

Abstract
Uterine leiomyomata (UL), the most prevalent pelvic tumors in women of reproductive age, pose a major public health problem given their high frequency, associated morbidities, and most common indication for hysterectomies. A genetic component to UL predisposition is supported by analyses of ethnic predisposition, twin studies, and familial aggregation. A genome-wide SNP linkage panel was genotyped and analyzed in 261 white UL-affected sister-pair families from the Finding Genes for Fibroids study. Two significant linkage regions were detected in 10p11 (LOD = 4.15) and 3p21 (LOD = 3.73), and five additional linkage regions were identified with LOD scores > 2.00 in 2q37, 5p13, 11p15, 12q14, and 17q25. Genome-wide association studies were performed in two independent cohorts of white women, and a meta-analysis was conducted. One SNP (rs4247357) was identified with a p value (p = 3.05 × 10−8) that reached genome-wide significance (odds ratio = 1.299). The candidate SNP is under a linkage peak and in a block of linkage disequilibrium in 17q25.3, which spans fatty acid synthase (FASN), coiled-coil-domain-containing 57 (CCDC57), and solute-carrier family 16, member 3 (SLC16A3). By tissue microarray immunohistochemistry, we found elevated (3-fold) FAS levels in UL-affected tissue compared to matched myometrial tissue. FAS transcripts and/or protein levels are upregulated in various neoplasms and implicated in tumor cell survival. FASN represents the initial UL risk allele identified in white women by a genome-wide, unbiased approach and opens a path to management and potential therapeutic intervention.
Introduction
Uterine leiomyomata (UL), commonly known as fibroids, are benign tumors of the uterine myometrium. They represent the most prevalent pelvic tumors in women and are found in more than 75% of women of reproductive age.1 Approximately 20%–25% of women with UL exhibit symptoms including menorrhagia, infertility, pelvic pain, and a range of complications during pregnancy.2 The leading cause for hysterectomy in the United States, UL account for >30% of all hysterectomies and >40% of hysterectomies among women aged 45–64 years.3 Annual health-care costs of UL are estimated at over two billion dollars, most of which is associated with hysterectomies.4 Although UL pose a major public health problem, little is known about the molecular basis for these tumors, and treatment options are limited.
Genes involved in UL have been discovered by cytogenetic analysis. Approximately 40% of UL have a nonrandom cytogenetic aberration, and several subgroups are recognized and include t(12;14)(q14-15;q23-24), del(7)(q22q32), trisomy 12, rearrangements involving 6p21 and 10q22, and deletions of 1p and 3q.5,6 Cytogenetic abnormalities have been correlated with tumor size, location, and histology, which indicates that genetic events play a fundamental role in UL biology.5,7,8 Cytogenetic heterogeneity of UL underlies phenotypic differences and supports involvement of different pathways in tumor development.
Several factors predispose women to developing UL. Age, obesity, parity, and race have all been associated with prevalence of UL. Black women are disproportionately affected by UL9—the incidence and prevalence rates for these women are at least three times greater than those for white women even after other known risk factors are controlled.10 Further, analyses of twin studies and familial aggregation indicate a genetic component to UL predisposition; first-degree relatives of affected women have a 2.5-fold higher risk of developing UL, and monozygotic twins’ concordance for UL diagnosis is almost twice that of dizygotic twins.11,12 Similarly, a study of a Finnish cohort found that monozygotic twins’ concordance for being hospitalized for UL was twice that of dizygotic twins.13 These findings support a genetic predisposition to developing UL, but no genome-wide study of UL in white women has been reported. Several candidate-gene association studies have been performed with limited success, although variants in the 5′ UTR of HMGA2 (MIM 600698), a gene involved in recurrent cytogenetic aberrations of UL and known to play a primary role,14 have been associated with UL diagnosis in a cohort of white sister pairs.15 Finding additional pathogenetic sequences that predispose women to UL will provide insight into tumor development and could lead to screening strategies or improved management and therapy.
Subjects and Methods
Finding Genes for Fibroid Linkage Analysis
Sister pairs affected by UL were recruited for the “Finding Genes for Fibroids” (FGFF) study. Approximately 385 sister pairs consented to this project. Both sisters in each pair had medical-record-confirmed UL, provided a blood sample, and completed a questionnaire on clinical, reproductive, sexual, and family history relating to UL. Other family members of the sisters also contributed samples and completed questionnaires. Study participants were recruited under an institutional-review-board (IRB) protocol approved by the Partners HealthCare System Human Research Committee. DNA was isolated with a Puregene Blood Kit (Gentra, Minneapolis, MN), and the DNA, pedigree information, and UL affection status were provided to the genotyping core at The Center for Inherited Disease Research at Johns Hopkins University. A whole-genome SNP linkage scan was performed with Illumina’s Human Linkage-12 Genotyping BeadChip (San Diego, CA). In addition to 14 SNP markers, two families affected by multiple Mendelian inconsistencies were excluded because of low-quality genotype calls.
Linkage analysis was performed with Genehunter software with samples from self-reported white sister pairs and family members, which composed 261 families for a total of 1,103 individuals. The minor allele frequency (MAF) of each SNP was calculated with the genotype information from one sister in each family. SNPs were pruned with PLINK software16 on the basis of an r2 of 0.2, resulting in a total of 4,196 SNPs for the final analysis. Sibling-pair analysis was carried out with UL status as the phenotype, and significant linkage with UL was defined as a LOD score greater than 3.6. Despite particular interest in recruitment of black women, the FGFF study has not yet reached an appropriate number of sister pairs for this important analysis.
Women’s Genome Health Study Association Study
Study participants were recruited under an IRB protocol approved by the Partners HealthCare System Human Research Committee. The Women’s Genome Health Study (WGHS)17 is a prospective cohort of female North American health-care professionals representing Women’s Health Study (WHS) participants who provided a blood sample at baseline and consent for blood-based analyses. Participants in the WHS were 45 years of age or older at enrollment and free of cardiovascular disease, cancer, or other major chronic illness. Additional information related to health and lifestyle was collected by questionnaires throughout the WHS trial and by continual observational follow up. WHS participants were asked whether they had ever been diagnosed with UL, their age at diagnosis, whether their mother or sister had ever been diagnosed with UL, and their history of hysterectomy.
Genotyping in the WGHS sample was performed with the HumanHap300 Duo “+” chips or the combination of the HumanHap300 Duo and iSelect chips (Illumina, San Diego, CA) according to the Infinium II protocol. In either case, the custom SNP content was the same; these custom SNPs were chosen without regard for MAF for saturating candidate genes for cardiovascular disease, as well as for increasing coverage of SNPs with known or suspected biological function, e.g., disease association, nonsynonymous changes, substitutions at splice sites, etc. For quality control, all samples were required to have successful genotyping with BeadStudio v.3.3 software (Illumina) for at least 98% of the SNPs. A subset of 23,294 individuals was identified to have self-reported European ancestry, verified on the basis of multidimensional scaling analysis of identity by state with the use of 1,443 ancestry informative markers in PLINK v.1.06.16 The final data set of these individuals included SNPs with a MAF > 1%, successful genotyping in 90% of subjects, and deviations not exceeding p = 10−6 from Hardy-Weinberg equilibrium. Among the final 23,294 individuals of verified European ancestry, genotypes for a total of 2,608,509 SNPs were imputed from the experimental genotypes and linkage disequilibrium (LD) relationships implicit in the HapMap r.22 CEU (Utah residents with ancestry from northern and western Europe from the CEPH collection) samples. Imputed SNPs were used for defining the LD region surrounding association signals found with genotyped SNPs.
UL status, age at diagnosis, mother or sister UL status, and history of hysterectomy were ascertained by recall in the 2009 WGHS questionnaire. UL cases and controls from the WGHS were stratified on the basis of these four variables for identifying women most and least likely to have a genetic basis for UL. Any participant who answered “not sure” for UL status or mother or sister UL status was excluded from the analysis. Participants who reported an age of UL diagnosis to be under 20 years or over 70 years were also excluded. Cases included women who answered “yes” for UL status and “yes” for mother or sister UL status and who either had an age of diagnosis under 40 years or had a hysterectomy. Controls included women who answered “no” to UL status and “no” to mother or sister UL status and who had not had a hysterectomy. Women not qualifying for either of these groups were excluded from the analysis. After stratification, there were 746 cases and 4,487 controls. Association analysis was performed on the set of 339,187 genotyped SNPs in PLINK with the standard case-control test, and p values less than 5 × 10−8 were considered significant.
Australian Cohort Association Study
Individuals composing the cohort from the Queensland Institute of Medical Research (QIMR) were women who had given consent and who had been genotyped previously on Illumina’s 317K, 370K, or 610K SNP platforms as part of a larger collection of genome-wide association studies conducted at the QIMR.18,19 Case samples (n = 484) were selected from among women originally recruited into a study of genetic factors underlying endometriosis20 and a twin study of gynecological health.21 For both studies, women completed questionnaires on various aspects of reproductive health, and cases answered “yes” to the “uterine fibroids” option of the question “Have you ever had any of the following conditions?” Controls (n = 610) were taken from among twin pairs from the gynecological health study in which both sisters answered “no” to the question on uterine fibroids (one sample per twin pair). A standard case-control association analysis was performed on the set of 269,629 SNPs genotyped in common between all samples and passing all quality-control metrics in PLINK.16 Approval for the studies was granted by the Human Research Ethics Committee at the QIMR and the Australian Twin Registry. All gene annotations and base-pair positions are derived from the human genome sequence hg18 (NCBI build 36.1).
Demographics
Demographics of the FGFF, WGHS, and Australian cohorts were considered with a focus on variables most relevant to UL diagnosis. In the WGHS and Australian cohorts, cases had a slightly higher body mass index (BMI) and lower stature than did controls, although the differences were not significant in the WGHS population (defined as a p > 0.05) (Table S1, available online). Previous studies also reported a correlation between UL diagnosis and a higher BMI.22,23 WGHS and Australian cases also had a younger age at menarche, which could be expected because UL are hormone-dependent neoplasms and because a younger age at menarche is associated with longer reproductive years and additional years of hormone exposure. Lastly, although Australian cases were not selected on the basis of a history of hysterectomy, UL cases had a very significant increase in hysterectomy prevalence, reflecting the fact that UL are the leading cause for this surgery. Overall, demographics of the WGHS and Australian cohorts were unremarkable and confirmed previously reported UL associations.
FAS Immunohistochemistry
Fatty-acid synthase (FAS [MIM 600212]) levels in UL and myometrium tissue were analyzed by immunohistochemistry of tissue microarrays (TMAs) consisting of tissue sections from 200 women: 36 myometrium samples and 337 UL samples. The TMA includes matched paraffin-embedded formalin-fixed myometrium and UL samples from 33 women. Two TMAs were constructed with sections from different areas of the same tissue. DNA corresponding to 106 tissue sections was isolated and used for genotyping rs4247357 in CCDC57. Immunostaining was performed on both TMAs with the primary monoclonal antibody against FAS (Transduction Laboratories, Lexington, KY) at a 1:100 dilution and with hematoxylin as a counterstain. Each core was evaluated for the ratio of stain to counterstain for accounting for variable cellularity in tissue sections. The average stain-to-counterstain ratio was determined and compared across myometrium and UL samples and also across samples with the major and minor allele of rs4247357. Errors bars and statistical differences were determined by standard error.
mRNA-Expression Analysis by Microarray
RNA was isolated from myometrium tissue and 28 UL from 14 women. Gene-expression levels were analyzed with the Affymetrix GeneChip system U133 plus 2.0. DNA was also extracted and used for genotyping rs4247357 in CCDC57. Gene-expression levels were compared between women with the major and minor allele of rs4247357.
Targeted mRNA-Expression Analysis by Quantitative PCR
RNA was isolated from myometrium tissue and 20 tumors from 12 women, 6 of whom had the major allele of rs4247357 and 6 of whom had the minor allele. cDNA was synthesized and used in quantitative PCR (qPCR) assessments of FASN, CCDC57, SLC16A3 (MIM 603877), DUS1L, CSNK1D (MIM 600864), and NARF (MIM 605349). Gene products were normalized to the internal reference gene, GAPDH, and averaged normalized ratios were compared across myometrium and UL samples and across women with the major and minor allele of rs4247357. Error bars and statistical differences were determined by standard error.
Results
Linkage Study
Linkage analysis with the FGFF population revealed two peaks with highly significant (>3.6) genome-wide LOD scores and five peaks with suggestive (>2.0) LOD scores (Figure 1). The highest LOD scores found were at 10p11 (LOD = 4.15) and 3p21 (LOD = 3.73). Both linkage regions are around 35 Mb and contain hundreds of genes. Of note, HMGA2 resides within the linkage region at 12q14 and has a suggestive LOD score of 2.62 (Figure S1).
Figure 1.
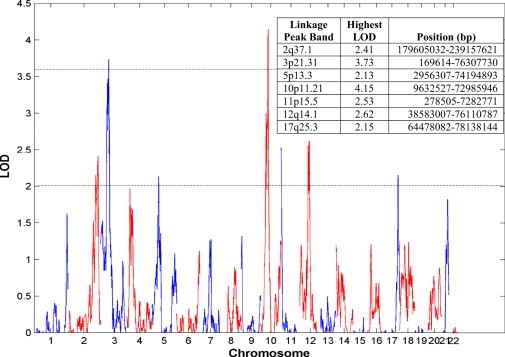
Summary of FGFF Sibling-Pair Linkage Analysis by Chromosome
Two peaks have significant (>3.6) LOD scores, and five peaks have suggestive (>2.0) LOD scores. Linkage-peak boundaries and the highest LOD score found under each peak are defined in the inserted table.
Association Analyses and Meta-analysis
Genome-wide association studies were undertaken with two independent cohorts of white women—the WGHS cohort and an Australian cohort. Analysis of the WGHS cohort revealed 45 SNPs with p values less than 10−4 (Table S2). Although none of the p values from this analysis are considered significant for identifying a genome-wide association, the quantile-quantile plot of the results provides evidence that there are more SNPs with small p values than are expected by chance (Figure S2A). In the Australian analysis, 25 SNPs were identified with p values less than 10−4, and the quantile-quantile plot reveals a small increase in low p values, although it is less striking than the WGHS results (Table S3 and Figure S2B). A meta-analysis was performed on the set of 344,655 genotyped SNPs from the WGHS and Australian cohorts with the use of an inverse-variance weighted method in METAL. One SNP, rs4247357, reached genome-wide significance and is considered significantly associated with UL status (Table 1). Five additional SNPs in the same location on chromosome 17 were identified with p values less than 10−6. The quantile-quantile plot of the meta-analysis results clearly shows that this group of SNPs has lower p values than what would be expected under the null hypothesis (Figure 2). The candidate SNPs on chromosome 17 are located in a large LD block, which contains three genes, fatty-acid synthase (FASN), coiled-coil-domain-containing 57 (CCDC57), and solute carrier family 16, member 3 (SLC16A3) (Figure 3 and Figure S3). Interestingly, this LD block lies under the FGFF linkage peak at 17q25 (Figure S4).
Table 1.
The Top SNP from Meta-analysis Results of WGHS and Australian Cohorts
| SNP | Locus Position (Gene) | Cohort | MAF | OR (95% CI) | p Value |
|---|---|---|---|---|---|
| rs4247357 | 17q25.3 | WGHS | 0.467 | 1.307 (1.170–1.459) | 1.95 × 10−6 |
| 77,760,277 | Australian | 0.432 | 1.282 (1.079–1.523) | 4.49 × 10−3 | |
| (CCDC57) | meta-analysis | − | 1.299 (1.184–1.426) | 3.05 × 10−8 |
The following abbreviations are used: MAF, minor allele frequency; OR, odds ratio; and CI, confidence interval.
Figure 2.
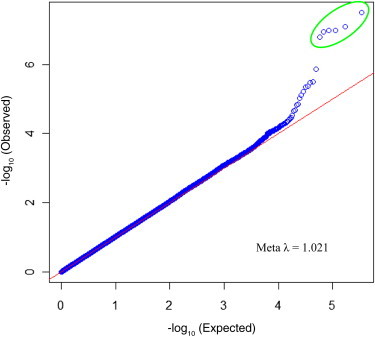
Quantile-Quantile Plot of Meta-analysis Results
The p values observed in the study are compared to p values expected under the null hypothesis. Top SNPs in the 17q25.3 region are circled in green.
Figure 3.
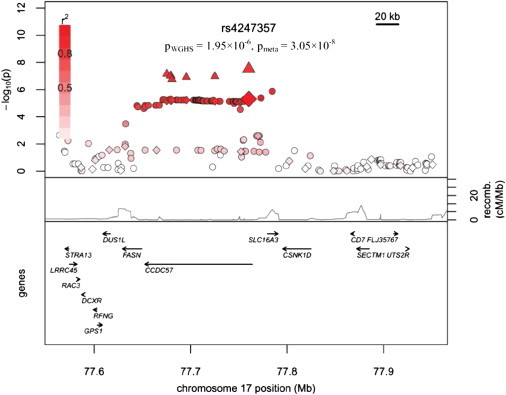
Candidate Region on Chromosome 17 Contains Significantly Associated Markers
The p values for WGHS genotyped SNPs are indicated by diamonds, the p values for WGHS imputed SNPs are indicated by circles, and the p values for SNPs included in the meta-analysis are indicated by triangles.
FAS Protein Levels
Little is known about CCDC57 or SLC16A3; however, FAS has been associated with several cancers and is often upregulated in cancer tissue.24 In order to investigate this finding in UL, we stained UL and myometrial tissue with a FAS antibody. FAS immunostaining revealed that FAS levels in UL were three times higher than those in matched, normal myometrium tissue (Figure 4). An increase in UL FAS was seen in 25 out of 33 (∼76%) matched samples (Figure S5). Stratifying matched samples by rs4247357 genotype showed an increase in FAS levels in myometrium tissue and UL with the minor (effect) allele compared to those with the major allele. Increased expression in myometrium samples with the minor allele was not substantial, but the increase in UL with the minor allele was about 2-fold (Figure 5A). When all UL samples were combined, the same pattern emerged, but the increase in FAS in UL with the minor allele was about 50% higher than in UL with the major allele (Figure 5B). Interestingly, in the matched analysis, FAS levels in both myometrium and UL samples heterozygous for major and minor alleles were between those of samples homozygous for either the major or minor alleles.
Figure 4.
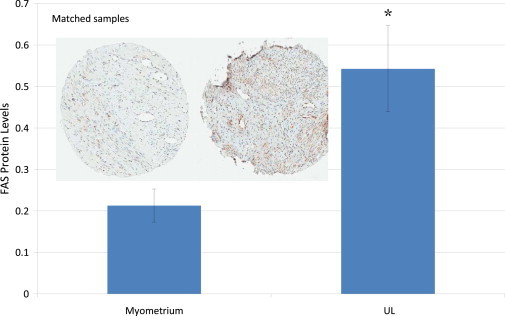
FAS levels in Myometrium Tissue and UL from Matched Samples
Representative tissue cores are shown from myometrium tissue (left) and UL (right). A significant increase compared to myometrium was determined by a t test (∗p = 0.004). The error bars were determined by standard error.
Figure 5.
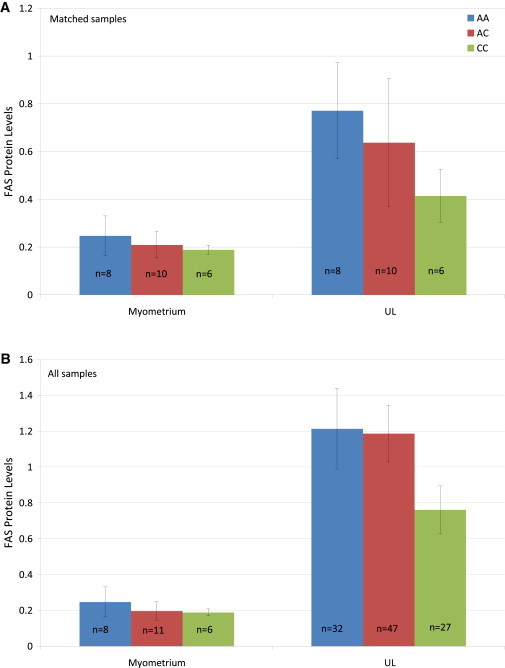
FAS Levels in Myometrium Tissue and UL from Matched Samples and All Samples Stratified by the rs4247357 Genotype
Matched samples (A) and all samples stratified by the rs4247357 genotype (B). The error bars were determined by standard error.
mRNA Expression
Analysis of mRNA-expression levels in myometrium tissue and UL by microarray of genes under the linkage peak on chromosome 17 revealed no genes with significant differential expression between women with the major (n = 3) and minor alleles (n = 6) of rs4247357. FASN, CCDC57, and SLC16A3 were found not to be expressed at a detectable level in the myometrium and UL samples analyzed. However, a more comprehensive analysis of 54,000 probes across the genome revealed that several genes were significantly more upregulated and downregulated in UL with the minor allele than in UL with the major allele (Table S4).
Targeted mRNA-expression analysis by qPCR detected no substantial expression differences in matched myometrium and UL samples for FASN, CCDC57, SLC16A3, and three genes (DUS1L, CSNK1D, and NARF) located directly nearby but outside of the candidate LD block (Figure S6A). Expression of FASN, CCDC57, and SLC16A3 was slightly higher in the matched UL samples, but expression of DUS1L and NARF was also higher in the matched UL samples, and the variation between samples was relatively large. It is unclear whether this expression difference has biological significance; however, it is clear that mRNA expression of FASN between myometrium and matched UL samples is not concordant with FAS levels because the same samples were used in both studies. This finding is not surprising because the correlation between mRNA and protein levels can be poor—some studies find that only 40% of variation in protein expression can be explained by mRNA expression.25 Also, expression of all six genes was higher in myometrium tissue and UL with the major allele than in those with the minor allele (Figures S6B and S6C). Similarly, it is unclear whether this difference is of relevance because it is relatively small and is observed for every gene analyzed and because the variation between samples is relatively large.
Discussion
The FGFF linkage study provides evidence of a genetic contribution to UL development, as seen by two significant linkage peaks and several other suggestive peaks. Although candidate genes have yet to be identified under five peaks, one gene (HMGA2) prominently recognized in UL biology is located under the peak on 12q14. Translocations involving HMGA2 are frequently found in UL tumors.14 Additionally, a recent study using the FGFF population found a significant association between UL status and a variant in the 5′ UTR of HMGA2.15 The discovery of several linkage peaks illustrates the genetic heterogeneity of these tumors and is consistent with the hypothesis that a variety of genes and pathways participate in UL development. Additionally, a genome-wide association study in a Japanese cohort found three loci (10q24.33, 22q13.1, and 11p15.5) significantly associated with UL diagnosis.26 These loci are not associated with UL in our cohorts of white women, and our locus at 17q25.3 was not identified in the Japanese study, supporting genetic heterogeneity in UL predisposition between ethnic groups.
Association analyses using the WGHS and Australian cohorts might have identified more precisely the locus underlying the linkage signal on chromosome 17. Six SNPs on chromosome 17 were in the top associated SNPs in the WGHS analysis and proved to be significantly associated with UL status after meta-analysis of the WGHS and Australian cohorts. At this time, it is unknown whether any of these markers are the causal SNP associated with UL affection status or whether they are in LD with the causal variant; however, these data show that the MAFs of the candidate SNPs are significantly higher in women with UL. The candidate SNPs are in LD across FASN, CCDC57, and SLC16A3. SLC16A3 is a member of the proton-linked monocarboxylate transporter family, which facilitates transport of substrates across the plasma membrane. CCDC57 contains a coiled-coil domain and might function by binding DNA. SLC16A3 and CCDC57 are minimally characterized, making it difficult to conjecture about their possible involvement in UL development. None of these genes or proteins has been indicated in disease, and our qPCR studies did not reveal any unusual mRNA expression in UL. Conversely, we found that FAS levels were much higher in UL than in matched myometrial tissue, as well as in myometrium and UL from women with the minor (effect) allele than in those from women with the major allele. FAS has been extensively characterized and is the enzyme responsible for de novo fatty-acid synthesis. It is most highly expressed in hormone-sensitive cells27 and has been found to be regulated at both transcriptional and posttranscriptional levels. Sterol-regulatory-element-binding transcription factor 1 (SREBP-1 [MIM 184756]) is the primary transcription factor of FASN and is activated downstream of growth-factor and hormone receptors.28 FAS is stabilized by USP2a, an isopeptidase that deubiquitinates and prevents protein degradation. USP2a is overexpressed in some prostate tumors with high FAS expression29 and could explain discordant levels of FASN mRNA and FAS levels in our UL samples.
Upregulation of FAS has been discovered in many cancers, including prostate, breast, and colon cancers.30–32 In some cancers, upregulation of FAS is correlated with poor prognosis, cancer progression, or specific tumor types.33 Inhibitors of FAS lead to growth arrest and apoptosis in cancer cell lines and have relatively minor effects in corresponding normal cells.34,35 Many studies have found a connection between FAS and the PI3K/Akt signaling pathway, one of the most frequently dysregulated pathways in human cancers.36,37 FAS inhibitors used in breast cancer animal models and several xenograft models have resulted in delayed development and slowed tumor progression.38,39 Knocking down FASN mRNA causes a host of changes in gene expression and protein activity in the cell,40 making it clear from these and many more studies that the role of FAS in neoplasia is much more complicated and probably more important than simply providing fatty acids. Although it remains to be known how the minor allele of the candidate LD block influences FASN and UL development, upregulation of FAS in these UL samples and the overwhelming evidence that FAS is a metabolic oncogene make it a compelling candidate gene for UL development, for which clinical trials with inhibitors might be warranted.
Acknowledgments
The authors thank all of the women and their families who participated in the Finding Genes for Fibroids (FGFF) study, the Women’s Genome Health Study (WGHS), and the Australian study. The FGFF study is supported by National Institute of Child Health & Human Development grants HD046226 and HD060530 (to C.C.M.). The WGHS is supported by HL043851 and HL69757 from the National Heart, Lung, and Blood Institute and CA047988 from the National Cancer Institute, the Donald W. Reynolds Foundation, and the Fondation Leducq; collaborative scientific support and funding for genotyping was provided by Amgen. The Australian cohort was supported by NIH Grants (AA07535, AA07728, AA13320, AA13321, AA14041, AA11998, AA17688, DA012854, and DA019951); by grants from the Australian National Health and Medical Research Council (339462, 389927, 389938, 442981, 443036, and 496739); by the 5th Framework Programme (FP-5) GenomEUtwin Project (QLG2-CT-2002-01254); by the Wellcome Trust (WT085235/Z/08/Z); and by funding provided by the Cooperative Research Centre for Discovery of Genes for Common Human Diseases, Cerylid Biosciences (Melbourne). G.W.M., S.E.M., and D.R.N. are supported by the National Health and Medical Research Council Fellowship Scheme, and K.T.Z. is supported by a Wellcome Trust Research Career Development Fellowship. Genotyping services for the FGFF study were provided by the Center for Inherited Disease Research (CIDR). The CIDR is funded through a federal contract from the NIH to The Johns Hopkins University (contract number HHSN268200782096C).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
METAL, http://www.sph.umich.edu/csg/abecasis/Metal/index.html
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
UCSC Human Genome Browser, http://genome.ucsc.edu
References
- 1.Cramer S.F., Patel A. The frequency of uterine leiomyomas. Am. J. Clin. Pathol. 1990;94:435–438. doi: 10.1093/ajcp/94.4.435. [DOI] [PubMed] [Google Scholar]
- 2.Buttram V.C., Jr., Reiter R.C. Uterine leiomyomata: Etiology, symptomatology, and management. Fertil. Steril. 1981;36:433–445. doi: 10.1016/s0015-0282(16)45789-4. [DOI] [PubMed] [Google Scholar]
- 3.Lepine L.A., Hillis S.D., Marchbanks P.A., Koonin L.M., Morrow B., Kieke B.A., Wilcox L.S. Hysterectomy surveillance—United States, 1980-1993. MMWR CDC Surveill. Summ. 1997;46:1–15. [PubMed] [Google Scholar]
- 4.Flynn M., Jamison M., Datta S., Myers E. Health care resource use for uterine fibroid tumors in the United States. Am. J. Obstet. Gynecol. 2006;195:955–964. doi: 10.1016/j.ajog.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 5.Rein M.S., Friedman A.J., Barbieri R.L., Pavelka K., Fletcher J.A., Morton C.C. Cytogenetic abnormalities in uterine leiomyomata. Obstet. Gynecol. 1991;77:923–926. [PubMed] [Google Scholar]
- 6.Gross K.L., Morton C.C. Genetics and the development of fibroids. Clin. Obstet. Gynecol. 2001;44:335–349. doi: 10.1097/00003081-200106000-00020. [DOI] [PubMed] [Google Scholar]
- 7.Brosens I., Deprest J., Dal Cin P., Van den Berghe H. Clinical significance of cytogenetic abnormalities in uterine myomas. Fertil. Steril. 1998;69:232–235. doi: 10.1016/s0015-0282(97)00472-x. [DOI] [PubMed] [Google Scholar]
- 8.Christacos N.C., Quade B.J., Dal Cin P., Morton C.C. Uterine leiomyomata with deletions of Ip represent a distinct cytogenetic subgroup associated with unusual histologic features. Genes Chromosomes Cancer. 2006;45:304–312. doi: 10.1002/gcc.20291. [DOI] [PubMed] [Google Scholar]
- 9.Huyck K.L., Panhuysen C.I., Cuenco K.T., Zhang J., Goldhammer H., Jones E.S., Somasundaram P., Lynch A.M., Harlow B.L., Lee H. The impact of race as a risk factor for symptom severity and age at diagnosis of uterine leiomyomata among affected sisters. Am J Obstet Gynecol. 2008;198:168.e1–168.e9. doi: 10.1016/j.ajog.2007.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marshall L.M., Spiegelman D., Barbieri R.L., Goldman M.B., Manson J.E., Colditz G.A., Willett W.C., Hunter D.J. Variation in the incidence of uterine leiomyoma among premenopausal women by age and race. Obstet. Gynecol. 1997;90:967–973. doi: 10.1016/s0029-7844(97)00534-6. [DOI] [PubMed] [Google Scholar]
- 11.Vikhlyaeva E.M., Khodzhaeva Z.S., Fantschenko N.D. Familial predisposition to uterine leiomyomas. Int. J. Gynaecol. Obstet. 1995;51:127–131. doi: 10.1016/0020-7292(95)02533-i. [DOI] [PubMed] [Google Scholar]
- 12.Treloar S.A., Do K.A., Martin N.G. Genetic influences on the age at menopause. Lancet. 1998;352:1084–1085. doi: 10.1016/S0140-6736(05)79753-1. [DOI] [PubMed] [Google Scholar]
- 13.Luoto R., Kaprio J., Rutanen E.M., Taipale P., Perola M., Koskenvuo M. Heritability and risk factors of uterine fibroids—the Finnish Twin Cohort study. Maturitas. 2000;37:15–26. doi: 10.1016/s0378-5122(00)00160-2. [DOI] [PubMed] [Google Scholar]
- 14.Schoenberg Fejzo M., Ashar H.R., Krauter K.S., Powell W.L., Rein M.S., Weremowicz S., Yoon S.J., Kucherlapati R.S., Chada K., Morton C.C. Translocation breakpoints upstream of the HMGIC gene in uterine leiomyomata suggest dysregulation of this gene by a mechanism different from that in lipomas. Genes Chromosomes Cancer. 1996;17:1–6. doi: 10.1002/(SICI)1098-2264(199609)17:1<1::AID-GCC1>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 15.Hodge J.C., T Cuenco K., Huyck K.L., Somasundaram P., Panhuysen C.I., Stewart E.A., Morton C.C. Uterine leiomyomata and decreased height: A common HMGA2 predisposition allele. Hum. Genet. 2009;125:257–263. doi: 10.1007/s00439-008-0621-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ridker P.M., Chasman D.I., Zee R.Y., Parker A., Rose L., Cook N.R., Buring J.E., Women’s Genome Health Study Working Group Rationale, design, and methodology of the Women’s Genome Health Study: a genome-wide association study of more than 25,000 initially healthy american women. Clin. Chem. 2008;54:249–255. doi: 10.1373/clinchem.2007.099366. [DOI] [PubMed] [Google Scholar]
- 18.Medland S.E., Nyholt D.R., Painter J.N., McEvoy B.P., McRae A.F., Zhu G., Gordon S.D., Ferreira M.A., Wright M.J., Henders A.K. Common variants in the trichohyalin gene are associated with straight hair in Europeans. Am. J. Hum. Genet. 2009;85:750–755. doi: 10.1016/j.ajhg.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Painter J.N., Anderson C.A., Nyholt D.R., Macgregor S., Lin J., Lee S.H., Lambert A., Zhao Z.Z., Roseman F., Guo Q. Genome-wide association study identifies a locus at 7p15.2 associated with endometriosis. Nat. Genet. 2011;43:51–54. doi: 10.1038/ng.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Treloar S.A., Wicks J., Nyholt D.R., Montgomery G.W., Bahlo M., Smith V., Dawson G., Mackay I.J., Weeks D.E., Bennett S.T. Genomewide linkage study in 1,176 affected sister pair families identifies a significant susceptibility locus for endometriosis on chromosome 10q26. Am. J. Hum. Genet. 2005;77:365–376. doi: 10.1086/432960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Treloar S.A., Do K.A., O’Connor V.M., O’Connor D.T., Yeo M.A., Martin N.G. Predictors of hysterectomy: An Australian study. Am. J. Obstet. Gynecol. 1999;180:945–954. doi: 10.1016/s0002-9378(99)70666-6. [DOI] [PubMed] [Google Scholar]
- 22.Kharazmi E., Fallah M., Luoto R. Cardiovascular diseases attributable to hysterectomy: A population-based study. Acta Obstet. Gynecol. Scand. 2007;86:1476–1483. doi: 10.1080/00016340701698633. [DOI] [PubMed] [Google Scholar]
- 23.Takeda T., Sakata M., Isobe A., Miyake A., Nishimoto F., Ota Y., Kamiura S., Kimura T. Relationship between metabolic syndrome and uterine leiomyomas: A case-control study. Gynecol. Obstet. Invest. 2008;66:14–17. doi: 10.1159/000114250. [DOI] [PubMed] [Google Scholar]
- 24.Menendez J.A., Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer. 2007;7:763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- 25.de Sousa Abreu R., Penalva L.O., Marcotte E.M., Vogel C. Global signatures of protein and mRNA expression levels. Mol. Biosyst. 2009;5:1512–1526. doi: 10.1039/b908315d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cha P.C., Takahashi A., Hosono N., Low S.K., Kamatani N., Kubo M., Nakamura Y. A genome-wide association study identifies three loci associated with susceptibility to uterine fibroids. Nat. Genet. 2011;43:447–450. doi: 10.1038/ng.805. [DOI] [PubMed] [Google Scholar]
- 27.Kusakabe T., Maeda M., Hoshi N., Sugino T., Watanabe K., Fukuda T., Suzuki T. Fatty acid synthase is expressed mainly in adult hormone-sensitive cells or cells with high lipid metabolism and in proliferating fetal cells. J. Histochem. Cytochem. 2000;48:613–622. doi: 10.1177/002215540004800505. [DOI] [PubMed] [Google Scholar]
- 28.Eberlé D., Hegarty B., Bossard P., Ferré P., Foufelle F. SREBP transcription factors: Master regulators of lipid homeostasis. Biochimie. 2004;86:839–848. doi: 10.1016/j.biochi.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 29.Graner E., Tang D., Rossi S., Baron A., Migita T., Weinstein L.J., Lechpammer M., Huesken D., Zimmermann J., Signoretti S., Loda M. The isopeptidase USP2a regulates the stability of fatty acid synthase in prostate cancer. Cancer Cell. 2004;5:253–261. doi: 10.1016/s1535-6108(04)00055-8. [DOI] [PubMed] [Google Scholar]
- 30.Alò P.L., Visca P., Trombetta G., Mangoni A., Lenti L., Monaco S., Botti C., Serpieri D.E., Di Tondo U. Fatty acid synthase (FAS) predictive strength in poorly differentiated early breast carcinomas. Tumori. 1999;85:35–40. doi: 10.1177/030089169908500108. [DOI] [PubMed] [Google Scholar]
- 31.Rossi S., Graner E., Febbo P., Weinstein L., Bhattacharya N., Onody T., Bubley G., Balk S., Loda M. Fatty acid synthase expression defines distinct molecular signatures in prostate cancer. Mol. Cancer Res. 2003;1:707–715. [PubMed] [Google Scholar]
- 32.Ogino S., Nosho K., Meyerhardt J.A., Kirkner G.J., Chan A.T., Kawasaki T., Giovannucci E.L., Loda M., Fuchs C.S. Cohort study of fatty acid synthase expression and patient survival in colon cancer. J. Clin. Oncol. 2008;26:5713–5720. doi: 10.1200/JCO.2008.18.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu H., Liu J.Y., Wu X., Zhang J.T. Biochemistry, molecular biology, and pharmacology of fatty acid synthase, an emerging therapeutic target and diagnosis/prognosis marker. Int J Biochem Mol Biol. 2010;1:69–89. [PMC free article] [PubMed] [Google Scholar]
- 34.Pizer E.S., Jackisch C., Wood F.D., Pasternack G.R., Davidson N.E., Kuhajda F.P. Inhibition of fatty acid synthesis induces programmed cell death in human breast cancer cells. Cancer Res. 1996;56:2745–2747. [PubMed] [Google Scholar]
- 35.Pizer E.S., Chrest F.J., DiGiuseppe J.A., Han W.F. Pharmacological inhibitors of mammalian fatty acid synthase suppress DNA replication and induce apoptosis in tumor cell lines. Cancer Res. 1998;58:4611–4615. [PubMed] [Google Scholar]
- 36.Van de Sande T., De Schrijver E., Heyns W., Verhoeven G., Swinnen J.V. Role of the phosphatidylinositol 3′-kinase/PTEN/Akt kinase pathway in the overexpression of fatty acid synthase in LNCaP prostate cancer cells. Cancer Res. 2002;62:642–646. [PubMed] [Google Scholar]
- 37.Porstmann T., Griffiths B., Chung Y.L., Delpuech O., Griffiths J.R., Downward J., Schulze A. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene. 2005;24:6465–6481. doi: 10.1038/sj.onc.1208802. [DOI] [PubMed] [Google Scholar]
- 38.Alli P.M., Pinn M.L., Jaffee E.M., McFadden J.M., Kuhajda F.P. Fatty acid synthase inhibitors are chemopreventive for mammary cancer in neu-N transgenic mice. Oncogene. 2005;24:39–46. doi: 10.1038/sj.onc.1208174. [DOI] [PubMed] [Google Scholar]
- 39.Lupu R., Menendez J.A. Pharmacological inhibitors of Fatty Acid Synthase (FASN)—Catalyzed endogenous fatty acid biogenesis: a new family of anti-cancer agents? Curr. Pharm. Biotechnol. 2006;7:483–493. doi: 10.2174/138920106779116928. [DOI] [PubMed] [Google Scholar]
- 40.Knowles L.M., Smith J.W. Genome-wide changes accompanying knockdown of fatty acid synthase in breast cancer. BMC Genomics. 2007;8:168. doi: 10.1186/1471-2164-8-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.