

Abstract
Tyrosine kinase inhibitors (TKIs) exhibit impressive activity against advanced renal cell carcinoma. However, recent clinical studies have demonstrated an equivocal response to sunitinib in patients with castration-resistant prostate cancer. The tumor suppressor phosphatase and tensin homolog (PTEN) acts as a gatekeeper of the PI3K/Akt/mTOR cell-survival pathway. Our experiments demonstrate that PTEN expression inversely correlates with sunitinib resistance in renal and prostate cancer cells. Restoration of PTEN expression markedly increases sensitivity of tumor cells to sunitinib both in vitro and in vivo. In addition, pharmacologic manipulation of PI3K/Akt/mTOR signaling with PI3K/mTOR inhibitor, GDC-0980, mTOR inhibitor, temsirolimus, or pan-Akt inhibitor, GSK690693, was able to overcome sunitinib resistance in cancer cells. Our findings underscore the importance of PTEN expression in relation to sunitinib resistance and imply a direct cytotoxic effect by sunitinib on tumor cells in addition to its anti-angiogenic actions.
Keywords: PTEN, mTOR, sunitinib, temsirolimus, cancer
INTRODUCTION
Small molecule multi-targeted receptor tyrosine kinase inhibitors (TKIs) represent a clinically relevant strategy for patients with advanced tumors such as renal cell carcinoma (RCC) and gastrointestinal stromal tumors (GISTs) (1, 2). It has been reported that sunitinib's tumoricidal activity is mediated mainly through inhibition of the platelet-derived growth factor receptor (PDGFR), the vascular endothelial growth factor receptor (VEGFR) 1 and 3, KIT, and fms-related tyrosine kinase 3 (FLT3) (3). In Phase III clinical trials with sunitinib, median survival for patients with metastatic RCC increased from 10 to 24 months (4). As such, sunitinib received approval by the Food and Drug Administration for the treatment of metastatic RCC in 2006. Due to its remarkable success with metastatic RCC, the drug is also under intense investigation as a treatment for several other cancers, including breast, colorectal, and non–small cell lung (3).
More recently, initial clinical experience with sunitinib in castration-resistant prostate cancer (CRPC) patients has been reported. In Phase II trials, a heterogeneous response to sunitinib was noted in both docetaxel-resistant and chemotherapy naïve patients with a significant proportion of tumors demonstrating at least a partial radiographic response. In the most recent study of subjects with docetaxel-resistant CRPC, 11.1% of patients demonstrated 30% tumor reduction by Response Evaluation Criteria In Solid Tumors (RECIST) criteria. Another 44.4% of patients had quantifiable but less significant tumor response (5).
Drug resistance to anti-cancer therapies remains a clinical challenge. Tumors may acquire stability to the drug during treatment or show intrinsic resistance from the outset of therapy. One possible mechanism implicated in TKI resistance may be the constitutive activation of various intracellular pro-tumorigenic pathways downstream of receptor signaling. Activation of the PI3K/Akt/mTOR pathway, chiefly due to redundant autocrine signaling rather than mutations, is clearly involved in tumor initiation and progression (6). The PI3K/Akt/mTOR axis is a central crossroad of many intracellular signaling pathways, exhibiting direct and indirect control over a multitude of cellular events including cell cycle progression, cellular proliferation/growth, autophagy, and angiogenesis (6).
The tumor suppressor phosphatase and tensin homolog (PTEN) acts as a gatekeeper of the PI3K/Akt/mTOR cell-survival signaling pathway. Under normal cellular conditions, PTEN is a negative regulator of tumorigenic activity; however, when lost or inactivated, the PI3K/Akt/mTOR pathway is enhanced. As such, loss of PTEN can be an indicator of aggressive behavior in tumors of various cell types (6). In our present study, we found that that the status of PTEN expression inversely correlates with sunitinib resistance in renal and prostate cancer cells. Induced expression of PTEN in PTEN-negative tumor cells markedly increased tumor sensitivity to sunitinib both in vitro and in vivo. Furthermore, we were able to circumvent sunitinib resistance in PTEN-negative tumor cells by manipulation of PI3K/Akt/mTOR signaling with pharmacologic agents such as GDC-0980, GSK690693 and temsirolimus. These results indicate that lack of PTEN expression may contribute to intrinsic sunitinib resistance, and the activity of the PI3K/Akt/mTOR pathway may be a marker of TKI response/resistance in genitourinary tumors. Furthermore, co-administration of sunitinib with PI3K/Akt/mTOR inhibitors may represent a rational treatment strategy for patients with resistant urologic tumors.
MATERIALS AND METHODS
Cells and culture conditions
Androgen-dependent LNCaP prostate cancer cells, androgen-independent PC-3 and DU-145 prostate cancer cells, and renal carcinoma 786-O cells were obtained from ATCC (Rockville, MD). The 769-P, HRC-51, HRC-45, HRC-78, and SK-45 human renal cell carcinoma cell lines were a kind gift of Dr. Joseph Testa (Fox Chase Cancer Center, Philadelphia, PA). The SK-26B cell line was obtained through the generosity of Dr. Finke (The Cleveland Clinic Foundation, Cleveland OH). Initial stocks were cryopreserved, and at every six-month interval a fresh aliquot of frozen cells was utilized for the experiments. No authentication was done by the authors. Cells were cultured in RPMI 1640 (Bio-Whittaker, Walkersville, MD) supplemented with 10% FCS (Hyclone, Logan, UT), gentamicin (50 mg/l), sodium pyruvate (1 mM) and non-essential amino acids (0.1 mM) under conditions indicated in the figure legends.
Antibodies and Reagents
Antibodies to PTEN, Akt, phospho-Akt(S473), phospho-Akt(T308), phospho-Tuberin/TSC2(T1462), Tuberin/TSC2, phospho-4EBP1(T37/46) and GAPDH were obtained from Cell Signaling Technology (Beverly, MA). Sunitinib and temsirolimus were obtained from LC Laboratories (Woburn, MA). GDC-0980 was obtained from ChemieTek (Indianapolis, IN). GSK690693 was obtained from Selleck (Houston, TX).
Stable Transfection with PTEN
To generate the PTEN expression vector PTEN ORF was amplified with specific forward 5‵-atcttgaagcttgccaccatgacagccatcatcaaag-3‵ and reverse 5‵-atcttggaattctcagacttttgtaatttgtgtatgctgatc-3‵ primers carrying HindIII and EcoR1 restriction sites (underlined) using as a template pOTB7 vector (clone ID#3937787) containing full-length PTEN cDNA (Thermo Scientific, Huntsville, AL) and then cloned into HindIII/EcoR1 sites of pcDNA3.1-puro vector. The sequence of the pcDNA3.1-PTEN-puro construct was validated in the DNA Sequencing Facility of the Fox Chase Cancer Center. Cells were transfected with either the PTEN expression vector or the pcDNA3.1-puro control vector using the TransIT-Prostate transfection kit (Mirus Bio, Madison, WI). Selection was performed using puromycin (1 μg/ml).
Western Blot Analysis
Whole cell lysates preparation and Western Blot Analysis were performed as described previously (7).
Measurement of apoptosis
DNA fragmentation was detected using the APO-BRDU kit (The Phoenix Flow Systems, Inc., San Diego, CA) according to manufacturer's instructions.
Measurement of IL-6 and IL8
IL-6 and IL-8 levels in cell culture supernatants were measured using ELISA kits (R&D Systems, Minneapolis, MN).
Assessment of in vivo tumor growth
For in vivo studies, 1 × 106 PC-3-PTEN or PC-3-puromycin cells were inoculated s.c. in the flank region of 6 week-old male C.B17/Icr-scid mice using a 27-gauge needle. All animal procedures were done in accordance with institutional guidelines on animal care and with appropriate institutional certification. Animals were fed an autoclaved AIN-93M diet (Harlan Teklad, Madison, WI) and water ad libitum. Two weeks after the injection of tumor cells, animals were randomly assigned to the control or experimental groups (n=5 mice/group). The mice were treated with either vegetable oil (control) or sunitinib (40 mg/kg in vegetable oil, 5 days/week, p.o.).
To examine the effect of co-administration of sunitinib and temsirolimus on tumor growth in vivo in PTEN-deficient cells, subcutaneous tumors were established using PTEN-negative PC-3 cells. Two weeks after tumor cell injection animals were randomly assigned to the control or experimental groups (n=7 mice/group). The mice were treated with (i) vegetable oil (control); (ii) sunitinib (40 mg/kg in vegetable oil, 5 days/week, p.o.); (iii) temsirolimus (10 mg/kg, once a week, i.v.); or with sunitinib and temsirolimus combined. Tumors were measured twice weekly and their volumes were calculated by the formula: [volume = 0.52 × (width)2 × length]. To examine sunitinib levels in plasma and tumor tissue, sunitinib (40 mg/kg in vegetable oil) was administered p.o. daily to C.B17/Icr-scid mice with PC-3 xenografts for 7 consecutive days. Animals were euthanized via CO2 asphyxiation 3 hours following last sunitinib administration. Levels of sunitinib in plasma and tumor samples were examined at Wolfe Laboratories (Watertown, MA).
Statistical analysis
Statistical analysis was performed using a two-sided Student's t-test. A p-value of <0.05 was considered statistically significant.
RESULTS
Lack of PTEN expression coincides with sunitinib resistance in renal and prostate cancer cells in vitro
To investigate the potential mechanisms underlying TKI resistance in vitro, we first analyzed the effect of sunitinib on the viability of several renal and prostate cancer cell lines. The findings presented in Figure 1A demonstrate that sunitinib was capable of inducing a significant level of apoptosis in seven out of the ten renal and prostate cancer cell lines tested with the notable exception of 786–0 (renal) and PC-3 and LNCaP (prostate) cancer cell lines. These three cell lines were nearly completely resistant to sunitinib-mediated apoptosis. Since constitutive activation of the PI3K/Akt/mTOR pathway due to deletion or mutation of PTEN expression has been detected in a large percentage of the most common human cancers (6, 8), we analyzed the status of PTEN expression in these ten cell lines. As shown in Figure 1B, PTEN expression was undetectable in the sunitinib-resistant 786–0, PC-3 and LNCaP cells. All other cell lines that were sensitive to sunitinib-mediated apoptosis manifested PTEN positivity.
Figure 1.
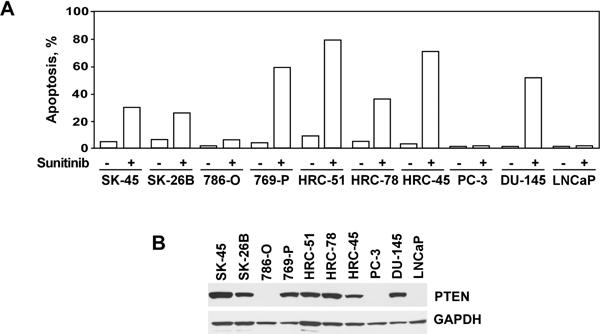
Differential induction of apoptosis by sunitinib in PTEN-positive and PTEN-negative renal and prostate cancer cell lines. (A) Cells were treated with sunitinib (25 μM) for 24 hours. The percentage of apoptotic cells was determined by the APO-BRDU DNA fragmentation assay followed by flow cytometry analysis as indicated in Materials and Methods. X-axis represents DNA content; Y-axis represents staining intensity. Representative data from one of five experiments are presented. (B) Western blot analysis of PTEN expression in renal and prostate cancer cell lines.
Makhov et al.
To validate our observation that PTEN expression modulates the sensitivity of malignant cells to sunitinib, we generated 786–0 renal and PC-3 prostate cancer cell lines capable of expressing PTEN (Fig. 2A). Consistent with the role of PTEN as a negative regulator of PI3K/Akt/mTOR signaling pathway, the level of Akt phosphorylation was markedly reduced in PTEN-expressing cells compared with 786-O and PC-3 cells transfected with the control vector (Fig. 2A). With successful restoration of PTEN expression in previously PTEN-deficient 786–0 and PC-3 cells, a pronounced sensitivity to sunitinib-mediated apoptosis was attained (Fig. 2B).
Figure 2.
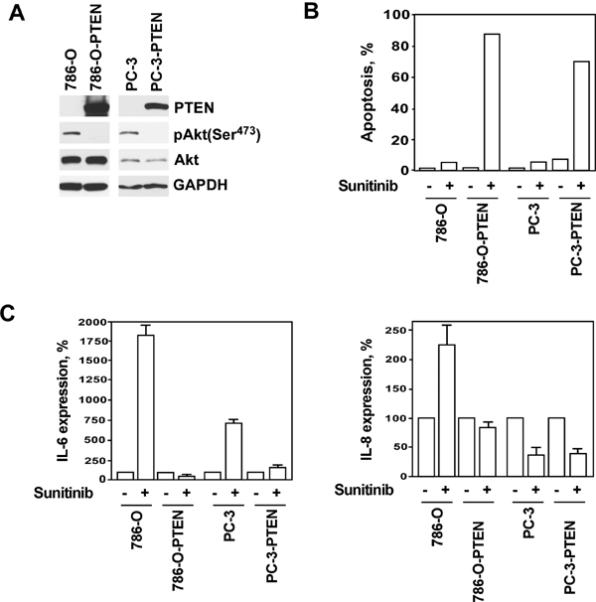
Expression of PTEN in PTEN-negative renal 786-O and prostate PC-3 cancer cells sensitizes them to sunitinib-mediated apoptosis. (A) Western blot analysis of PTEN expression in 786-O and PC-3 cells stably transfected with either PTEN or control vectors. (B) PTEN and control vector transfected 786-O and PC-3 cells were treated with sunitinib (25 μM) for 24 hours. The percentage of apoptotic cells was determined by APO-BRDU assay followed by flow cytometry analysis. Data is representative of one of three experiments. (C) IL-6 and IL-8 levels in cell culture supernatants of 786-O and PC-3 cells treated with sunitinib for 12 hours. The assay was performed in triplicates. Results are expressed as the mean ± SEM.
Makhov et al.
Restoration of PTEN expression suppresses the production of pro-tumorigenic cytokines, IL-6 and IL-8 by PC-3 and 786-O cells
IL-6 and IL-8 production is up-regulated following treatment with sunitinib in sunitinib-resistant hepatocellular carcinoma and renal cell carcinoma (9, 10). To investigate the potential impact of PI3K/Akt/mTOR signaling on the production of IL-6 and IL-8, we measured IL-6 and IL-8 levels in cell culture supernatants of PTEN-negative and PTEN-positive 786–0 and PC-3 cells treated with sunitinib for 12 hours. As shown in Figure 2C, treatment with sunitinib significantly increased expression levels of IL-6 in parental but not PTEN-positive cells. In PC-3 cells, this effect was most pronounced in the presence of TNF-alpha (unpublished observation). Interestingly, administration of sunitinib up-regulated IL-8 expression solely in 786-O but not PC-3 cells. Nevertheless, restitution of PTEN expression completely blocked sunitinib-mediated increase of IL-8 production by 786-O cells. It is important to note that IL-6 and IL-8 levels were measured before the onset of cell death at a point when cell viability was greater than 95%.
Sunitinib exhibits a significant anti-tumor effect in vivo on PTEN expressing PC-3 prostate cancer cells
To extend our in vitro findings to the in vivo setting, xenograft tumors were established in SCID mice using PC-3 cells that exhibited stable expression of PTEN. Control tumors were established using PTEN-negative PC-3 cells transfected with the control vector as described above. In addition, a xenograft model was created using 786-O and 786-O-PTEN tumors, however it was severely limited by the slow and unpredictable growth of both populations of cells to make its use practical. Animals were treated with vehicle alone or sunitinib (40 mg/kg) through oral gavage. Vehicle-treated animals uniformly showed a progressive increase in primary tumor growth. In contrast, tumor growth was consistently and markedly reduced in animals carrying PC-3-PTEN xenografts treated with sunitinib (p<0.001) (Fig. 3A). Treatment with sunitinib also caused an observable decrease in the growth of tumors established from control PC-3 cells (Fig. 3A), however this decrease was not statistically significant (p=0.08). At the termination of the study, tumors were resected and weighed. The weight of the tumors in mice injected with PC-3-PTEN tumors and treated with sunitinib were significantly lighter compared to mice injected with PC-3 tumors (p=0.002). There was no significant difference in the tumor weights between mice injected with PC-3 cells treated with vehicle or sunitinib (p=0.065) (Fig. 3B). It is important to underscore that whereas plasma levels of sunitinib were within a low micromolar range, intratumoral sunitinib concentrations were significantly higher (mean ± SEM: 5.4 ± 0.8μM versus 66.7 ± 15.8μM respectively) (Table 1). Our data confirms recent findings by Gotink et. al. demonstrating that intratumor sunitinib concentrations in the murine Renca RCC model and tumor tissue specimens obtained from cancer patients were 10-fold higher than the corresponding plasma concentrations (11). Indeed, our in vivo findings confirm the crucial role of PTEN status as a marker of sensitivity to sunitinib and provide further validation that sunitinib has a direct effect on tumor cells, which extends beyond the established angiogenic inhibitory actions.
Figure 3.
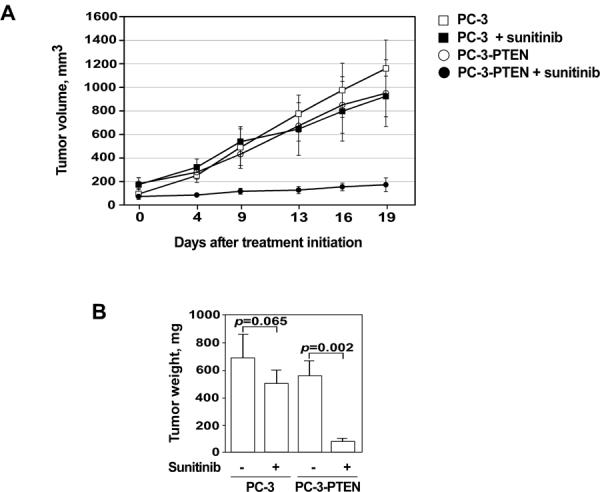
The effect of PTEN expression on tumor growth in vivo. PC-3-puro (squares) or PC-3-PTEN (circles) cells were inoculated s.c. in the flank region of 6 week old male C.B17/Icr-scid mice. Animals were treated with either sunitinib or control (vegetable oil) as detailed in Materials and Methods. (A) Dynamics of tumor growth in the experimental and control groups of animals. (B) Weights of xenograft tumors obtained from the experimental and control groups of animals. Data shown are mean of 5 mice in each group; bars, SEM.
Makhov et al.
Table 1.
Sunitinib levels in mouse plasma and xenograft tumor specimens. Sunitinib concentrations were examined as indicated in Materials and Methods section.
| Mean sunitinib concentrations | ||
|---|---|---|
| Plasma | Tumor | |
| Experimental group #1 (n = 3) | 4.63 μM | 54 μM |
| Experimental group #2 (n = 3) | 4.63 μM | 48 μM |
| Experimental group #3 (n = 3) | 7.08 μM | 98 μM |
| Mean (SEM) | 5.4 (0.8) | 66.7 (15.8) |
Pharmacological manipulation of PI3K/Akt/mTOR signaling enhances anti-tumor efficacy of sunitinib in vitro and in vivo
In order to explore a clinically relevant strategy to manipulate the PI3K/Akt/mTOR pathway in cells exhibiting sunitinib resistance, we pre-incubated parental PTEN-negative 786-O and PC-3 cells with pharmacologic inhibitors of PI3K/Akt/mTOR signaling - GDC-0980, a selective, dual PI3K/mTOR inhibitor, GSK690693, a pan-Akt inhibitor, and temsirolimus, an mTOR inhibitor (Fig. 4A). Temsirolimus is currently approved by the FDA for the treatment of advanced RCC (12). Phase I clinical trials for GDC-0980 and GSK690693 are currently recruiting participants (13, 14). The cytotoxic effect of sunitinib in combination with either GDC-0980, GSK690693 or temsirolimus was evaluated using the DNA fragmentation assay (Fig. 4B). As previously shown, treatment of PTEN-deficient renal or prostate cancer cells with sunitinib alone for 24 hours induced a negligible level of apoptosis. Similarly, treatment with GDC-0980, GSK690693 or temsirolimus alone similarly failed to induce a significant level of cell death in both cell lines. However, concomitant treatment of PC-3 and 786–0 cells with sunitinib and either GDC-0980, GSK690693 or temsirolimus resulted in a synergistic induction of DNA fragmentation in these previously resistant cell lines (Fig. 4B).
Figure 4.

The effect of concomitant treatment with sunitinib and temsirolimus, GDC-0980 or GSK690693 on tumor growth in vitro and in vivo. (A) Chemical structures of sunitinib, temsirolimus, GDC-0980 and GSK690693. (B) Parental PTEN-negative 786-O and PC-3 cells were pre-treated with GDC-0980 (10 μM), GSK690693 (20μM) or temsirolimus (25 μM) for 1 hour followed by treatment with sunitinib (25 μM) for 24 hours. The percentage of apoptotic cells was determined by APO-BRDU assay. Representative data are from one of three experiments. (C) Parental PC-3 and 786-O cells were cultured with various combinations of sunitinib (25μM), temsirolimus (25μM), GDC-0980 (10 μM), and GSK690693 (20μM) for 12 hours. Expression and phosphorylation status of PI3K/Akt/mTOR signaling proteins was evaluated by western blotting using specific antibodies. (D) IL-6 and IL-8 levels in cell culture supernatants of parental 786-O and PC-3 cells treated with sunitinib (25μM) and temsirolimus (25μM) for 12 hours. The assay was performed in triplicate. Results are expressed as the mean ± SEM. (E) Subcutaneous PC-3 tumors were established in 6 week old male C.B17/Icr-scid mice. Treatment with sunitinib and/or temsirolimus and assessment of tumor growth were performed as described in Materials and Methods.
Makhov et al.
Next, we examined the effect of various treatment modalities on the status of PI3K/Akt/mTOR pathway activity. Cells incubated with sunitinib, GDC-0980, GSK690693 or temsirolimus alone did not demonstrate any significant decrease in Akt phosphorylation. Of note, treatment with sunitinib alone resulted in modest down-regulation of p-AktT308 levels in PC-3 cells, however overall Akt activity was not affected, elicited by the fact that phosphorylation level of its direct target, Tuberin/TSC-2 (phospho-T1462), remained unchanged. Concomitant treatment with sunitinib and either GDC-0980, GSK690693 or temsirolimus resulted in complete inhibition of PI3K/Akt/mTOR signaling, as indicated by lack of phosphorylation of TSC2 and 4EBP1 targets (Fig. 4C). Pharmacologic inhibition of mTORC1 signaling with temsirolimus also blocked sunitinib-mediated up-regulation of IL-6 and IL-8 expression in PC-3 and 786-O cells (Fig. 4D).
In light of our encouraging in vitro data, we next examined the anti-tumor effects of sunitinib in combination with temsirolimus using a mouse xenograft model bearing PTEN-negative PC-3 tumors. Three weeks after the initiation of therapy, single agent treatment with temsirolimus or sunitinib showed a notable decrease in the growth of xenograft tumors (Fig. 4E). However, combination treatment with sunitinib and temsirolimus resulted in vastly more impressive inhibition of tumor growth among the experimental groups. These results confirm the importance of PTEN as an important surrogate for tumor cell resistance to TKIs but also provide pre-clinical data to suggest a readily available clinical strategy to circumvent resistance patterns in PTEN-deficient tumors.
DISCUSSION
Treatment with multi-targeted TKIs has dramatically altered the landscape in the treatment of patients with advanced tumors such as kidney and certain gastrointestinal tumors (1, 2). Despite encouraging results, clinical studies of certain solid organ malignancies, i.e. CRPC, have shown a suboptimal response to the TKI, sunitinib (5, 15–17). Our present work with urologic malignancies features sunitinib resistance as a factor of PTEN expression, which serves the role of a gatekeeper of the PI3K/Akt/mTOR signaling pathway. Re-establishment of PTEN expression in PTEN deficient cancer cells restores sensitivity to TKI therapy. Furthermore, suppression of PI3K/Akt/mTOR signaling with clinically relevant agents such as temsirolimus, GDC-0980 and GSK690693, serves to reinstate sensitivity of PTEN-deficient cancer cells to TKI-mediated apoptosis.
Treatment with sunitinib completely abrogates Akt-dependent and Akt-independent survival signaling in PTEN-positive cells. Importantly, PTEN-negative cells exhibit constitutively active Akt signaling downstream of sunitinib's cellular targets (Fig. 5). As such, additional pharmacologic modulation is required to suppress aberrant Akt activity. To elaborate further, we conclude that inhibition of Akt signaling alone is incapable of inducing apoptosis due to the existence of Akt-independent survival pathways. Thus, only concomitant administration of sunitinib and PI3K/Akt/mTOR inhibitors is capable of triggering apoptosis in PTEN-negative cancer cells.
Figure 5.

Potential mechanisms of interaction between sunitinib and PI3K/Akt/mTOR inhibitors.
Makhov et al.
The ability of multi-targeted TKIs to inhibit angiogenesis, and thereby affect tumor progression, has been well-established (3, 18). Recent studies make evident that sunitib's sole influence stems from its anti-angiogenic effect, further emphasizing its lack of ability to exert a direct anti-tumor effect. These observations are based on the notion that pharmacologically relevant plasma concentrations of sunitinib are insufficient to inhibit RCC tumor cell proliferation (19). Alternatively, other investigations have elucidated that actions of sunitinib are not limited to inhibition of angiogenesis, but additionally show clear evidence of tumor growth inhibition (20, 21) and anti-tumor immune response modulation (22). Our present findings are consistent with latter, providing compelling evidence of its multi-targeted effects, i.e. direct anti-tumor effect in conjunction with the previously established anti-angiogenic activity. These observations are made evident by the fact that intratumor sunitinib levels are vastly higher than the corresponding plasma concentrations as previously reported (23, 24) and confirmed by our own results (Table 1). In fact, sunitinib modulates a series of complex interactions between tumor and the surrounding tissues, thereby influencing multiple cellular processes, such as angiogenesis, cell growth, and cell survival (Fig. 5).
Our in vitro experiments make evident that PTEN expression inversely correlates with sunitinib resistance in renal and prostate cancer cells. Our in vitro findings were further validated in vivo: resistance to sunitinib was demonstrated in PTEN-negative PC-3 xenograft tumors, while profound sensitivity to sunitinib was gained when xenograft tumors were established from PTEN-positive PC-3 cells. Furthermore, we successfully overcame sunitinib resistance in PTEN-negative xenograft tumors using temsirolimus, a pharmacological inhibitor of mTORC1 signaling. Although our findings require clinical validation, PTEN tumor status could be translated into a biomarker to assist guide selection of therapeutic agents presently approved by the FDA for treatment of advanced urologic malignancies.
ACKNOWLEDGEMENTS
We acknowledge the support of the Laboratory Animal and Cell Sorting Facilities at Fox Chase Cancer Center.
GRANT SUPPORT This work was supported in part by the National Institutes of Health Grant RO1 CA134463 (VMK); the American Institute for Cancer Research Grant (RGU); the Department of Defense, Physician Research Training Award (AK).
This work was supported in part by the National Institutes of Health Grant RO1 CA134463 (VMK); the American Institute for Cancer Research Grant (RGU); the Department of Defense, Physician Research Training Award (AK).
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
REFERENCES
- 1.Goodman VL, Rock EP, Dagher R, Ramchandani RP, Abraham S, Gobburu JV, et al. Approval summary: sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma. Clin Cancer Res. 2007;13:1367–73. doi: 10.1158/1078-0432.CCR-06-2328. [DOI] [PubMed] [Google Scholar]
- 2.Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115–24. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 3.Gotink KJ, Verheul HM. Anti-angiogenic tyrosine kinase inhibitors: what is their mechanism of action? Angiogenesis. 2010;13:1–14. doi: 10.1007/s10456-009-9160-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, et al. Sunitinib versus Interferon Alfa in Metastatic Renal-Cell Carcinoma. N Engl J Med. 2007;356:115–24. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 5.Sonpavde G, Periman PO, Bernold D, Weckstein D, Fleming MT, Galsky MD, et al. Sunitinib malate for metastatic castration-resistant prostate cancer following docetaxel-based chemotherapy. Ann Oncol. 2010;21:319–24. doi: 10.1093/annonc/mdp323. [DOI] [PubMed] [Google Scholar]
- 6.Ciuffreda L, Di Sanza C, Incani UC, Milella M. The mTOR pathway: a new target in cancer therapy. Curr Cancer Drug Targets. 2010;10:484–95. doi: 10.2174/156800910791517172. [DOI] [PubMed] [Google Scholar]
- 7.Golovine K, Makhov P, Uzzo RG, Shaw T, Kunkle D, Kolenko VM. Overexpression of the zinc uptake transporter hZIP1 inhibits nuclear factor-kappaB and reduces the malignant potential of prostate cancer cells in vitro and in vivo. Clin Cancer Res. 2008;14:5376–84. doi: 10.1158/1078-0432.CCR-08-0455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Menon S, Manning BD. Common corruption of the mTOR signaling network in human tumors. Oncogene. 2008;(Suppl 2):S43–51. doi: 10.1038/onc.2009.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang D, Ding Y, Zhou M, Rini BI, Petillo D, Qian CN, et al. Interleukin-8 mediates resistance to antiangiogenic agent sunitinib in renal cell carcinoma. Cancer Res. 2010;70:1063–71. doi: 10.1158/0008-5472.CAN-09-3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu AX, Sahani DV, Duda DG, di Tomaso E, Ancukiewicz M, Catalano OA, et al. Efficacy, safety, and potential biomarkers of sunitinib monotherapy in advanced hepatocellular carcinoma: a phase II study. J Clin Oncol. 2009;27:3027–35. doi: 10.1200/JCO.2008.20.9908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gotink K, Broxterman HJ, Labots M, de Haas RR, Dekker H, Honeywell RJ, et al. Lysosomal sequestration of sunitinib: a novel mechanism of drug resistance. Clin Cancer Res. 2011;17:7337–46. doi: 10.1158/1078-0432.CCR-11-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kwitkowski VE, Prowell TM, Ibrahim A, Farrell AT, Justice R, Mitchell SS, et al. FDA approval summary: temsirolimus as treatment for advanced renal cell carcinoma. Oncologist. 2010;15:428–35. doi: 10.1634/theoncologist.2009-0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–62. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 14.Wallin JJ, Edgar KA, Guan J, Berry M, Prior WW, Lee L, et al. GDC-0980 is a novel class I PI3K/mTOR kinase inhibitor with robust activity in cancer models driven by the PI3K pathway. Mol Cancer Ther. 2011;10:2426–36. doi: 10.1158/1535-7163.MCT-11-0446. [DOI] [PubMed] [Google Scholar]
- 15.Aragon-Ching JB, Dahut WL. About tyrosine kinase inhibitors (TKIs) in prostate cancer: where do we go from here? Ann Oncol. 2010;21:183–4. doi: 10.1093/annonc/mdp467. [DOI] [PubMed] [Google Scholar]
- 16.Dror Michaelson M, Regan MM, Oh WK, Kaufman DS, Olivier K, Michaelson SZ, et al. Phase II study of sunitinib in men with advanced prostate cancer. Ann Oncol. 2009;20:913–20. doi: 10.1093/annonc/mdp111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sonpavde G, Hutson TE, Berry WR, Boehm KA, Asmar L. Phase II trial of sunitinib for the therapy of progressive metastatic castration-refractory prostate cancer after previous docetaxel chemotherapy. Clin Genitourin Cancer. 2008;6:134–7. doi: 10.3816/CGC.2008.n.023. [DOI] [PubMed] [Google Scholar]
- 18.Chow LQ, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. J Clin Oncol. 2007;25:884–96. doi: 10.1200/JCO.2006.06.3602. [DOI] [PubMed] [Google Scholar]
- 19.Huang D, Ding Y, Li Y, Luo WM, Zhang ZF, Snider J, et al. Sunitinib acts primarily on tumor endothelium rather than tumor cells to inhibit the growth of renal cell carcinoma. Cancer Res. 2010;70:1053–62. doi: 10.1158/0008-5472.CAN-09-3722. [DOI] [PubMed] [Google Scholar]
- 20.Seandel M, Shia J, Linkov I, Maki RG, Antonescu CR, Dupont J. The activity of sunitinib against gastrointestinal stromal tumor seems to be distinct from its antiangiogenic effects. Clin Cancer Res. 2006;12:6203–4. doi: 10.1158/1078-0432.CCR-06-1292. [DOI] [PubMed] [Google Scholar]
- 21.Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. 2009;69:2506–13. doi: 10.1158/0008-5472.CAN-08-4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heine A, Held SA, Bringmann A, Holderried TA, Brossart P. Immunomodulatory effects of anti-angiogenic drugs. Leukemia. 2011;25:899–905. doi: 10.1038/leu.2011.24. [DOI] [PubMed] [Google Scholar]
- 23.Gotink KJ, Broxterman HJ, Labots M, de Haas RR, Dekker H, Honeywell RJ, et al. Lysosomal sequestration of sunitinib: a novel mechanism of drug resistance. Clin Cancer Res. 2011;17:7337–46. doi: 10.1158/1078-0432.CCR-11-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hammers HJ, Verheul HM, Salumbides B, Sharma R, Rudek M, Jaspers J, et al. Reversible epithelial to mesenchymal transition and acquired resistance to sunitinib in patients with renal cell carcinoma: evidence from a xenograft study. Mol Cancer Ther. 2010;9:1525–35. doi: 10.1158/1535-7163.MCT-09-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]