

Abstract
Cytochrome P450 mono-oxygenases (2UUQ) enzyme from Mycobacterium tuberculosis catalyzes oxidation of organic compounds such as lipids and steroidal hormones therefore remain as potential drug target. Currently available first line anti-tuberculosis drugs have been caused several side effects in the body as well as resistance development by mycobacterium against these drugs, necessitates the considerable need for finding new drugs. Therefore, we propose a structure based computational method to find a new potential inhibitor for cytochrome P450 mono-oxygenases enzyme. Compounds from several ligand databases were docked against the functional sites of 2UUQ (A) through the standard GEMDOCK v2.0 and AUTODOCK4.0 molecular docking tools. Commercially available chemical compound ZINC00004165 (5-[3-(2-nitroimidazol-1-yl) propyl] phenanthridine) has produced top rank with lowest interaction energy of -113.2 (via GEMDOCK) and lowest docking energy of -9.80 kcal/mol (via AUTODOCK) as compared to first line anti TB compounds. Z score and normal distribution analysis verified that the ZINC00004165 compound has more affinity towards 2UUQ in comparison to large number of random population of compounds. ZINC00004165 is also in agreement with the drug likeness properties of Lipinski rule of five without any violation. Therefore, our finding concludes that the commercial compound ZINC00004165 can act as a potential inhibitor against cytochrome P450 mono-oxygenases enzyme of Mycobacterium tuberculosis.
Keywords: Cytochrome P450 mono-oxygenases, Tuberculosis, Ligand database, Docking, Gemdock, Autodock
Background
The P450 (cytochrome P450 mono-oxygenases) belongs to super family of haem-containing mono-oxygenase enzymes [1]. This enzyme participates in the oxygen transfer reaction, where it catalyzes the reduction of di-oxygen atoms that bound to the haem iron of the P450 [3]. This step leads to removable of single oxygen atom from di-oxygen and transfer it to an organic substrate along with the formation of a water molecule.
RH+O2+2e- +2H+ = ROH + H2O The cytochrome P450 monooxygenases may catalyze diverse sets of oxidation reactions. However the important step is to introduce the oxygen atoms in the substrate [3, 4]. This step is important in the survival of the pathogenic microorganisms like Mycobacterium tuberculosis. In addition, the reaction catalyzed by the cytochrome P450 monooxygenases helps in regulation of important pathways that are essential for the infectivity or pathogenicity of Mycobacterium tuberculosis [5]. Therefore, this enzyme is always a potential drug target against the Mycobacterium tuberculosis [6]. Many drugs are available in the market for the treatment of tuberculosis disease and have been classified in to different categories based on their activity as well as effectiveness against inhibiting the growth of the Mycobacterium such as first line, second line and third line drugs [7].
Some of the first line drugs like Ethambutol are known to be a bacteriostatic agent and function effectively by inhibiting the growth of actively growing TB bacilli [8]. It mainly targets the cell wall of the tuberculosis bacteria and inhibits its formation during the cell division. Another drug, Isoniazid is bactericidal in nature and acts on the mycobacterium cell when it rapidly divides in the human body. On the other hand, it becomes bacteriostatic when the mycobacterium grows slowly and manifests the human body for pathogenicity [9, 10]. Isoniazid performs its action by inhibiting the P450 system. One of the major first line anti-tuberculosis drug, Rifampicin is typically used to treat Mycobacterium tuberculosis infections, including tuberculosis and leprosy. The chemical composition of Rifampicin provides lipophilic nature which makes it more active drug to treat the meningitis form of tuberculosis [11, 12]. In addition, the lipophilic nature of the Rifampicin helps in smooth distribution of the drug into the central nervous system and crossing the blood-brain barrier. However, it caused several side effects and toxicity in the human body, thereby creating the limitations for using as an effective antituberculosis drug therapy. For example, the Rifampicin may cause hepatotoxicity of the liver after prolonged use [13]. The most common (approximately 1%) side effect of Pyrazinamide is joint pains (arthralgia), Ethambutol may cause stomach upset, dizziness, fatigue, or headache. Isoniazid can cause allergy also and the symptoms of an allergic reaction include: rash, itching, swelling, dizziness, trouble breathing [13]. Due to above sever complications of the existing anti-tuberculosis drugs; there is a need to develop new drug therapy against tuberculosis disease. Here, we proposed the use of computational drug designing methods to find the alternative potential inhibitor against P450 enzyme of mycobacterium which can act as an anti-tuberculosis drug compound. We also compared the efficacy of our compounds with the existing first line anti-tuberculosis drugs. The new drug was also tested for drug likeness properties derived by Lipinski rule of five
Methodology
Input receptor file:
The CYP130 protein (pdb id 2UUQ) from Mycobacterium tuberculosis has been chosen as potential drug target for our docking study. From its original research paper we enumerated the active site pattern and prepared as input file for docking simulations.
Setting anti-tuberculosis compound libraries:
Ligand databases such as ZINC [14], DRUG BANK (www.drugbank.ca) and PUBCHEM [15] were searched for obtaining putative anti-tuberculosis compounds for our docking study. The compounds searching strategies were divided in to three steps: (1) obtained the first line antituberculosis drugs which are known to target Mycobacterium tuberculosis. Compound obtained from DRUG BANK databases. (2) Obtained natural (plant origin) anti-tuberculosis compounds from PUBCHEM databases. The information for natural antituberculosis compounds was obtained from published literature’s that describing the effectiveness of natural antituberculosis compounds against Mycobacterium tuberculosis. The SMILES strings [16] of DRUG BANK and natural compounds were downloaded and converted in to 3D structure via CORINA server (http://www.molecularnetworks.com/online_demos/corina_demo). (3) The SMILES strings of first line anti-tuberculosis drugs were virtually screened against ZINC database. We constructed different compound libraries based on the similarity with SMILES strings and set as input files for docking simulations.
Docking Simulation:
The compound libraries (First line, Natural and ZINC compounds) were virtually screened individually against the active site pattern of 2UUQ via GEMDOCKv2.0 software [17]. This docking tool works effectively based on the genetic algorithm (GA) with default parameters of virtual screening. Furthermore, the top ten compounds that produced lower fitness values were selected for next cycle of docking simulation via GEMDOCK. In the second phase of docking cycle, 29 compounds were submitted into docking simulation via GEMDOCK v2.0. Note that the several successive simulations were performed in order to find all possible poses of the compounds. After completion of docking, the top ten compounds were selected and subjected for further final validation via AUTODOCK 4.0 [18]. The automated molecular docking simulations were performed by Genetic Algorithm- Local Search (GA-LS) with standard parameters. The compounds were ranked based on the docking energy. The compound with the highest affinity for the 2UUQ active site pattern with lowest docking energy was selected. Here, we examined the performance of these docking software to select best compound without and biased.
Statistical analysis:
The histogram was generated for analyzing the number of compounds showing fitness values in the specific range. Furthermore, standard score known as Z score was calculated to evaluate the fitness value of the compounds and to find unique compound showing affinity for 2UUQ which was unlikely to produce by other compounds in the random population. Here, the Z score value will determine the best compound fitness value that may be far from the mean of the fitness values generated by total number of compounds. The affinity of selected compound was further tested by normal distribution analysis. The compounds P values were calculated for each Z score and normal distribution analysis was performed. The standard P value was set to 0.05 at 95% confidence interval and it was assumed that compounds with more negative Z score and lesser P value than < 0.05 are better than the other random population.
Drug likeness calculation:
The compound that produced lowest docking energy was subjected to MOLINSPIRATION (www.molinspiration.com) drug likeness analyzing server. Their molecular and drug likeness properties were measured and compared with properties of first line anti tuberculosis drugs against Mycobacterium tuberculosis. Please refer (Figure 1) for the overall methodology.
Figure 1.
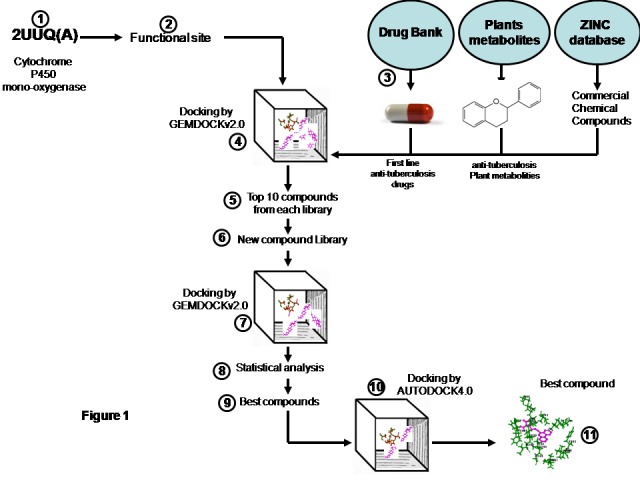
Overall methodology: (1) downloaded pdb file of cytochrome P450 mono-oxygenase enzyme (2UUQ) from RCSB protein data bank (2) Extracted the functional site residues from 2UUQ, reported in the literature of 2UUQ (3) downloaded the first line anti-tuberculosis compounds from DRUG BANK, plant metabolites from PUBCHEM and ZINC compounds similar to first line compounds from ZINC database (4) Docked the compounds against the functional sites of 2UUQ via GEMDOCKv2.0 (5) Ranked the compounds based on the fitness values and selected the top ten compounds from each category (6) made new compound library (7) Docked against the functional site of 2UUQ via GEMDOCKv2.0 (8) Performed the statistical analysis (9) Selected the best compounds (10) Docked the best compounds against the site via AUTODOCK4.0 for final validation (11) Selected the best hit compound with lowest docking energy.
Discussion
The CYP130 protein (2UUQ) from Mycobacterium tuberculosis was selected for our docking study. A total of nine first-line antituberculosis drugs were selected from DRUG BANK databases Table 1 (see supplementary material) and were screened against the functional site of 2UUQ (Chain A) through GEMDOCKv2.0 software. Drug Rifampicin was produced lowest interaction energy (fitness value) of -89.819 with 2UUQ (A) Table 1 (see supplementary material). Furthermore, 45 plants origin natural anti tuberculosis compounds were submitted for the docking analysis against the protein 2UUQ (A). Compounds Artonin E and Artonin B have obtained highest rank with the lowest fitness value of -90.67 and -84.92 respectively. We have selected top ten compounds for further study. Additionally, the screening of 637 commercial available chemical compounds from ZINC databases against functional site of 2UUQ (A) obtained ranking of best hit compounds Table 1(Supplementary material). The top ten ZINC compounds were filtered. The selected 29 compounds (nine from first line + ten compounds from natural inhibitors + ten from ZINC) were included into a single set of compound library. Subsequently, these compounds were set as input for further docking simulation.
The chemical compound from ZINC database ZINC00004165 (5-[3-(2-nitroimidazol-1-yl) propyl] phenanthridine) (Figure 2) was obtained highest rank with lowest interaction energy of - 113.255 as compared to first line anti-tuberculosis compounds (Table 1). Furthermore, results were verified via AUTODOCK4.0. Further, ZINC00004165 produced lowest docking energy of -9.80 kcal/mol Table 1 (see supplementary material), (Figure 3). Statistical analysis also revealed that ZINC00004165 has lowest Z score of -2.681 as compared to other compounds (Graph 1). It indicates that the affinity of the compound ZINC00004165 is far from the mean of the population of other drug compounds fitness values and not able to produce by other compounds. Simultaneously, normal distribution and P value analysis (Graph 2) follow a bell shaped curve. The compound ZINC00004165 exhibits a Z score of -2.681 with P value of 0.003, which is lesser than P value of 0.05 (represents 95% confidence interval). It signifies that there is less chance of getting similar affinity by any other compound for 2UUQ as compare to ZINC00004165. Drug likeness analysis for selected ZINC and first line anti-tuberculosis compounds through MOLINSPIRATION server revealed that ZINC00004165 has no violation for the Lipinski rule of five. On the other hand, the first line compounds, namely Streptomycin and Rifampicin have three violations Table 2 (see supplementary material). Additionally, Structure-Structure superposition between 2UUQ (with docked compound ZINC00004165) and 2UVN (with bound ligand) is produced the r.m.s.d of 0.96 Å (Figure 4). Interestingly, the catalytic residues that surrounds the compound ZINC00004165 (5-[3-(2- nitroimidazol-1-yl) propyl] phenanthridine) have matched with the literature reported for 2UUQ as well as with the catalytic residues that bound to the first line anti-tuberculosis compounds (Figure 3).
Figure 2.
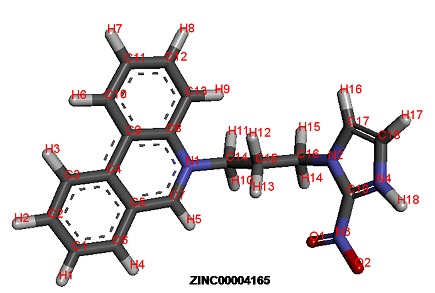
Compound ZINC00004165 from ZINC database.
Figure 4.
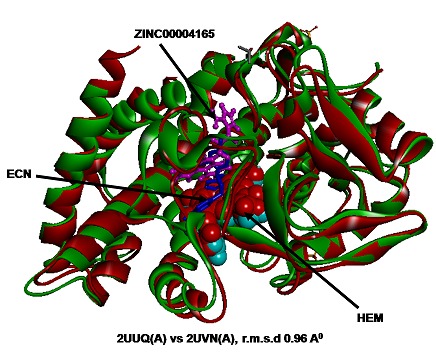
Structure-structure superimposition between 2UUQ (A) (green) with docked ZINC00004165 compound (magenta) and of 2UVN (A) (red) with bound ligand ECN, 1-[ (2S)-2-[ (4- Chlorobenzyl) Oxy]-2-(2,4-Dichloropheyl) Ethyl]- 1H-Imidazole (blue) produced r.m.s.d. of 0.96 Å (No. of atoms 1456).
Figure 3.
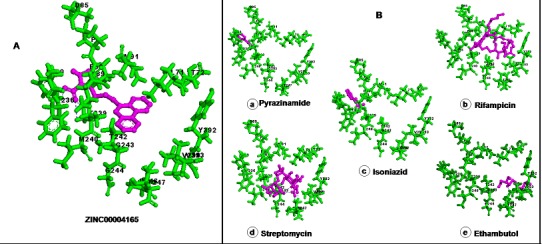
Docking simulation analysis for ZINC compound and first line anti tuberculosis compounds. (A) Compound ZINC00004165 docked with functional site of 2UUQ (A) docking energy of -9.80 kcal/mol. (B): docking of first line anti tuberculosis compounds (a) Pyrazinamide (DE -4.18 kcal/mol) (b) Rifampicin (DE -8.00 kcal/mol) (c) Isoniazid (DE -4.53 kcal/mol) (d) Streptomycin (DE -8.19 kcal/mol) (e) Ethambutol (DE -5.84 kcal/mol) with functional site of 2UUQ (A).
Conclusion
The Molecular docking analysis concludes that the novel compound ZINC00004165 (5-[3-(2-nitroimidazol-1-yl) propyl] phenanthridine) produces lowest docking energy and fitness value than the first line anti-tuberculosis drug compounds. This unequivocally shows that the compound ZINC00004165 has more affinity towards the Protein 2UUQ in comparison to the commercially available anti-tuberculosis compounds. Further, the Z score statistical test has verified the uniqueness of the above compound by exhibiting top Z score than the rest antituberculosis compounds. The normal distribution analysis for the calculated Z scores has further concluded that the percentage of affinity for the ZINC00004165 compound is comparatively higher than any other compounds from the drug database. Significantly, the drug likeness test also confirmed that the compound ZINC00004165 follows no violation of “Lipinski rule of five” and possess the criteria of standard drug like properties. Therefore, it is likely that ZINC00004165 can act as a potential inhibitor for the CYP130 protein (2UUQ). In addition, they can serve as an effective model compound for designing novel drugs against the harmful pathogenic bacterium Mycobacterium tuberculosis.
Supplementary material
Acknowledgments
AS and A jointly conceived the idea for the current research work. AS, IC, NS, KKS and OR are great full to New Era Proteomics, India as well as France for all financial support. AN kindly acknowledges DBT, India for all financial resources. PPC kindly acknowledges Bioroutes life sciences for all funds.
Footnotes
Citation:Sharma et al, Bioinformation 8(19): 931-937 (2012)
References
- 1.KJ McLean, et al. Biochem Soc Trans. 2005;33:796. doi: 10.1042/BST0330796. [DOI] [PubMed] [Google Scholar]
- 2.DW Reichhart, et al. Genome Biology. 2000;1:3003. [Google Scholar]
- 3.FP Guengerich, et al. J Biol Chem. 1991;266:10019. [PubMed] [Google Scholar]
- 4.VB Urlacher, et al. Trends Biotechnol. 2006;24:324. doi: 10.1016/j.tibtech.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 5.KJ McLean, et al. Trends Microbiol. 2006;14:220. doi: 10.1016/j.tim.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 6.D Leys, et al. J Biol Chem. 2003;278:5141. doi: 10.1074/jbc.M209928200. [DOI] [PubMed] [Google Scholar]
- 7.SB Masters, et al. Lange Medical Books/McGraw Hill, Medical Pub. Division. 2005 ISBN 0-07-142290-0. [Google Scholar]
- 8.R Yendapally, et al. Bioorg Med Chem Lett. 2008;18:1607. doi: 10.1016/j.bmcl.2008.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.HM Blumberg, et al. Am J Respir Crit Care Med. 2003;167:603. doi: 10.1164/rccm.167.4.603. [DOI] [PubMed] [Google Scholar]
- 10.A Jindani, et al. Am Rev Respir Dis. 1980;121:939. doi: 10.1164/arrd.1980.121.6.939. [DOI] [PubMed] [Google Scholar]
- 11.R Tripathi, et al. Med Res Rev. 2005;25:93. doi: 10.1002/med.20017. [DOI] [PubMed] [Google Scholar]
- 12.AL Demain, S Sanchez. J Antibiot (Tokyo) 2009;62:5. [Google Scholar]
- 13.CC Leung, et al. Am J Respir Crit Care Med. 2005;169:1168. [Google Scholar]
- 14.JJ Irwin, BK Shoichet. J Chem Inf Model. 2005;45:117. [Google Scholar]
- 15.WD Ihlenfeldt, et al. J Cheminform. 2009;1:20. doi: 10.1186/1758-2946-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.D Weininger. J Chemical Inform Comput Sci. 1998;28:31. [Google Scholar]
- 17.JM Yang, et al. Proteins. 2004;55:288. [Google Scholar]
- 18.GM Morris, et al. J Comput Chem. 1998;19:1639. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.