Type 2 fibers |
| Internal nuclei | ||
| Chromosome | 19q13.3 | 3q21 |
| Mutated gene | DMPK | ZNF9 |
| Mutation type | CTG repeats | CCTG repeats |
| Repeat size | 100 to 4,000 | Mean ~5,000 |
| CNS MRI Δ | White & Gray matter | White matter |
From Alan Pestronk, Neuromuscular Disease Center Website, Washington University, St. Louis, MO USA, 6/20/2011; http://neuromuscular.wustl.edu
Myotonia Congenita
Myotonia congenita (Thomsen’s disease) presents in infancy and is inherited as an autosomal dominant condition. An abnormality of the muscle chloride channel is observed, and the disease is linked to the 7q35 loci. There is variable penetrance. Symptoms can be present from birth but usually develop later. The myotonia is relatively mild and can be manifest as difficulty in releasing objects or by difficulty walking or climbing stairs. Most patients do not show overt weakness. Functional difficulties in climbing stairs can be present. The myotonia is exacerbated by prolonged rest or inactivity. A “warm-up” phenomenon with reduced myotonia is noted after repeated activity. The myotonia can be aggravated by cold, hunger, fatigue, and emotional upset. Patients can demonstrate grip myotonia or lid lag after upward gaze or squint, and diplopia after sustained conjugate movement of the eyes in one direction. Nearly all have electrical myotonia by EMG, but there is a warm-up phenomenon with the myotonia reduced after a period of maximal contraction. Half of individuals also have percussion myotonia. Patients can be symptom free for weeks to months. The other common feature of myotonia congenita is muscle hypertrophy. Patients can exhibit a “Herculean” appearance. Patients have shown some benefit from treatment with quinine, mexiletine, phenytoin, procainamide, carbamazepine, and acetazolamide.
A recessive form of myotonia congenita (Becker form) also exists with later onset (ages 4 to 12), more marked myotonia, more striking hypertrophy of muscles, and associated weakness of muscles, particularly with short bouts of exercise. EMG shows myotonia in distal muscles and less myotonia after maximal contraction. On repetitive stimulation there is a decremental CMAP response at high stimulation frequency (30 Hz) and after exercise. The recessive form seems less prone to aggravation of the myotonia by cold. Diagnosis is suspected based on clinical information and the presence of classical myotonic discharges on EMG. Diagnosis is confirmed with molecular genetic testing.
Paramyotonia Congenita
Paramyotonia congenita is an autosomal dominant myotonic condition with at least two distinct genetic etiologies. One involves the sodium channel – α subunit located at chromosome 17q35, and the other a muscle chloride channel located at chromosome 7q35. The worsening of the myotonia with exercise is referred to as paradoxical myotonia. Weakness or stiffness can occur together or separately; there is cold and exercise aggravation, hypertrophy of musculature, and more severe involvement of hands and muscles of the face and neck. Myotonic episodes usually subside within a matter of hours but can last for days. Some patients become worse with a potassium load. On electrodiagnostic studies there is a drop in CMAP amplitude with cooling. Dense fibrillations disappear below 28° C; myotonic bursts disappear below 20° C; and electrical silence can occur below 20° C.
Treatment has involved mexiletine or tocainide.
Schwartz-Jampel Syndrome (Chondrodystrophic Myotonia)
Schwartz-Jampel syndrome is an autosomal recessive disorder with myotonia, dwarfism, diffuse bone disease, narrow palpebral fissures, blepharospasm, micrognathia, and flattened facies. Onset is usually before age 3. Patients have respiratory and feeding difficulties with impaired swallowing. Limitation of joint movement can be present along with skeletal abnormalities, including short neck and kyphoscoliosis. Muscles are typically hypertrophic and clinically stiff. A characteristic facies with pursed lips, micrognathia, and small mouth is seen. Patients can be difficult to intubate. Ocular changes include myopia and cataracts. Hirsutism and small testes can also be seen. The symptoms are not progressive. The protein perlecan with gene loci at chromosome 1p34-p36 has been implicated.
Electrodiagnostic studies show continuous electrical activity with electrical silence being difficult to obtain. Relatively little waxing and waning in either amplitude or frequency of complex repetitive discharges is observed. Abnormal sodium channel kinetics in the sarcolemma of muscle has been demonstrated. Some therapeutic benefit has been reported with procainamide and carbamazepine.
Inflammatory Myopathies
The hallmark of an inflammatory myopathy is the predominance of inflammatory cells on muscle biopsy. The three primary types are polymyositis, dermatomyositis, and inclusion body myositis (IBM). Although each is distinct, this group of myopathies is thought to involve immune mediated processes possibly triggered by environmental factors in genetically susceptible individuals. Dermatomyositis and and polymyositis can be associated with disorders of the heart and lung, as well as neoplasms. An inflamatory myopathy can also be present as part of a multisystem disorder in other connective tissue diseases, most commonly scleroderma, systemic lupus erythematosus, mixed connective tissue disease, and Sjogren’s syndrome. Overall, the age of onset for idiopathic inflammatory myopathies is bimodal, with peaks between 10 and 15 years of age in children and between 45 and 60 years of age in adults. Women are affected twice as often, with the exception of IBM, which is twice as common in men. It is important to diagnose accurately and in a timely fashion for both dermatomyositis and polymyositis because treatment is available and the prognosis depends on early initiation of immunotherapy.
Dermatomyositis
Characteristic features of dermatomyositis include muscle weakness that can present acutely, subacutely, or insidiously, along with a characteristic rash. This violaceous, scaling rash typically involves the eyelids and occurs with periorbital edema, termed a heliotrope rash. Other common locations for the rash are the dorsum of the hands, extensor surfaces of the knees and elbows, and ankles. Myalgias might or might not be present. The weakness initially involves the proximal musculature and can progress to the distal muscles. Pharyngeal muscle involvement is evident from the frequent finding of dysphagia or dysphonia. Other manifestations include cardiac dysrhythmias and cardiomyopathy, joint arthralgias, and interstitial lung disease. There appears to be an association between dermatomyositis and occult carcinoma in adults, and a judicious workup for carcinoma is advisable in newly diagnosed adult patients. Childhood dermatomyositis differs somewhat from the adult version because of the higher incidence of vasculitis, ectopic calcification in the subcutaneous tissues or muscle, and lipodystrophy. Corticosteroids alone are often highly effective in both inducing a remission and preventing a recurrence, and can usually be gradually withdrawn. Adults with dermatomyositis do not respond to corticosteroids so predictably, and other immunosuppressive agents are often required. It can be difficult to fully discontinue pharmacologic treatment.
Polymyositis
The diagnosis of polymyositis is often more difficult to make than dermatomyositis because no distinctive rash is present. It rarely occurs before age 20. Proximal limb and neck flexor muscle weakness presenting subacutely or insidiously should raise suspicion for polymyositis. Myalgias are present in as many as one third of patients but are not generally the predominant symptom. CK elevation usually occurs at some point in the disease and is generally a reasonable indicator of disease severity. CK can be normal in advanced cases with significant muscle atrophy. Needle EMG shows a classic triad of abnormal spontaneous rest activity, myopathic motor unit action potentials with early myopathic recruitment, and complex repetitive discharges. Increasingly MRI of affected muscles with both T2 and short tau inversion recovery images is used diagnostically for polymyositis and dermatomyositis.91 Muscle biopsies must be interpreted with caution because of the potential for sampling error. Potential cardiac and pulmonary manifestations are similar to dermatomyositis. Underlying carcinoma might less commonly occur than with dermatomyositis in adults. Treatment is primarily with corticosteroids supplemented by other immunosuppressive medications.
Inclusion Body Myositis
A third type of inflammatory myopathy with a different pattern of involvement is termed inclusion body myositis because of the presence of both inflammatory cells and vacuolated muscle fibers with nuclear and cytoplasmic fibrillary inclusions. IBM is now recognized as the most common myopathy in patients aged more than 50 years.6 Males are affected more than females. IBM has distinctive involvement of both proximal and distal musculature. In particular, the wrist and finger flexors are often more affected than the extensors, and the quadriceps can be affected out of proportion to other muscle groups. About one third have dysphagia, and the disease can be mistaken for amyotrophic lateral sclerosis because age of onset is frequently after 50. IBM is relentlessly progressive in most cases, sometimes to the point of requiring a wheelchair for mobility. Unfortunately it is not responsive to immunosuppressive medications, and treatment primarily involves appropriate rehabilitation interventions such as provision of assistive devices. For sporadic non-hereditary IBM, clinical trials are on the horizon using small molecules that produce anabolic effects through varied approaches to induce inhibition of myostatin. In addition, follistatin gene therapy will also soon be evaluated in trials.
Metabolic Myopathies
Inborn errors of glycogen metabolism and fatty acid metabolism can result in neuromuscular disorders. The major clinical presentations include fixed and progressive weakness, or exercise intolerance, cramps, myalgias, and myoglobinuria. Fixed and progressive weakness can be caused by glycogenoses (acid maltase deficiency or Pompe disease, debrancher deficiency, brancher deficiency, and aldolase A deficiency) or disorders of lipid metabolism (primary systemic carnitine deficiency, primary myopathic carnitine deficiency, secondary carnitine deficiency, short-chain acylocoenzyme A synthetase deficiency, medium-chain acylocoenzyme A synthetase dehydrogenase deficiency, etc.). Exercise intolerance, cramps/myalgias, and myoglobinuria can be caused by glycogenoses (myophosphorylase deficiency or McArdle’s disease, phosphorylase kinase deficiency, phosphofructokinase deficiency, phosphoglycerate mutase deficiency, etc.), disorders of lipid metabolism (CPT2 deficiency, VLCAD deficiency, and TP deficiency, etc.), and respiratory chain defects (coenzyme Q10 deficiency, complex I deficiency, complex III deficiency, and complex IV deficiency). Thre prototypical metabolic myopathies—McArdle’s disease, Pompe disease, and CPT2 deficiency—deserve mention.
Myophosphorylase Deficiency (McArdle’s Disease)
The most common glycogen storage disease is myophosphorylase deficiency, also known as McArdle’s disease or glycogenosis type 5. The autosomal recessive disorder has been linked to chromosome 11q13, and more than 65 different disease-causing mutations have been identified. Initial onset of symptoms often occurs during childhood and consists of poor endurance, fatigue, and exercise-induced cramps and myalgia that mainly affect active muscle groups. Myoglobinuria can also be absent during childhood with prevalence of fixed muscle weakness increasing as the patient ages. Symptoms can be precipitated by activities such as lifting heavy weights or climbing long flights of stairs. The “second wind” phenomenon is characteristic of this disorder. With the onset of myalgia, patients who rest briefly are then able to continue their physical activity with few or no symptoms. The normal function of muscle myophosphorylase is to catalyze the removal of 1,4-glycosyl residues from glycogen to produce glucose-1-phosphate.
Its absence leads to decreased metabolic substrate for glycolysis to produce adenosine triphosphate. CK is persistently elevated between episodes of myoglobinuria. EMG is normal when patients are asymptomatic but can show myotonic discharges and fibrillation potentials during an acute attack. Nonischemic forearm exercise testing shows only an increase in ammonia and stable levels of lactic acid and pyruvate. The diagnosis is made by demonstrating absence of myophosphorylase on muscle biopsy or by genetic mutation analysis. Possible treatments include high protein diet, pyridoxine, and creatine monohydrate.
Acid Maltase Deficiency (Glycogenosis Type 2, Pompe Disease)
Acid maltase deficiency is also referred to as glycogenosis type 2 or Pompe disease. It is caused by a deficiency of acid α-1,4-glucosidase (GAA). Inheritance is autosomal recessive with linkage to chromosome 17q23. Disease incidence is 1 in 40,000 to 50,000 live births. The level of residual enzyme activity correlates with the severity of disease. The GAA activity is less than 1% for those with infantile onset (birth to 1 year), 2% to 6% for childhood and juvenile onset (1 year to teens), and 1% to 29% in those with adult onset (third decade or later). All patients have glycogen accumulation in tissues. In those with infantile onset, clinical symptoms and signs usually include hypotonia, weakness, cardiomegaly, congestive heart failure, and arrhythmia. Liver and pulmonary involvement is also noted. Death occurs within the first year of life in 80% to 95% of untreated patients. In childhood onset there is mildly enlarged tongue, symmetric proximal weakness, and calf hypertrophy. Death occurs between 3 and 24 years as a result of respiratory failure. Glycogen accumulation is observed mainly in muscle. Patients with adult-onset Pompe disease present with proximal lower extremity weakness and restrictive lung disease. Sleep-disordered breathing is common. Expiration is more involved than inspiration because of chest wall muscle involvement. Nocturnal noninvasive ventilation is occasionally necessary. Atrophy of paraspinous muscles and scapular winging is seen. The disease course is one of slow progression over years. Pain, fatigue, and cramps are common complaints. There can be mild calf hypertrophy and diffuse muscle atrophy more proximally. Progressive disability is related to disease duration rather than age of onset. Eventually respiratory involvement is common, and many need wheelchairs or walking devices. Death is most often to the result of respiratory failure.
This is a diagnosis the neuromuscular specialist, neurologist, or physiatrist does not want to miss because it is a potentially treatbale disorder. The diagnosis of Pompe disease is confirmed with either molecular genetic studies or biochemical analysis of acid maltase activity with muscle biopsy. New methods using blood samples to measure GAA activity, however, are rapidly becoming adopted because of their speed and convenience.60,68 Typically serum CK is elevated (<10 times) in infants and is less elevated in adults. The EMG findings include an irritative myopathy with fibrillations, complex repetitive discharges, and myotonic discharges. Treatment now involves enzyme replacement with intravenous administration of recombinant α-glucosidase (Myozyme). Better outcomes are seen with earlier initiation of therapy. Myozyme has been shown to benefit infantile disease and possibly late-onset disease. Improvement is noted in strength of distal and proximal muscles, pulmonary function, cardiomyopathy, and increased survival.91,97
Carnitine palmitoyltransferase II (CPT2) deficiency
CPT2 is a rare autosomal recessive disorder of mitochondrial fatty acid oxidation and represents the most common metabolic cause of repeated myoglobinuria. The CPT2 protein mediates transport of Fatty acid-CoA across the inner mitochondrial membrane and is Involved in fatty acid β-oxidation. The metabolic defect promotes glycogen depletion. In the adolescent and adult later onset form of this recessive and semidominant disease (linked to Chromosome 1p32.3). The disease is characterized by muscle stiffness, myalgia, cramps, and exercise intolerance. Rhabdomyolysis is triggered by activities requiring fatty acid oxidation, prolonged exercise, cold, a low-carbohydrate - high-fat diet; fasting, infections, and treatment with Valproate. Other symptoms include malaise and asthenia. The attack frequency has been shown to be reduced by behavior modification. With regard to overt myopathy early in the disease course patients show normal strength between attacks, but later in the disease course patients may show weakness on examination. Males are more commonly symptomatic than females (80% of presenting patients are typically males). Patients may develop renal failure with rhabdomyolysis episodes. Laboratory studies show the serum CK to be normal or mildly elevated (50%) between episodes and high with rhabdomyolysis. Serum long chain acylcarnitines shows a high ratio of (Palmitoylcarnitine (C16:0) + Oleoylcarnitine (C18:1))/Acetylcarnitine (C2), and the serum carnitine is usually normal. When fasting there is a normal rise of ketone bodies and no myoglobinuria. IV glucose administration improves exercise tolerance, however, oral glucose is not effective. The EMG is myopathic or normal. Muscle biopsy shows normal or varied fiber size (Small type 1) and type 2 muscle fiber predominance. Lipid is increased in muscle fibers (50% increased). The CPT activity is reduced by 80% to 90% in homozygotes.
Treatment emphasizes a low fat; high carbohydrate diet with frequent meals. In addition, patients should avoid exercise with fasting or infection. General anesthesia should provide IV glucose before & during procedures. For specific treatment a diet treatment with triheptanoin (anaplerotic) at 30% to 35% of total daily caloric intake is recommended. In one study none experienced rhabdomyolysis or hospitalizations while on the diet. All patients returned abnormal SF-36 physical composite scores returned to normal levels that persisted for the duration of the therapy in all symptomatic patients.73 In a pilot9 study of six patients, it was found that bezafibrate, a commonly used hypolipidemic drug, restored the capacity for normal fatty acid oxidation in muscle cells from patients with a mild form of CPT2 deficiency by stimulating the expression of the mutated gene. Bezafibrate was administered for 6 months (at a dose of three 200-mg tablets per day) and the primary end point was the level of fatty acid oxidation in skeletal muscle biopsy. After bezafibrate treatment, the values of fatty acid oxidation increased significantly in the six patients (by 60 to 284%), CPT2 messenger RNA in skeletal muscle increased in all the patients (by 20 to 93%), as did the CPT2 protein level.9 These findings that were consistent with the increased oxidation levels. Patient-reported outcomes in physical funcyion and bodily pain also improved.
Mitochondrial Encephalomyopathies
Mitochondrial encephalomyopathies, also referred to as “mitochondrial cytopathy,” represent a complex group of disorders that affect multiple organ systems. Mitochondria are essential cellular organelles that convert carbohydrates, lipids, and proteins into usable energy in the form of adenosine triphosphate via an aerobic metabolism. Although the human mitochondrial genome is only 16.5 kilobase pairs and encodes 13 proteins, many different clinical syndromes can result from mutations of these genes. Mutant mitochondrial DNA can be present in different proportions in various cell populations in a phenomenon known as heteroplasmy. The pathogenic effect of the mutation is only manifested when a critical level of mutation is reached. Mutant and normal mitochondrial DNA segregate randomly during cell division, changing the proportion of mutant DNA in different cells over time. All mitochondria and mitochondrial DNA are derived from the mother’s oocyte. A family history compatible with maternal inheritance is strong evidence for a primary mitochondrial DNA mutation. Different family members in the maternal lineage can be asymptomatic or oligospermatic. Of the many clinical features of mitochondrial disorders that involve multiple organ systems, some are frequently present together and should alert the clinician to a mitochondrial etiology. Ptosis and progressive external ophthalmoplegia (PEO) are hallmarks of Kearns-Sayre syndrome, which produces diplopia and blurred vision. Myopathy is common among patients with mitochondrial disorders. Neck flexors can be affected earlier and more severely than neck extensors. Progressive fixed proximal weakness is more common, and patients can develop decreased muscle bulk. Premature fatigue, exercise intolerance, myalgia, and recurrent myoglobinuria can be symptoms of mitochondrial disorders. Serum lactate and pyruvate often are elevated at rest, and these levels can increase significantly after moderate exercise. Sensorineural hearing loss is frequently associated with mitochondrial encephalomyopathies. The hearing loss can be asymmetric and fluctuating in severity. Maternally inherited deafness and diabetes is another phenotypic combination in patients with mitochondrial DNA mutations. Dementia can be a prominent feature in mitochondrial cytopathy.
The diagnostic workup of a mitochondrial disorder often includes a complete blood count, serum electrolytes (including calcium and phosphate), liver function tests, blood urea nitrogen, creatinine, blood lactate and pyruvate, ECG, lumbar puncture for CSF protein, glucose, lactate, and pyruvate, EMG and nerve conduction study, brain imaging with MRI, and muscle biopsy for histology and electron microscopy. Histochemical stains for mitochondrial enzymes (succinate dehydrogenase, NADH-tetrazolium reductase, and cyclooxygenase) can be obtained, and the activities of mitochondrial respiratory chain enzymes can be measured in muscle tissue. The identification of numerous mitochondrial DNA mutations provides specific genetic diagnoses, including duplications, deletions, multiple deletions, and more than 100 pathogenic point mutations. Treatment is symptomatic for seizures (with avoidance of valproic acid, which is contraindicated because of depletion of carnitine and direct inhibitory effects on the mitochondrial respiratory chain). Electrolyte disturbances related to hypoparathyroidism and diabetes mellitus are corrected. Thyroid replacement alleviates hypothyroidism, and cardiac pacemaker placement prolongs life in those with Kearns-Sayre syndrome with conduction defects. Impairments in the oxidative phosphorylation pathway can generate increased amounts of free radical; consequently, antioxidants are prescribed (which include β-carotene, vitamin C, vitamin E, and CoQ 10 ). CoQ 10 shuttles electrons from complex I and II to complex III and can stabilize the oxidative phosphorylation enzyme complexes within the inner mitochondrial membrane. The dose for CoQ 10 in adults is 50 to 100 mg, three times per day. LCarnitine is also recommended. Dichloroacetate increases the pyruvate dehydrogenase complex and reduces lactate. Aerobic training is recommended for those with some mitochondrial conditions. Brief descriptions of common mitochondrial disorders follow.
Kearns-Sayre Syndrome
These patients show progressive external ophthalmoplegia, retinitis pigmentosa on fundoscopic examination, and complete heart block. Onset is usually before 20 years of age. Cerebellar findings can be present on physical examination, and patients can show limb weakness, hearing loss, diabetes mellitus, hypoparathyroidism, irregular menses, and growth hormone deficiency. Dementia can be progressive. CSF protein is frequently greater than 100 mg/dL.
Myoclonus Epilepsy With Ragged-Red Fibers
This clinical syndrome is defined by the presence of myoclonus, generalized seizures, ataxia, and ragged-red fibers on muscle biopsy. Symptoms usually begin in childhood. Other common clinical manifestations include hearing loss, dementia, exercise intolerance, and lactic acidosis. Multiple lipomatosis is common. Multiple members of a pedigree usually show the full syndrome.
Mitochondrial Encephalopathy, Lactic Acidosis, and Strokelike Episodes (MELAS)
One particular mitochondrial cytopathy the clinician does not want to miss is MELAS. This clinical syndrome is characterized by strokelike episodes at a young age (typically before 40 years), lactic acidosis, and encephalopathy evident as seizures, dementia, or both. Muscle biopsy shows ragged-red fibers as a result of respiratory chain defects. Other frequent clinical features include normal early development, limb weakness, ataxia, myoclonus, migraine-like headaches, recurrent nausea and vomiting, and hearing loss. The abrupt-onset strokes often affect the occipital cortex but can involve other regions of the brain. These patients often describe an antecedent history of migraine headaches that often occur before the strokelike event. Patients can experience improvement over weeks to months, but these events virtually always recur. The lesions do not conform to territories of large vessels, a finding that favors the term strokelike episodes. Based on the hypothesis that MELAS is caused by impaired vasodilation in an intracerebral artery, oral L-arginine, a nitric oxide precursor, has been administered acutely within 30 minutes of a stroke, and this treatment was shown to significantly decrease the frequency and severity of strokelike episodes.38
Neuropathy, Ataxia, and Retinitis Pigmentosa
This disorder consists of the variable combinations of proximal neurogenic limb weakness, sensory neuropathy, ataxia, pigmentary retinopathy, developmental delay, dementia, and seizures. The onset occurs in teens and young adults, and the course is gradually progressive.
Mitochondrial Neurogastrointestinal Encephalomyopathy
This syndrome is clinically recognized by the unusual combination of six features: PEO, severe gastrointestinal dysmotility, cachexia, peripheral neuropathy, diffuse leukoencephalopathy on MRI, and evidence of mitochondrial dysfunction (histologic, biochemical, or genetic). The peripheral neuropathy and the prominent gastrointestinal dysmotility are defining features. Lactic acidosis at rest is present in two thirds of patients. Both axonal and demyelinating polyneuropathy is frequent. Muscle biopsy reveals ragged-red fibers and neurogenic changes.
SUMMARY
This article has reviewed the clinical approach to the diagnostic evaluation of progressive neuromuscular diseases with an emphasis on relevant neuromuscular history, family history, clinical examination findings, laboratory studies and a brief discussion of the role of muscle biopsy. Molecular genetic and immunocytochemistry studies of muscle have been major advances in the diagnostic evaluation of the neuromuscular disease patient, however, all diagnostic information needs to be interpreted within the context of relevant clinical information. In some instances, a precise diagnosis is not medically possible, however, the accurate characterization of an individual patient within the most appropriate NMD clinical syndrome often allows the clinician to provide the patient and family with accurate prognostic information and anticipatory guidance for the future. After synthesizing all available clinical and diagnostic information, the physiatrist or neurologist may at times determine that an NMD patient has an inappropriate diagnosis warranting further diagnostic evaluation.
The current and subsequent issue focuses on the management and rehabilitation of progressive neuromuscular diseases with an emphasis on optimization of health, prevention or minimization of complications and enhancement of quality of life. Appropriate rehabilitation approaches and novel therapeutics require an accurate and timely diagnosis. In addition, patient education in NMD is dependent upon access to current and accurate diagnostic information. The first step in providing accurate information and appropriate treatment is constantly ensuring that all NMD patients have appropriate diagnoses based on a thorough evaluation of both clinical information, physical examination, and appropriate application of current medical science and available diagnostic technology. Thorough discussions of those diagnostic technologies are reviewed in the following three articles.5,37,43
Key Points.
Progressive acquired or hereditary neuromuscular diseases (NMDs) are disorders caused by an abnormality of any component of the lower motor neuron - anterior horn cell, peripheral nerve, neuromuscular junction (pre-synaptic or post-synaptic region), or muscle.
Many neuromuscular diseases are multi-system disorders affecting multiple organ systems.
In the context of a neuromuscular disease diagnostic evaluation, the clinician still must be able to obtain a relevant patient and family history and perform focused general, musculoskeletal, neurologic and functional physical examinations to direct further diagnostic evaluations.
Laboratory studies often include relevant molecular genetic studies in certain instances, however, specific genetic entities need to be strong diagnostic considerations, as these studies may be expensive with limited sensitivity.
Early diagnosis is facilitated by knowledge of the common initial clinical presentations of specific NMDs, and in many cases the early diagnosis has potential implications for treatment and prevention of secondary conditions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aitkens S, Lord J, Bernauer E, Fowler WM, Jr, Lieberman JS, Berck P. Relationship of manual muscle testing to objective strength measurements. Muscle Nerve. 1989;12:173–177. doi: 10.1002/mus.880120302. [DOI] [PubMed] [Google Scholar]
- 2.Aitkens SG, McCrory MA, Kilmer DD, Bernauer EM. Moderate resistance exercise program: Its effect in slowly progressive neuromuscular disease. Arch Phys Med Rehabil. 1993;74:711–715. doi: 10.1016/0003-9993(93)90031-5. [DOI] [PubMed] [Google Scholar]
- 3.Andersen PM, Nilsson P, Keranen M-L, et al. Phenotypic hetrogeneity in motor neuron disease patiens with CuZn-superoxide dismutase mutations in Scandanavia. Brain. 1997;120:1723–37. doi: 10.1093/brain/120.10.1723. [DOI] [PubMed] [Google Scholar]
- 4.Arkin AM. Absolute muscle power: The internal kinesiology of muscle, thesis. Department of Orthopedic Surgery. State University of Iowa; 1939. [Google Scholar]
- 5.Arnold WD, Flanigan KM. A Practical Approach to Molecular Diagnostic Testing in Neuromuscular Diseases, Physical Medicine and Rehabilitation Clinics of North America. 2012. [DOI] [PubMed] [Google Scholar]
- 6.Askanas V, Engel WK. Inclusion body myositis, a multifactorial muscle disease associated with aging: current concepts of pathogenesis. Curr Opin Rheumatol. 2007;19 (6):550–559. doi: 10.1097/BOR.0b013e3282efdc7c. [DOI] [PubMed] [Google Scholar]
- 7.Bach JR, Want TG. Noninvasive Long-Term Ventilatory Support for Individuals with Spinal Muscular Atrophy and Functional Bulbar Musculature. Arch Phys Med Rehabil. 1995;76:213. doi: 10.1016/s0003-9993(95)80603-2. [DOI] [PubMed] [Google Scholar]
- 8.Barisic N, Claeys KG, Sirotkoviæ-Skerlev M, Löfgren A, Nelis E, De Jonghe P, Timmerman V. Charcot-Marie- Tooth disease: a clinico-genetic confrontation. Ann Hum Genet. 2008 May;72(Pt 3):416–41. doi: 10.1111/j.1469-1809.2007.00412.x. Epub 2008 Jan 23. Review. [DOI] [PubMed] [Google Scholar]
- 9.Bonnefont JP, Bastin J, Behin A, Djouadi F. Bezafibrate for an inborn mitochondrial beta-oxidation defect. N Engl J Med. 2009 Feb 19;360(8):838–40. doi: 10.1056/NEJMc0806334. [DOI] [PubMed] [Google Scholar]
- 10.Bradshaw DY, Jones HR., Jr Guillain-Barre Syndrome in Children: Clinical Course, Electrodiagnosis and Prognosis. Muscle Nerve. 1992;15:4, 500. doi: 10.1002/mus.880150415. [DOI] [PubMed] [Google Scholar]
- 11.Brooke MH, Fenichel GM, Griggs RC, et al. Clinical investigation in Duchenne dystrophy. 2. Determination of the “power” of therapeutic trials based on the natural history. Muscle Nerve. 1983;6:91–103. doi: 10.1002/mus.880060204. [DOI] [PubMed] [Google Scholar]
- 12.Carter GT, Abresch RT, Fowler WM, Jr, Johnson ER, Kilmer DD, McDonald CM. Profiles of neuromuscular diseases: hereditary motor and sensory neuropathy, types I and II. Am J Phys Med Rehabil. 1995;749 (Suppl):S140–S149. doi: 10.1097/00002060-199509001-00008. [DOI] [PubMed] [Google Scholar]
- 13.Carter GT, Abresch RT, Fowler WM, Jr, Johnson ER, Kilmer DD, McDonald CM. Profiles of neuromuscular diseases: spinal muscular atrophy. Am J Phys Med Rehabil. 1995;74(Suppl):S150–S159. doi: 10.1097/00002060-199509001-00009. [DOI] [PubMed] [Google Scholar]
- 14.Carter GT, Weiss MD, Han JJ, Chance PF, England JD. Charcot-marie-tooth disease. Curr Treat Options Neurol. 2008 Mar;10(2):94–102. doi: 10.1007/s11940-008-0011-3. [DOI] [PubMed] [Google Scholar]
- 15.Coleman RA, Stajich JM, Pact VW, Pericak-Vance MA. The ischemic exercise test in normal adults and in patients with weakness and cramps. Muscle Nerve. 1986;9:216. doi: 10.1002/mus.880090305. [DOI] [PubMed] [Google Scholar]
- 16.Cros D, Harnden P, Pellisier JF, Serratrice G. Muscle hypertrophy in Duchenne muscular dystrophy: a pathological and morphometric study. J Neurol. 1989;236:43–47. doi: 10.1007/BF00314217. [DOI] [PubMed] [Google Scholar]
- 17.D’Amico A, Bertini E. Congenital myopathies. Curr Neurol Neurosci Rep. 2008 Jan;8(1):73–79. doi: 10.1007/s11910-008-0012-3. Review. [DOI] [PubMed] [Google Scholar]
- 18.de Greef JC, Lemmers RJLF, Camaño P, Day JW, Sacconi S, Dunand M, van Engelen BGM, Kiuru-Enari S, Padberg GW, Rosa AL, Desnuelle C, Spuler S, Tarnopolsky M, Venance SL, Frants RR, van der Maarel SM, Tawil R. Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology. 2010;75:1548–1554. doi: 10.1212/WNL.0b013e3181f96175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Emery AH. Population Frequencies of Inherited Neuromuscular Diseases—A World Survey. Neuromuscul Disord. 1991;1:19. doi: 10.1016/0960-8966(91)90039-u. [DOI] [PubMed] [Google Scholar]
- 20.Eng GD, Binder H, Koch B. Spinal muscular atrophy: Experience in diagnosis and rehabilitation in management of 60 patients. Arch Phys Med Rehabil. 1984;65:549–553. [PubMed] [Google Scholar]
- 21.Epstein MA, Sladky JT. The Role of Plasmapheresis in Childhood Guillain-Barre Syndrome. Ann Neurol. 1990;28:65. doi: 10.1002/ana.410280112. [DOI] [PubMed] [Google Scholar]
- 22.Finanger EL, Russman B, Forbes SC, Roonney WD, Walter GA, Vandenborne K. Use of skeletal muscle MRI in diagnosis and monitoring disease progression in Duchenne muscular dystrophy. Phys Med Rehabil Clin N Am. 2012;23:1–10. doi: 10.1016/j.pmr.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fischer AQ, Carpenter DW, Hartlage PL, et al. Muscle imaging in neuromuscular disease using computerized realtime sonography. Muscle & Nerve. 1988;11:270–275. doi: 10.1002/mus.880110313. [DOI] [PubMed] [Google Scholar]
- 24.Fowler WM, Jr, Abresch RT, Aitkens S, Carter GT, Johnson ER, Kilmer DD, McCrory MA, Wright NC. Profiles of neuromuscular diseases: design of the protocol. AM J Phys Med and Rehabil. 1995;74 (Suppl):S62–S69. doi: 10.1097/00002060-199509001-00002. [DOI] [PubMed] [Google Scholar]
- 25.Fowler WM, Gardner GW. Quantitative strength measurements in muscular dystrophy. Arch Phys Med Rehabil. 1968;48:629–629. [PubMed] [Google Scholar]
- 26.Gospe SM, Lozaro RP, Lava NS, et al. Familial X-linked myalgia and cramps: a non-progressive myopathy associated with a deletion in the dystrophin gene. Neurology. 1989;39:1277–1280. doi: 10.1212/wnl.39.10.1277. [DOI] [PubMed] [Google Scholar]
- 27.Guenther UP, Handoko L, Laggerbauer B, Jablonka S, Chari A, Alzheimer M, Ohmer J, Plöttner O, Gehring N, Sickmann A, von Au K, Schuelke M, Fischer U. IGHMBP2 is a ribosome-associated helicase inactive in the neuromuscular disorder distal SMA type 1 (DSMA1) Hum Mol Genet. 2009 Apr 1;18(7):1288–300. doi: 10.1093/hmg/ddp028. Epub 2009 Jan 20. [DOI] [PubMed] [Google Scholar]
- 28.Heckmatt JZ, Dubowitz V. Ultrasound imaging and directed needle biopsy in the diagnosis of selective involvement in neuromuscular disease. Journal of Child Neurology. 1987;2:205–213. doi: 10.1177/088307388700200307. [DOI] [PubMed] [Google Scholar]
- 29.Heckmatt JZ, Leeman S, Dubowitz V. Ultrasound imaging in the diagnosis of muscle disease. Journal of Pediatrics. 1982;101:656–660. doi: 10.1016/s0022-3476(82)80286-2. [DOI] [PubMed] [Google Scholar]
- 30.Heckmatt JZ, Pier N, Dubowitz V. Real-time ultrasound imaging of muscles. Muscle & Nerve. 1988;11:56–65. doi: 10.1002/mus.880110110. [DOI] [PubMed] [Google Scholar]
- 31.Hoffman WH, Hat ZH, Frank RN. Correlates of Delayed Motor Nerve Conduction and Retinopathy in Juvenile- Onset Diabetes Mellitus. J Pediatr. 1983;102:351. doi: 10.1016/s0022-3476(83)80647-7. [DOI] [PubMed] [Google Scholar]
- 32.Huang Y, Majumdar S, Genant HK, Chan WP, Sharma KR, Yu P, Mynhier M, Miller RG. Quantitative MR relaxometry study of muscle composition and function in Duchenne muscular dystrophy. Journal of Magnetic Resonance Imaging. 1994;4(1):59–64. doi: 10.1002/jmri.1880040113. [DOI] [PubMed] [Google Scholar]
- 33.Innaccone ST, Browne RH, Samaha FJ, Buncher CR DCN/SMA Group. Prospective study of spinal muscular atrophy before age 6 years. Pediatr Neurol. 1993;9:187–193. doi: 10.1016/0887-8994(93)90082-n. [DOI] [PubMed] [Google Scholar]
- 34.Ianassecu V. Charcot-Marie-Tooth neuropathies: From clinical description to molecular genetics. Muscle & Nerve. 1995;18:267–275. doi: 10.1002/mus.880180302. [DOI] [PubMed] [Google Scholar]
- 35.Johnson ER, Abresch RT, Carter GT, Kilmer DD, Fowler WM, Jr, Sigford BJ, Wanlass RL. Profiles of neuromuscular diseases: myotonic dystrophy. Am J Phys Med Rehabil. 1995;74(Suppl):S104–S116. [PubMed] [Google Scholar]
- 36.Jones HR, Jr, Bradshaw DY. Guillain-Barre Syndrome and Plasmapheresis in Childhood. Ann Neurol. 1991;29:688. doi: 10.1002/ana.410290622. [DOI] [PubMed] [Google Scholar]
- 37.Joyce N, Oskarsson B, Jin L. Muscle Biopsy Evaluation in Neuromuscular Disorders. Phys Med Rehabil Clin North Am. doi: 10.1016/j.pmr.2012.06.006. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koga Y, Akita Y, Nishioka J, et al. L-arginine improves the symptoms of strokelike episodes in MELAS. Neurology. 2005 Feb 22;64(4):710–712. doi: 10.1212/01.WNL.0000151976.60624.01. [DOI] [PubMed] [Google Scholar]
- 39.Kilmer DD, Abresch RT, McCrory MA, Carter GT, Fowler WM, Jr, Johnson ER, McDonald CM. Profiles of neuromuscular diseases: facioscapulohumeral muscular dystrophy. Am J Phys Med Rehabil. 1995;74:S131–S139. doi: 10.1097/00002060-199509001-00007. [DOI] [PubMed] [Google Scholar]
- 40.Kilmer DD, McCrory MA, Wright NC, Aitkens SG, Bernauer EM. The effect of high resistance exercise program in slowly progressive neuromuscular disease. Arch Phys Med Rehabil. 1994;75:560–563. [PubMed] [Google Scholar]
- 41.Lamont PJ, Johnston HM, Berdoukas VA. Plasmapheresis in Children with Guillain-Barre Syndrome. Neurology. 1991;41:12, 1928. doi: 10.1212/wnl.41.12.1928. [DOI] [PubMed] [Google Scholar]
- 42.Lavenstein BL, Shin W, Watkin T, Keller S. Four-Year Followup Study of Use of IVIG in Childhood Acute Inflammatory Demyelinating Polyneuropathy (GBS) Neurology. 1994;44:A169. [Google Scholar]
- 43.Lipa BM, Han JJ. Electrodiagnosis in Neuromuscular Disease. Phys Med Rehabil Clin North Am. doi: 10.1016/j.pmr.2012.06.007. this issue. [DOI] [PubMed] [Google Scholar]
- 44.Liu GC, Jong YJ, Chiang CH, Jaw TS. Duchenne muscular dystrophy: MR grading system with functional correlation. Radiology. 1993;186(2):475–80. doi: 10.1148/radiology.186.2.8421754. [DOI] [PubMed] [Google Scholar]
- 45.Liu M, Chino N, Ishihara T. Muscle damage progression in Duchenne muscular dystrophy evaluated by a new quantitative computed tomography method. Archives of Phys Med & Rehab. 1993;74(5):507–14. doi: 10.1016/0003-9993(93)90115-q. [DOI] [PubMed] [Google Scholar]
- 46.Lord JP, Aitkens S, McCrory M, Bernauer EM. Isometric and isokinetic measurement of hamstring and quadriceps strength. Arch Phys Med Rehabil. 1992;73:324–330. doi: 10.1016/0003-9993(92)90004-g. [DOI] [PubMed] [Google Scholar]
- 47.McDonald CM, Abresch RT, Carter GT, Fowler WM, Jr, Johnson ER, Kilmer DMD. Profiles of neuromuscular diseases: Becker’s muscular dystrophy. Am J Phys Med Rehabil. 1995;74 (Suppl):S93–S103. doi: 10.1097/00002060-199509001-00004. [DOI] [PubMed] [Google Scholar]
- 48.McDonald CM, Abresch RT, Carter GT, Fowler WM, Johnson ER, Kilmer DMD, Sigford Profiles of neuromuscular diseases: Duchenne muscular dystrophy. Am J Phys Med Rehabil. 1995;74 (Suppl):S70–S92. doi: 10.1097/00002060-199509001-00003. [DOI] [PubMed] [Google Scholar]
- 49.McDonald CM, Jaffe KM, Shurtleff DB. Clinical assessment of Muscle strength in children with meningomyelocele: Accuracy and stability of measurements over time. Archives of Physical Medicine and Rehabilitation. 1986;67:855–861. [PubMed] [Google Scholar]
- 50.McDonald CM, Johnson ER, Abresch RT, Carter GT, Fowler WM, Jr, Kilmer DMD. Profiles of neuromuscular diseases: limb-girdle syndromes. Am J Phys Med Rehabil. 1995;74(Suppl):S117–S130. doi: 10.1097/00002060-199509001-00006. [DOI] [PubMed] [Google Scholar]
- 51.Meyerson MD, Lewis E, ILL K. Facioscapulohumeral muscular dystrophy and accompanying hearing loss. Archives of Otolaryngology. 1984 Apr;110(4):261–6. doi: 10.1001/archotol.1984.00800300053012. [DOI] [PubMed] [Google Scholar]
- 52.Mills KR, Edwards RHT. Investigative strategies for muscle pain. J Neurol Sci. 1983;58:73. doi: 10.1016/0022-510x(83)90111-9. [DOI] [PubMed] [Google Scholar]
- 53.Muchir A, Worman HJ. Emery-Dreifuss muscular dystrophy. Curr Neurol Neurosci Rep. 2007 Jan;7(1):78–83. doi: 10.1007/s11910-007-0025-3. [DOI] [PubMed] [Google Scholar]
- 54.Munsat TL, Davies KE. Meeting report: International SMA consortium meeting. Neuromuscul Disord. 1992;2:423–428. doi: 10.1016/s0960-8966(06)80015-5. [DOI] [PubMed] [Google Scholar]
- 55.Munsat TL. Standardized forearm ischemic exercise test. Neurology. 1970;20:1171. doi: 10.1212/wnl.20.12.1171. [DOI] [PubMed] [Google Scholar]
- 56.Munsat TL. Workshop report: International SMA collaboration. Neuromuscul Disord. 1991;1:81. [Google Scholar]
- 57.Muntoni F, Valero de Bernabe B, et al. 8th Workshop of the International Consortium on CMD; 3rd Workshop of the MYO-CLUSTER project GENRE. Neuromuscul Disord; 114th ENMC International Workshop on Congenital Muscular Dystrophy (CMD); 17–19 January 2003; Naarden, The Netherlands. 2003. Sep, pp. 579–588. [DOI] [PubMed] [Google Scholar]
- 58.Neuromuscular disorders: gene location. Neuromuscul Disord. 2006 Jan;16(1):64–90. [PubMed] [Google Scholar]
- 59.Norris F, Sheperd R, Denys E, et al. Onset, natural history and outcome in idiopathic adult motor neuron disease. J Neurol Sci. 1993;118 (1): 48–55. doi: 10.1016/0022-510x(93)90245-t. [DOI] [PubMed] [Google Scholar]
- 60.Okumiya T, Keulemans JL, Kroos MA, et al. A new diagnostic assay for glycogen storage disease type II in mixed leukocytes. Mol Genet Metab. 2006 May;88(1):22–28. doi: 10.1016/j.ymgme.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 61.Ouvrier RA, McLeod JG, Pollard JD. Peripheral Neuropathy in Childhood. International Child Neurology Association, Cambridge University Press; 1999. Acute Inflammatory Demyelinating Polyradiculoneuropathy. [Google Scholar]
- 62.Padberg GW, Brouwer OF, deKeizer RJ, Dijkman G, Wijmenga C, Grote JJ, Frants RR. On the significance of retinal vascular disease and hearing loss in facioscapulohumeral muscular dystrophy. Muscle and Nerve. 1995;2:S73–80. [PubMed] [Google Scholar]
- 63.Parano E, Fiumara A, Falsaperla R, Pavone L. A clinical study of childhood spinal muscular atrophy in Sicily: A review of 75 cases. Brain Dev. 1994;16(2):104–107. doi: 10.1016/0387-7604(94)90043-4. [DOI] [PubMed] [Google Scholar]
- 64.Parsons DW, McAndrew PE, Iannaccone ST, et al. Intragenic telSMN mutations: frequency, distribution, evidence of a founder effect, and modification of the spinal muscular atrophy phenotype by cenSMN copy number. Am J Hum Genet. 1998;63(6):1712–1723. doi: 10.1086/302160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat Genet. 2006;7:710–23. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- 66.Phoenix J, Betal D, Roberts N, Helliwell TR, Edwards RH. Objective quantification of muscle and fat in human dystrophic muscle by magnetic resonance image analysis. Muscle & Nerve. 1996;19(3):302–10. doi: 10.1002/(SICI)1097-4598(199603)19:3<302::AID-MUS4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 67.Pineda M, Arpa J, Montero R, Aracil A, Domínguez F, Galván M, Mas A, Martorell L, Sierra C, Brandi N, García-Arumí E, Rissech M, Velasco D, Costa JA, Artuch R. Idebenone treatment in paediatric and adult patients with Friedreich ataxia: long-term follow-up. Eur J Paediatr Neurol. 2008 Nov;12(6):470–5. doi: 10.1016/j.ejpn.2007.11.006. Epub 2008 Jan. [DOI] [PubMed] [Google Scholar]
- 68.Winchester B, Bali D, et al. Pompe Disease Diagnostic Working Group. Methods for a prompt and reliable laboratory diagnosis of Pompe disease: report from an international consensus meeting. J Mol Genet Metab. 2008 Mar;93(3):275–281. doi: 10.1016/j.ymgme.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 69.Pradas J, Finison L, Andres PL, et al. The natural history of amyotrophic lateral sclerosis and the use of natural history controls in therapeutic trials. Neurology. 1993;43 (4): 751–5. doi: 10.1212/wnl.43.4.751. [DOI] [PubMed] [Google Scholar]
- 70.Pradhan S. New clinical sign in Duchenne muscular dystrophy. Pediatr Neurol. 1994;11:298–300. doi: 10.1016/0887-8994(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 71.Reimers CD, Schlotter B, Eicke BM, Witt TN. Calf enlargement in neuromuscular diseases: a quantitative ultrasound study in 350 patients and review of the literature. Journal of the Neurological Sciences. 1996;143(1–2):46–56. doi: 10.1016/s0022-510x(96)00037-8. [DOI] [PubMed] [Google Scholar]
- 72.Ringel SP, Murphy JR, Alderson MK, et al. The natural history of amyotrophic lateral sclerosis. Neurology. 1993;43 (7): 1316–22. doi: 10.1212/wnl.43.7.1316. [DOI] [PubMed] [Google Scholar]
- 73.Roe CR, Yang BZ, Brunengraber H, Roe DS, Wallace M, Garritson BK. Carnitine palmitoyltransferase II deficiency: successful anaplerotic diet therapy. Neurology. 2008 Jul 22;71(4):260–4. doi: 10.1212/01.wnl.0000318283.42961.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 75.Shahar E, Murphy EG, Roifman CM. Benefit of Intravenously Administered Immune Serum Globulin in Patients with Guillain-Barre Syndrome. J Pediatr. 1990;116:1, 141. doi: 10.1016/s0022-3476(05)81667-1. [DOI] [PubMed] [Google Scholar]
- 76.Shaw CE, Al-Chalabi A. Susceptibility genes in sporadic ALS: Separating the wheat from the chaff by international collaboration. Neurology. 2006;67:738–39. doi: 10.1212/01.wnl.0000238979.73142.cd. [DOI] [PubMed] [Google Scholar]
- 77.Sharma KR, Miller RG. Electrical and mechanical properties of skeletal muscle underlying increased fatigue in patients with amyotrophic lateral sclerosis. Muscle Nerve. 1996;19:1391–1400. doi: 10.1002/(SICI)1097-4598(199611)19:11<1391::AID-MUS3>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 78.Sigford B. Psychosocial, cognitive and educational issues in neuromuscular disease. Phys Med Rehabil Clinics N America. 1998 [PubMed] [Google Scholar]
- 79.Sinkeler SPT, Daanen HAM, Wevers RA, et al. The relation between blood lactate and ammonia in ischemic handgrip exercise. Muscle Nerve. 1985;8:523. doi: 10.1002/mus.880080608. [DOI] [PubMed] [Google Scholar]
- 80.Skalsky AJ, Han JJ, Abresch RT, McDonald CM. Regional and whole-body dual-energy X-ray absorptiometry to guide treatment and monitor disease progression in neuromuscular disease. Phys Med Rehabil Clin N Am. 2012 Feb;23(1):67–73. x. doi: 10.1016/j.pmr.2011.11.007. Epub 2011 Dec 11. Review. [DOI] [PubMed] [Google Scholar]
- 81.Skalsky AJ, McDonald CM. The prevention and management of limb contractures in progressive neuromuscular diseases. Phys Med Rehabil Clin North Am. doi: 10.1016/j.pmr.2012.06.009. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Skalsky AJ, Oskarsson B, Richman D. Current pharmacologic management in selected neuromuscular diseases. Phys Med Rehabil Clin North Am. doi: 10.1016/j.pmr.2012.09.003. this issue. [DOI] [PubMed] [Google Scholar]
- 83.Sobue I, Saito N, Iida M, Ando K. Juvenile type of distal and segmental muscular atrophy of the upper extremities. Ann Neurol. 1978;3:429–432. doi: 10.1002/ana.410030512. [DOI] [PubMed] [Google Scholar]
- 84.Suput D, Zupan A, Sepe A, Demsar F. Discrimination between neuropathy and myopathy by use of magnetic resonance imaging. Acta Neurologic Scandinavica. 1993;87(2):118–23. doi: 10.1111/j.1600-0404.1993.tb04089.x. [DOI] [PubMed] [Google Scholar]
- 85.Swoboda KJ, Prior TW, Scott CB, et al. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann Neurol. 2005;57(5):704–712. doi: 10.1002/ana.20473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Swash M, Brown MM, Thakkar C. CT muscle imaging and the clinical assessment of neuromuscular disease. Muscle and Nerve. 1995;18(7):708–14. doi: 10.1002/mus.880180706. [DOI] [PubMed] [Google Scholar]
- 87.Tekgul H, Serdaroglu G, Tutuncuoglu S. Outcome of axonal and demyelinating forms of Guillain-Barré syndrome in children. Pediatr Neurol. 2003 Apr;28(4):295–9. doi: 10.1016/s0887-8994(02)00626-4. [DOI] [PubMed] [Google Scholar]
- 88.Tomasová Studynková J, Charvát F, Jarosová K, et al. The role of MRI in the assessment of polymyositis and dermatomyositis. Rheumatology (Oxford) 2007 Jul;46(7):1174–1179. doi: 10.1093/rheumatology/kem088. [DOI] [PubMed] [Google Scholar]
- 89.Topalogu H, Gucuyener K, Yalaz K, et al. Selective involvement of the quadriceps muscle in congenital muscular dystrophies: an ultrasonographic study. Brain and Development. 1992;14:84–87. doi: 10.1016/s0387-7604(12)80091-x. [DOI] [PubMed] [Google Scholar]
- 90.Udd B, Meola G, Krahe R, et al. 140th ENMC International Workshop: Myotonic Dystrophy DM2/PROMM and other myotonic dystrophies with guidelines on management. Neuromuscul Disord. 2006 Jun;16(6):403–413. doi: 10.1016/j.nmd.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 91.Van den Hout JM, Kamphoven JH, Winkel LP, et al. Long-term intravenous treatment of Pompe disease with recombinant human alpha-glucosidase from milk. Pediatrics. 2004 May;113(5):e448–e457. doi: 10.1542/peds.113.5.e448. [DOI] [PubMed] [Google Scholar]
- 92.van Engelen BG, Eymard B, Wilcox D. Neuromuscul Disord; 123rd ENMC International Workshop: management and therapy in myotonic dystrophy; 6–8 February 2004; Naarden, The Netherlands. 2005. May, pp. 389–94. [DOI] [PubMed] [Google Scholar]
- 93.Verhagen WI, Huygen PL, Padberg GW. The auditory, vestibular and oculomotor system in facioscapulohumeral dystrophy. Acta Oto-Laryngologic (Suppl) 1995;520(Pt 1):140–2. doi: 10.3109/00016489509125212. [DOI] [PubMed] [Google Scholar]
- 94.Voit T, Krogmann O, Lennard HG, et al. Emery-Dreifuss Muscular Dystrophy: Disease Spectrum and Differential Diagnosis. Neuropediatrics. 1988;19:62. doi: 10.1055/s-2008-1052404. [DOI] [PubMed] [Google Scholar]
- 95.Von Recklinghausen H. Gliedermechanik and Lahmungsprothesen. Berlin: Springer-Verlag; 1920. [Google Scholar]
- 96.Wijesekera LC, Leigh PN. Amyotrophic lateral sclerosis. Orphanet J Rare Dis. 2009;4:3. doi: 10.1186/1750-1172-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Winkel LP, Van den Hout JM, Kamphoven JH, et al. Enzyme replacement therapy in late-onset Pompe’s disease: a three-year follow-up. Ann Neurol. 2004 Apr;55(4):495–502. doi: 10.1002/ana.20019. [DOI] [PubMed] [Google Scholar]
- 98.Young ID, Harper PS. Hereditary distal spinal muscular atrophy with vocal cord paralysis. J Neurol Neurosurg Psychiatry. 1980;43:413–418. doi: 10.1136/jnnp.43.5.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zaidman CM, Connolly AM, Malkus EC, Florence JM, Pestronk A. Quantitative ultrasound using backscatter analysis in Duchenne and Becker muscular dystrophy. Neuromuscul Disord. 2010 Dec;20(12):805–9. doi: 10.1016/j.nmd.2010.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zerres K, Rudnik-Schoneborn S. Natural History in Proximal Spinal Muscular Atrophy. Arch Neurol. 1995;52:518. doi: 10.1001/archneur.1995.00540290108025. [DOI] [PubMed] [Google Scholar]
- 101.Zerres K, Wirth B, Rudnik-Schöneborn S. Spinal muscular atrophy–clinical and genetic correlations. Neuromuscul Disord. 1997;7(3):202–207. doi: 10.1016/s0960-8966(97)00459-8. [DOI] [PubMed] [Google Scholar]