

To the Editor: In their report, Christoforou et al.1 demonstrate the effect that linkage-disequilibrium (LD)-based binning has on the interpretation of genome-wide association studies (GWASs) and conclude that “ignoring LD can result in the misinterpretation of the GWAS findings and have an impact on subsequent genetic and functional studies.” Although this conclusion is true and trivial, we argue that their proposed LD-based binning approach uses the LD information incorrectly and will lead to increased type 1 error (resulting in the misinterpretation of GWAS findings) and will hence have a negative impact on subsequent genetic and functional studies. The LD-based binning approach assigns SNPs to genes or bins by using pairwise LD data calculated from reference data, such as that from the 1000 Genomes Project or HapMap or other user-provided data. It can assign a SNP to more than one gene. After the bins have been defined, standard gene-based approaches, such as taking the minimum SNP p value in a bin after the application of a modified Sidak’s correction,2 are used. Thus, the essence of this method is to include as “hits” not only those genes in (or around) which extreme p values for SNPs are found but also those genes that include SNPs found to be in significant LD with them. This approach will result in increased correlations among genes because a SNP’s p value can be repeatedly represented in different genes.
Christoforou et al.1 assessed their method of LD binning with respect to (1) gene converge, (2) the interpretation of findings, and (3) pairwise concordance of the findings among three GWASs. We first summarize their results for (1) and (3). On comparing LD binning with positional binning, their Tables 1 and 2 clearly show an increase in the number of post-quality-control-binned SNPs and a decrease in the number of SNPs binned to only one gene, indicating an increased number of SNPs assigned to more than one gene. This automatically increases the correlations among genes in any subsequent pathway analysis. For the genotyped Welcome Trust Case Control Consortium (WTCCC) SNPs in their Table 1, the number of SNPs binned to more than one gene increases from 16% with positional binning to 36% with LD-based binning; similar results are seen for the Norwegian Thematically Organized Psychosis bipolar disorder (BP) GWAS and German BP GWAS data. For imputed genotype data, although the increase is not as large as for actually genotyped data, the absolute percentage is much larger—it increases from 55.5% with positional binning to 63% with LD-based binning in the case of the WTCCC BP data and increases from 59% to 61.5% for the German BP data. Because LD-based binning results in spurious correlations among genes, it is not surprising that when Christoforou et al. used LD-based binning, 15.5%–34% new genes moved into the top-ranked 2,000 genes. Thus, many of the top genes are selected because of their LD with a common SNP rather than because of association evidence attributable to the gene itself. In other words, the same association evidence is used repeatedly but is assumed to be independent. To show that LD-based binning improves the concordance of results across studies, Christoforou et al. present in their Table 4 the pairwise correlations of the SNP ranks between studies (as determined by their p values) and, similarly, the correlation of the gene ranks by comparing positional binning with LD-based binning. However, as we have already explained, these correlations between different studies arise mostly from the correlations among genes, and the fact that the correlations with LD-based binning have a higher significance than the correlations with positional binning is attributable solely to the increase in correlations among genes caused by LD-based binning rather than to any consistency of association results across studies.
Table 1.
Mean Spearman Rank Correlation, Based on 280 Replications, between Study Groups 1 and 2
|
Positional Binning |
LD-Based Binning |
|||||
|---|---|---|---|---|---|---|
|
Bins Defined by r2= 0.5 Cutoff |
Bins Defined by r2= 0.8 Cutoff |
|||||
| Mean Spearman Rank r (SEM) | Median of p Values | Mean Spearman Rank r (SEM) | Median of p Values | Mean Spearman Rank r (SEM) | Median of p Values | |
| Gene Level | ||||||
| 0 kb window | 0.045 (0.01) | 9.51 × 10−8 | 0.085 (0.011) | 7.33 × 10−34 | 0.09 (0.011) | 1.35 × 10−35 |
| 1 kb window | 0.041 (0.009) | 6.86 × 10−8 | 0.089 (0.011) | 1.10 × 10−49 | 0.093 (0.01) | 1.69 × 10−50 |
| 10 kb window | 0.045 (0.009) | 1.58 × 10−14 | 0.082 (0.012) | 1.66 × 10−49 | 0.089 (0.011) | 9.15 × 10−57 |
| SNP Level | 3 × 10−5 (0.003) | 0.284 | - | - | - | - |
Window sizes were extended either 0, 1, or 10 kb upstream and downstream of a gene. The following abbreviations are used: LD, linkage disequilibrium; and SEM, standard error of the mean.
Table 2.
Pairwise Concordance between GWASs at the SNP and Gene Levels
| WTCCC versus TOP | WTCCC versus German | TOP versus German | TOP Imputed versus German Imputed | |
|---|---|---|---|---|
| SNP level | 0.0066 (0.00018) | 0.0037 (0.31) | −0.0018 (0.51) | −0.00023 (0.83) |
| Gene level (positional binning) | 0.030 (1.78 × 10−7) | −0.0017 (0.78) | 0.023 (4.78 × 10−5) | 0.068 (<2.2 × 10−16) |
| Gene level (LD-based binning) | 0.077 (<2.2 × 10−16) | 0.027 (7.24 × 10−7) | 0.053 (<2.2 × 10−16) | 0.098 (<2.2 × 10−16) |
This table was adapted from Table 4 in Christoforou et al., 2012.1 The Spearman rank correlation and p value (in parentheses) are shown for each pairwise comparison. The following abbreviations are used: WTCCC, Welcome Trust Case Control Consortium; TOP, Norwegian Thematically Organized Psychosis; and LD, linkage disequilibrium.
To verify this conclusion, we randomly assigned the WTCCC3 1,868 BP individuals and 2,938 common controls to form two separate study groups, each comprising 2,403 individuals. In each study group, an individual was randomly assigned as a case or control with equally probability. Thus, there was no genetic contribution to the phenotype in either study group. Quality control of the genotype data was performed as in Feng and Zhu.4 We used PLINK5 to calculate the association p value for each SNP and then applied the LDsnpR software, developed by Christoforou et al.,1 with the modified Sidak correction as suggested in Christoforou et al. to obtain gene-based p values in each bin. We did this for both the positional and the LD-based binning approaches. We converted the p values to corresponding chi-square values with 1 degree of freedom, and from these, we calculated the genomic control value λ. We varied the window size by extending the gene size by 0, 1, and 10 kb both upstream and downstream. For the LD-based binning method, we also varied the cutoff r2 by using 0.5 and 0.8. We observed that both the SNP-level analysis (Figure 1) and the positional binning procedure give genomic control values close to 1 (Figures 2A–2C). We also found that the LD-based binning approach results in substantially smaller medians of test statistics; λ ranged from 0.643 to 0.716 for different LD levels and window sizes (Figures 2D–2I). We explain below that these small λ values for LD-based binning are probably caused by overcorrection for multiple tests at the gene level with the use of the modified Sidak correction, which does not properly correct for linkage disequilibrium among SNPs in a bin. These small values are also probably caused by the same SNP being assigned to multiple genes and the subsequent increased correlation among genes.
Figure 1.
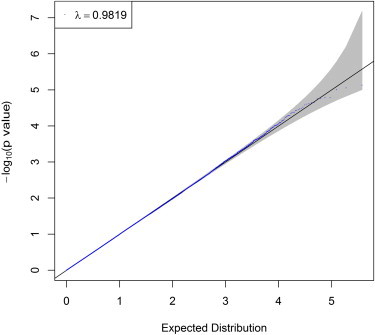
Quantile-Quantile Plot of the Single-SNP Analysis Using the 1,868 WTCCC BP Individuals and 2,938 Common Controls Randomly Assigned to Case-Control Status
Figure 2.
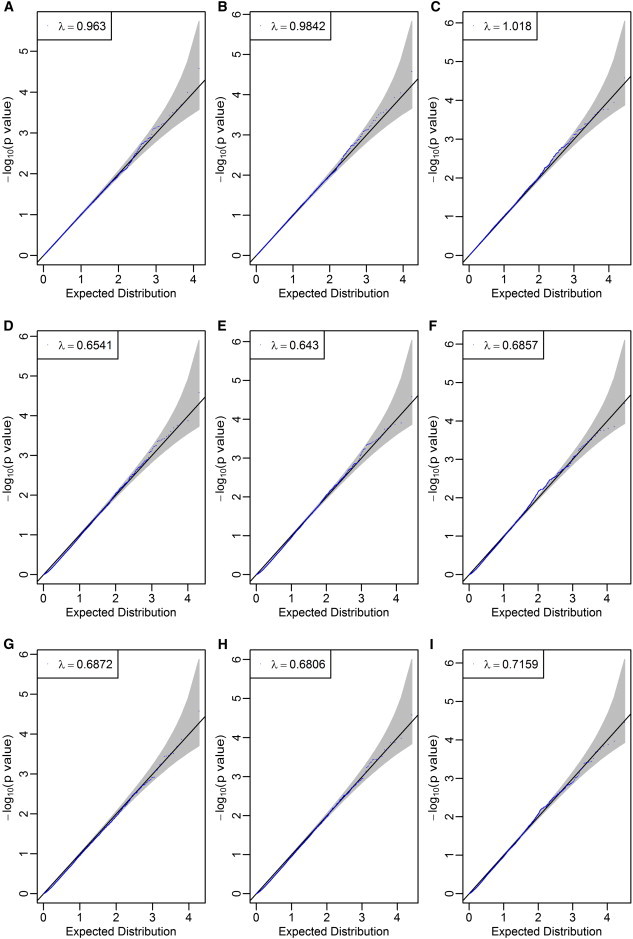
Quantile-Quantile Plots after Positional Binning and LD-Based Binning Using the 1,868 WTCCC BP Individuals and 2,938 Common Controls Randomly Assigned to Case-Control Status
(A) Positional binning with no extension.
(B) Positional binning with 1 kb extension upstream and downstream of a gene.
(C) Positional binning with 10 kb extension upstream and downstream of a gene.
(D) LD-based binning with no extension (r2 = 0.5).
(E) LD-based binning with 1 kb extension upstream and downstream of a gene (r2 = 0.5).
(F) LD-based binning with 10 kb extension upstream and downstream of a gene (r2 = 0.5).
(G) LD-based binning with no extension (r2 = 0.8).
(H) LD-based binning with 1 kb extension upstream and downstream of a gene (r2 = 0.8).
(I) LD-based binning with 10 kb extension upstream and downstream of a gene (r2 = 0.8).
The small λ values in (D–I) are caused by substantially more observed p values close to 1.
Out of concern for statistical noise, we performed 280 replicate random assignments to case-control status of the WTCCC BP cases and controls. Because there was no association between any of the genes and the phenotype, we expected there to be no pairwise correlation of the gene-based p values between the two study groups in these simulated data. However, we observed an association (Table 1) similar to that observed in their Table 4 (Table 2 in this letter) for both the positional and the LD-based binning procedures. Although we observed significant correlations for both methods, the results with LD-based binning yielded correlations about twice as large as those with positional-based binning (Table 1). We observed smaller correlations at the SNP-level analysis (Table 1). Among the 280 replications, we still observed 90 for which the Spearman-rank-test p value was less than 0.05 at the SNP level. It has been suggested that the WTCCC BP samples might have much higher rates of recent identity by descent than do participants collected for the rest of the WTCCC cohorts,6 and this could cause such an association. We therefore studied two groups each comprising the same 4,806 individuals and randomly assigned disease status in each. We calculated the p value for each SNP in each group and the Spearman rank correlation between their ranks in the two groups in exactly the same way. When we did this, we did not observe an increased Spearman rank correlation, suggesting that the observed correlation at the SNP-level analysis was not caused by any cryptic relatedness in the WTCCC BP data. Thus, this association is most likely due to the linkage disequilibrium among SNPs.
For positional-based binning, the observed correlation was due to (1) the correlation among genes induced by linkage disequilibrium among SNPs and (2) inaccurate modification of the Sidak correction. The first reason is similar to what occurs in the SNP-level analysis. Regarding the second reason, the modified Sidak correction replaces the number of SNPs in a bin, m, with (m + 1) / 2 to adjust for linkage disequilibrium.2 This modified correction might be either liberal or conservative in the calculation of a gene-based p value, which only depends on the linkage disequilibrium among SNPs. As a result, a gene can consistently either improve or drop in rank across studies, and this leads to a pairwise rank correlation between studies. However, the excess of correlation observed in LD-based binning is caused largely by the uncorrected assignment of a SNP to multiple genes.
In summary, we conclude that LD-based binning will most likely “discover” gene correlations that are due to the way the SNPs are assigned to genes rather than improve the interpretation of GWASs. Therefore, LD-based binning, as implemented in LDsnpR, will have a negative impact on subsequent genetic and functional studies, and this method should not be used. For example, suppose that an initial pathway analysis detects genes associated with a phenotype by using the LD-based binning procedure. A similarly performed replication analysis using independent samples might detect the same genes associated with the phenotype. However, this replication might be attributed to the spurious correlation caused by applying LD-based binning. Thus, we suggest that this method not be used in practice. Caution should also be taken when the positional-binning approach is used, especially regarding correlations caused by LD; these correlations can be addressed in various ways.7–9 It should be pointed out that our study does not deny the usefulness of binning-based methods for pathway analyses. However, better methods for obtaining gene-level or pathway-level association evidence are still needed in practice.
Acknowledgments
This work was supported by National Institutes of Health grants HL086718 (from the National Heart, Lung, and Blood Institute), HG003054 and HG005854 (from the National Human Genome Research Institute), and U01HG006382 (from the National Human Genome Research Institute), as well as a National Research Foundation of Korea grant (NRF-2011-220-C00004) funded by the Korean government. The content of this letter is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Funding for the original Welcome Trust Case Control Consortium project was provided by the Wellcome Trust under award 076113. We thank the two anonymous reviewers for their constructive comments, which improved the paper significantly.
Web Resources
The URL for data presented herein is as follows:
References
- 1.Christoforou A., Dondrup M., Mattingsdal M., Mattheisen M., Giddaluru S., Nöthen M.M., Rietschel M., Cichon S., Djurovic S., Andreassen O.A. Linkage-disequilibrium-based binning affects the interpretation of GWASs. Am. J. Hum. Genet. 2012;90:727–733. doi: 10.1016/j.ajhg.2012.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saccone S.F., Hinrichs A.L., Saccone N.L., Chase G.A., Konvicka K., Madden P.A., Breslau N., Johnson E.O., Hatsukami D., Pomerleau O. Cholinergic nicotinic receptor genes implicated in a nicotine dependence association study targeting 348 candidate genes with 3713 SNPs. Hum. Mol. Genet. 2007;16:36–49. doi: 10.1093/hmg/ddl438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feng T., Zhu X. Genome-wide searching of rare genetic variants in WTCCC data. Hum. Genet. 2010;128:269–280. doi: 10.1007/s00439-010-0849-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Browning S.R., Browning B.L. Population structure can inflate SNP-based heritability estimates. Am. J. Hum. Genet. 2011;89:191–193. doi: 10.1016/j.ajhg.2011.05.025. author reply 193–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conneely K.N., Boehnke M. So many correlated tests, so little time! Rapid adjustment of P values for multiple correlated tests. Am. J. Hum. Genet. 2007;81:1158–1168. doi: 10.1086/522036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dudbridge F., Koeleman B.P. Efficient computation of significance levels for multiple associations in large studies of correlated data, including genomewide association studies. Am. J. Hum. Genet. 2004;75:424–435. doi: 10.1086/423738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Won S., Morris N., Lu Q., Elston R.C. Choosing an optimal method to combine P-values. Stat. Med. 2009;28:1537–1553. doi: 10.1002/sim.3569. [DOI] [PMC free article] [PubMed] [Google Scholar]