

Abstract
Background
Despite improvement in therapeutic techniques, patients with early-stage laryngeal cancer still recur after treatment. Gene expression prognostic models could suggest which of these patients would be more appropriate for testing adjuvant strategies.
Materials and methods
Expression profiling using whole-genome DASL arrays was carried out on 56 formalin-fixed paraffin-embedded tumor samples of patients with early-stage laryngeal cancer. We split the samples into a training and a validation set. Using the supervised principal components survival analysis in the first cohort, we identified gene expression profiles that predict the risk of recurrence. These profiles were then validated in an independent cohort.
Results
Gene models comprising different number of genes identified a subgroup of patients who were at high risk of recurrence. Of these, the best prognostic model distinguished between a high- and a low-risk group (log-rank P < 0.005). The prognostic value of this model was reproduced in the validation cohort (median disease-free survival: 38 versus 161 months, log-rank P = 0.018), hazard ratio = 5.19 (95% confidence interval 1.14–23.57, P < 0.05).
Conclusions
We have identified gene expression prognostic models that can refine the estimation of a patient's risk of recurrence. These findings, if further validated, should aid in patient stratification for testing adjuvant treatment strategies.
Keywords: early stage, expression profiling, laryngeal cancer, recurrence
introduction
Laryngeal cancer is one of the most common subtypes among head and neck malignancies [1, 2]. In about half of the cases, laryngeal cancer is located on the vocal cords. These patients frequently present in early stages with persistent hoarseness [3]. Early-stage (T1NOMO, T2NOMO) disease can be treated with radiotherapy or partial laryngectomy, endoscopic or open, according to the NCCN guidelines [4]. Five-year survival of patients with early-stage laryngeal cancer is 80%–85% after potentially curative treatment [5, 6]. Despite the improvement of the therapeutic techniques, some patients still recur after treatment. Known prognostic factors for early-stage disease are tumor differentiation [7, 8], involved margins of the dissection [9] and perineural invasion [10, 11]. However, these factors cannot adequately predict which patients will recur or not and the assessment of risk needs further refinement. Expression profiling has been successfully used in other types of cancer to identify subgroups of patients with early-stage disease who would potentially benefit from the administration of more aggressive treatments [12–15]. Molecular profiling, in early-stage cancer of the larynx, is a field that has not been extensively studied. Some microarray studies, focusing mostly on advanced laryngeal cancer, have revealed candidate molecular markers of laryngeal cancer, genes predictive of metastasis and genes differentially expressed between cancer and non-neoplastic tissue or between early and advanced cancer of the larynx [16–20].
In this study, we carried out expression profiling using paraffin samples on a panel of patients with early-stage laryngeal cancer. Splitting our cohort randomly in half, we identified genes prognostic of disease-free survival (DFS) in the first set and validated their performance in the second independent set of samples. We were able to identify several gene sets with similar performance, which were able to distinguish patients at high risk of recurrence.
materials and methods
study population
Our cohort includes 61 early-stage squamous cell laryngeal cancer samples from patients diagnosed between 1994 and 2007. These patients were part of two longitudinal cohorts of 78 and 37 early-stage (T1N0M0, T2N0M0) laryngeal cancer patients treated between 1994 and 2007 at two institutions, the AHEPA hospital of Thessaloniki and the ‘Metaxas’ Cancer Hospital, Piraeus, respectively. Among these 115 patients, we focused on the ones for whom we had available associated tissue material. All patients were treated locally either with surgery or radiotherapy. None of the patients received chemotherapy. Follow-up examinations included physical examination and endoscopy every 6 months. Additional examinations were carried out as indicated.
tumor specimens
Formalin-fixed paraffin-embedded (FFPE) samples were collected from the Department of Pathology of the Aristotle University of Thessaloniki and from the Department of Pathology of the ‘Metaxas’ Cancer Hospital in Piraeus. We used paraffin blocks with a tumor cell content of >50%, which is consistent with recent literature [21]. All slides were once more reviewed by a single specialist pathologist (MB). The present study was approved by the Bioethics Committee of the Aristotle University of Thessaloniki, School of Medicine. Waiver of consent was obtained from this committee for all patients included in the study before 2003. All patients included in the study after 2003 provided their informed consent for the provision of biological material for future research studies.
RNA isolation and whole-genome DASL profiling
FFPE samples were cut into 1- to 3-mm cores. Isolation of total RNA was carried out using the Qiagen RNeasy FFPE protocol. Profiling included whole-genome 24K DASL (c-DNA-mediated, Annealing, Selection and Ligation) arrays (Illumina, San Diego, CA). The DASL assay is a bead-based method for high-throughput expression profiling of degraded RNA, frequently found in FFPE samples [22–25]. Profiling experiments were carried out at the Molecular Genetics Core, Children's Hospital (Boston) and Harvard Medical School. Outlier exclusion was based on the percent present call of the samples and their ratio signal to background noise. Our exclusion criteria included either a ‘signal to background noise ratio value’ <2 or >100 or a detection rate <3000 genes, P < 0.05. Detailed values can be found in the supplemental Table S6 (available at Annals of Oncology online).
Normalization was carried out following commercially available manufacturer instructions (Genome StudioTM, Gene Expression Module v1.0 User Guide; Illumina). Sample intensities are scaled by a factor equal to the ratio of average intensity of a virtual reference sample to the average intensity of a given sample. Background is subtracted before the scaling.
The gene expression data have been deposited in National Center for Biotechnology Information Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) and are available through GEO series accession number GSE25727. The following link has been created to allow review of record GSE25727: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=jbkdfscswekwgdw&acc=GSE25727.
statistical analysis
First, we randomly split our cohort into a training and a validation set. Survival models were developed exclusively on the training set. We focus on genes univariately associated with DFS (P < 0.05, Cox proportional hazard model). We ranked these genes based on their hazard ratio (HR) and chose several sets with different numbers of top ranking genes. Using the supervised principal component survival algorithm [26], we developed prognostic models to assign a risk of recurrence to each clinical sample. We directly applied these models to the validation set without any modification. Kaplan–Meier (DFS) curves were plotted for two risk groups, with higher or lower than median risk of death. Statistical significance was assessed using a random permutation-corrected log-rank test. All reported P values are two-sided. Univariate and multivariate adjustment for known prognostic factors was carried out using Cox proportional hazards analysis. Analyses were carried out using BRB-ArrayTools developed by Dr Richard Simon and BRB-ArrayTools Development Team and SPSS software, version 18, (IBM Corporation, NY).
We used Subclass Mapping (Submap) methodology (Gene Pattern Software, Version 3.0; Broad Institute) [27] to assess whether high- and low-risk groups of the training set correspond molecularly to the respective groups of the validation set. Submap is an unsupervised method which assesses the molecular similarity of groups of samples in multiple independent datasets despite their technical differences. This algorithm assesses the enrichment of the expression profile of each predefined phenotype in the first dataset for marker genes identified in the other dataset. A P value is also provided to show the likelihood that these different phenotypes share similar underlying molecular properties.
We also carried out gene set analysis (GSA) [28] to assess whether the expression profiles of groups with high- or low-risk patients were enriched for biological themes or functional groups of genes.
Finally, we applied publicly available, experimentally developed and validated gene expression ‘readouts’ of oncogenic pathway activation. These readouts were utilized to train Bayesian probit regression models to estimate the probability of activation of each of those pathways [29]. We applied these models on each sample of our study, after correcting for nonbiological experimental variation between the two different datasets, using the batch effect adjustment algorithm [30]. Each individual sample was assigned a probability value (from zero to one) of pathway activation. In order to afford high confidence, a probability value >0.8 was used as a cut-off for predicted pathway activation. We used these predictions to explore associations between known oncogenic pathways and the different subgroups, employing 2 × 2 table statistics and odds ratios.
results
identification and validation of multigene prognostic classifiers
The flow of our study can be seen in Figure 1. First we excluded five technical outliers. The clinical characteristics of the remaining patients are listed in Table 1. We then randomly split the remaining 56 samples into two equal groups, training and validation sets. In the training set, we identified different prognostic models choosing genes associated with DFS (Cox regression P = 0.05) after ranking them based on their absolute HR value. Models with a range of different number of genes (40–100) carried out well in the training set. A gene set comprising 70 top ranking genes was the best prognostic model in the training set (median DFS: 92 versus 123 months, log-rank P = 0.003, permutation P = 0.04; Figure 2). Gene models with as few as 40 genes gave similar and significant results. We then applied these models (40–100 genes) to the validation set and demonstrated that they could distinguish between high- and low-risk samples (median DFS: 38 versus 161 months for the 70-gene model, log-rank P = 0.018). HR of recurrence for the high-risk group versus the low-risk group in the validation set was HR = 5.19 [95% confidence interval (CI) 1.14–23.57, P = 0.033] (an HR for the training set could not be estimated because of the small number of events). Results for the additional gene models demonstrated statistical discrimination of low- and high-risk samples and can be found in the supplemental Table S1 (available at Annals of Oncology online). Concordance between the risk assignments using different gene sets was high, 87% (Cramer V test = 0.729), suggesting that the risk assessments based on the different prognostic models represent a stable genomic phenotype. Detailed identification of the genes in the 70-gene model can be found in the supplemental Table S2 (available at Annals of Oncology online).
Figure 1.
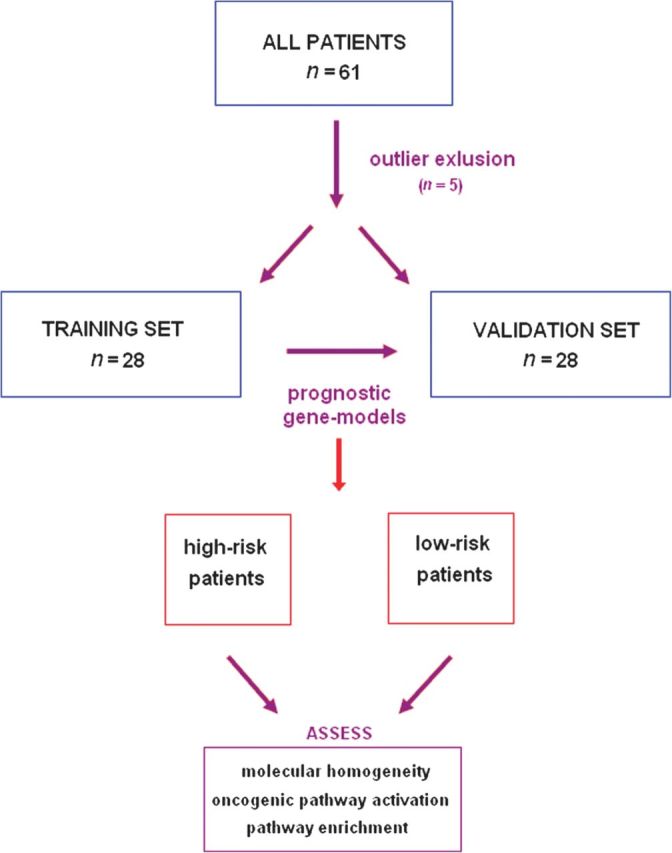
Consort diagram. The initial cohort comprises 61 samples. After excluding outlier samples, we randomly split the cohort into two sets of 28 patients each. The first set of patients was used to develop prognostic gene models, which were then independently validated in the second set of patients. We used SubMap to assess the molecular correspondence of low- and high-risk phenotypes in the training and validation sets.
Table 1.
Clinical and pathological characteristics of training and validation cohorts
| Clinical and pathological characteristics |
|||
|---|---|---|---|
| Training set | Validation set | ||
| Patients (n = 28) | Patients (n = 28) | ||
| Characteristics | No. (%) | No. (%) | P value |
| Age | |||
| Median (range) | 62.5 (48–77) | 63 (40–87) | 0.902 |
| Gradea | |||
| 1 | 11 (47.83) | 10 (38.46) | 0.925 |
| 2 | 9 (39.13) | 12 (46.15) | |
| 3 | 3 (13.04) | 4 (15.38) | |
| Gender | |||
| Male | 26 (92.86) | 26 (92.86) | 0.681 |
| Female | 2 (7.14) | 2 (7.14) | |
| Smoking | |||
| Yes | 23 (82.14) | 26 (92.86) | 0.197 |
| No | 5 (17.86) | 2 (7.14) | |
| Alcohol consumption | |||
| Yes | 15 (53.5) | 14 (50) | 0.449 |
| No | 13 (46.5) | 14 (50) | |
| Treatment modality | |||
| Surgery | 23 (82.1) | 24 (85.7) | 1.000 |
| Radiation therapy | 5 (17.9) | 4 (14.3) | |
| Disease-free survival (months) | |||
| Median (range) | 48 (1–148) | 59 (0–161) | 0.376 |
| Recurrence site | |||
| Primary site | 7 (25) | 7 (25) | 1.000 |
| Lymph nodes | 2 (7.14) | 1 (3.57) | |
| Distant site | 0 (0) | 0 (0) | |
Differences between patients of the training and validation sets were assessed using t-statistics for grade, gender, smoking status (defined as any smoking versus no smoking), treatment modality, recurrence site and alcohol consumption. Differences between patients of the training and validation sets were assessed using Mann–Whitney U test for age and disease-free survival. There was no statistically significant difference between training and validation sets in age, grade, gender, smoking status, alcohol consumption, treatment modality used and recurrence site or disease-free survival.
aGrade was not available for seven patients.
Figure 2.

Association between 70-gene model and disease-free survival (DFS) in the training and validation sets. Based on the 70-gene model, 13 patients of the training set and 8 patients of the validation set were predicted as high risk, while 15 patients of the training set and 20 patients of the validation set were predicted as low risk. The 70-gene model distinguished between a high- and a low-risk group in the training set with a median DFS of 92 and 123 months, respectively (log-rank P < 0.003, permutation P < 0.05), and a high- and a low-risk group for DFS in the validation set (median DFS 38 161 months, log-rank P = 0.018).
We were interested in evaluating whether the effect of the 70-gene model was independent of known clinicopathological prognostic factors. We first determined that high- and low- risk groups were well balanced for grade, smoking status and alcohol consumption. Using the entire dataset, we indeed observed that the 70-gene model maintained its prognostic significance, while grade did not. Specifically, for the 70-gene model, the univariate and multivariate HR of recurrence for the high-risk group versus the low-risk group was 8.33 (95% CI 2.29–30.32, P = 0.001) and 8.33 (95% CI 2.25–30.75, P = 0.001), respectively. For the histological differentiation, the univariate and multivariate HR was 1.86 (95% CI 0.96–3.60, P = 0.07) and 1.41 (95% CI 0.75–2.65, P = 0.28). Similarly, several models ranging from 40–100 genes maintained independent prognostic significance (details in supplemental TableS3, available at Annals of Oncology online).
For further external validation, we looked at publicly available gene expression datasets. Very few, if any, were comparable to our collection of samples. We used gene expression data for 24 laryngeal cancer samples, published by Chung et al. in 2004 [31]. Focusing on the 34 genes of our predictor that were present in that platform, we used hierarchical clustering and identified two groups with a statistically significant difference in DFS (median DFS: 16 months versus not yet reached, log-rank P = 0.01). Thus, despite the technical differences and the disparate nature of this second independent dataset (Agilent versus Illumina, different number of probe sets; advanced-stage versus early-stage laryngeal cancer samples, different biological material), we were able to reproduce the power of our gene signature.
Furthermore, we explored whether possible heterogeneity in the characteristics and treatment patterns in our cohort may have influenced our results. First, we split our patients into two groups based on the date of diagnosis, treated before and after 2000 (median time point). We carried out multivariate analysis using a Cox proportional hazards regression, with the gene model and the chronologically defined subsets as covariates and demonstrated that our signature was independent of the time effect on our cohort (HR = 7.97, 95% CI 2.18–29.14, P = 0.002). Additionally, we considered that two different modalities, surgery and radiation therapy were used to treat these patients. We examined whether our training and validation sets as well as high- and low-risk patients, as defined by our 70-gene model, were enriched for patients treated either with surgery or radiation therapy. We indeed observed that there was no statistically significant difference in the therapeutic modality used either between the training and validation sets or in the high- and low-risk groups (Fisher's exact test, P = 1.000 and P = 1.000, respectively). Furthermore, we assessed whether patients treated with radiation had a survival difference compared with the ones treated with surgery. There was not a statistically significant difference between the two groups (P = 0.36).
molecular match of high- and low-risk groups between the training and validation sets
We then sought to assess whether the high- and low-risk samples in the training set were molecularly homologous to the respective groups in the validation set above and beyond the expression patterns of the 70-gene model. For this reason, we used Subclass Mapping (Submap), a method that assesses the molecular correspondence (molecular match) of predefined subtypes in different datasets above and beyond the collection of specific genes. Results are shown in Figure 3, demonstrating good molecular match of the high-risk samples in the two datasets. Low-risk samples seem to be more heterogeneous and with no obvious strong correspondence.
Figure 3.

Genome-wide molecular match of high- and low-risk groups between training and validation sets. Analysis of molecular correspondence between high- and low-risk groups in the training and validation sets. Red color denotes high confidence for correspondence; blue color denotes lack of correspondence. False Discovery Rate-adjusted P values are noted in the corresponding box.
pathway analysis in high- and low-risk disease groups
In order to explore the pathways and biological themes enriched in the gene profiles of high- and low-risk groups, we carried out pathway analysis. GSA was carried out between high- and low-risk groups based on the 70-gene model assignments and revealed 39 statistically, significantly differentially expressed pathways (Efron–Tibshirani GSA test P < 0.05). Selected pathways, some of them not previously associated with outcome in head and neck cancer, such as tumor necrosis factor/stress-related signaling pathway and phospholipase C signaling pathway are shown at Table 2, while the full list can be found in the supplemental Table S4 (available at Annals of Oncology online).
Table 2.
Functional gene set analysis in high- and low-risk disease samples
| Pathways–gene sets | ||
|---|---|---|
| GSA | Up-regulated in high-risk group | Up-regulated in low-risk group |
| VEGF signaling pathway | TGF-b signaling pathway | |
| Phospholipase C signaling pathway | CD40L signaling pathway | |
| Shh pathway | TNF/stress-related signaling | |
| Trka receptor signaling pathway | Role of EGF receptor transactivation by GPCRs in cardiac hypertrophy | |
| Signal transduction through IL1R | ||
| NF-kB signaling pathway | ||
| Downregulated in high-risk group | Downregulated in low-risk group | |
| Basal transcription factors | Cell cycle: G1/S check point | |
| TNFR2 signaling pathway | ||
GSA demonstrated pathways that were statistically, significantly differentially expressed in either high- or low-risk patients (Effron–Tibshirani GSA, P < 0.05). Selected pathways of interest are shown in this table. The full list of these pathways is found in the supplementary data.
EGF, endothelial growth factor; GSA, gene set analysis; Shh, Sonic Hedgehog; TGF-b, transforming growth factor-beta; VEGF, vascular endothelial growth factor; GPCR, G protein-coupled receptor; TNFR2, tumour necrosis factor receptor.
prognostic gene expression models reflect activation of RAS oncogenic pathway in individual tumor samples
The previous analysis was designed to assess pathway activation status in a group of patients not in individual samples. Therefore, we used experimentally validated gene expression readouts and identified individual tumor samples with activation of known oncogenic pathways. First, we obtained the probability of activation for pathways ras, src and b-catenin for each laryngeal cancer sample. Of the 21 high-risk patients, based on the 70-gene model risk assignments, 13 had ras pathway and 4 b-catenin pathway activated. Of the 35 low-risk patients, 10 appeared to have ras pathway activated and 15 b-catenin pathway activated. Furthermore, we observed that the odds ratio of the ras pathway activation in the unfavorable group was 4.063 (95% CI 1.29–12.78, P = 0.02), while the odds ratio for b-catenin pathway activation was 0.22 (95% CI 0.59–0.82, P= 0.04). The odds ratio for src pathway was not statistically significant.
In multivariate analysis including the 70-gene model, the two oncogenic pathways, ras and b-catenin and the histological differentiation, the 70-gene model maintained its independent prognostic significance (details in the supplemental Table S5, available at Annals of Oncology online).
discussion
While it has been previously shown that gene profiles are associated with survival of patients suffering from different types of cancer [32–37], such analysis in early-stage laryngeal cancer is lacking. In this paper, we sought to identify a robust gene expression profile in patients with early-stage laryngeal cancer in order to address an important dilemma in this disease. Patients at high risk of recurrence, if identified, could be treated either with more aggressive locoregional approaches or even with a combination of local and systemic therapy. Patients at low risk of recurrence may be spared the side-effects of such unnecessary treatments.
We identified several gene models that can distinguish between high- and low-risk patients. We then used a separate cohort to validate these models as suggested in the literature [38]. We observed high reproducibility of our results in the validation cohort despite the small sample size. Additionally, we established the independent prognostic significance of our profile, demonstrating that it was not confounded by grade. While larger number of patients could perhaps result in even more robust findings, one should take into consideration that early-stage laryngeal cancer is a rare disease and access to larger numbers of patients can be challenging.
Our study comprises of patients treated either with surgery or radiation therapy over a 13-year period. We have to acknowledge that progress in the treatment modalities might have influenced the results of this study. To address this issue, we carried out multivariate analysis and observed that our prognostic profile maintained its independent prognostic significance despite the various changes that might have influenced treatment over this long period of sample collection. While it would be ideal to have focused on patients treated uniformly during a short time period, access to such a cohort of a large sample size would be very difficult.
Interestingly, we found that not only our 70-prognostic model was reproducible in the validation set but so was the molecular phenotype of high risk (above and beyond the 70-gene model) as evidenced by Subclass Mapping analysis (Figure 3). This suggests that there is a unique molecular program of high risk and recurrence in early-stage laryngeal cancer, which could perhaps be targeted for additional therapies. In contrast, the lack of strong molecular match between the low-risk groups may either be a function of small sample size or possibly a higher molecular heterogeneity in good prognosis larynx cancer. Furthermore, we used a publicly available dataset [31] to externally validate our signature. Despite the technical variation of this second dataset, we were able to demonstrate the prognostic relevance of our signature.
After distinguishing patients at high risk and low risk of recurrence, we looked into the biological characteristics of these groups in an attempt to identify pathways or biological themes hidden behind the expression profiles of these tumors. We used GSA in order to explore pathways up- or downregulated in either high- or low-risk patients. We indeed identified several pathways with clear evidence of relevance to head and neck cancer, such as vascular endothelial growth factor (VEGF) signaling pathway, Sonic Hedgehog (Shh) pathway, transforming growth factor-beta (TGF-b) signaling pathway and NF-kB signaling pathway. For example, it has been previously shown that VEGF positivity, determined by immunohistochemistry, is a negative prognostic marker for patients with early-stage laryngeal cancer [39]. Another pathway which was found to be up-regulated in high-risk tumors, based on the 70-gene model, Shh pathway, has been found to be associated with poor prognosis in patients with head and neck cancer [40]. Furthermore, downregulation of TGF-b has been associated with loss of differentiation in oral carcinomas [41] and has also been correlated with depth of invasion, lymph node metastasis, pathological stage and poor prognosis in esophageal squamous cell carcinomas [42]. These findings, which are consistent with our results, suggest that downregulation of TGF-b is characteristic of more aggressive tumors such as the ones in patients with recurrence. All these reports in the literature further strengthen the reliability of the associations identified in our analysis. In addition, we identified several pathways, known to be associated with survival in other types of cancer, such as phospholipase C, CD40L signaling pathway [43–45]. These pathways can potentially serve as novel areas of research in laryngeal cancer.
Another novel finding of this study is that RAS pathway activation is statistically significantly higher in the high-risk patients. The fact that the unfavorable group is enriched with tumors characterized by RAS pathway activation, suggests that these patients can benefit from drugs targeting this pathway. However, these data originate from in silico analysis and need further experimental and prospective validation in order to be incorporated in daily clinical practice. Additional clinical studies are mandatory to investigate whether patients with tumors characterized by a specific phenotype will gain benefit from more aggressive surveillance or more aggressive treatment.
Our study purposely utilized paraffin-embedded tissue for the gene expression experiments. The use of FFPE tissue has clear advantages over the use of fresh frozen tissue, since it is abundant and easily accessible both in the academic community and in local hospitals. Conversely, performing microarray studies can be challenging due to technical limitations, such as degradation and chemical modification of genetic material (DNA, RNA) of the tissue stored in paraffin. Recent developments in technology have allowed the use of FFPE tissue samples for gene expression profile with reliable and reproducible results [22, 24, 25, 47–50], as it was done in our study.
Previous studies in laryngeal cancer have focused mostly on identifying biomarkers of carcinogenesis, e.g. Markowski et al. [16] profiled 14 patients and identified novel biomarkers of laryngeal cancer. Additional information originates from very small numbers of laryngeal samples embedded in larger studies with different head and neck cancer subtypes [30, 51–54]. To our knowledge, this is the first study discovering and validating multigene expression profiles associated with outcome and potentially useful for clinical management in early-stage laryngeal cancer. We anticipate that further prospective validation of this approach in a multicenter setting will aid in developing refined management patterns. Finally, some of the themes implicated in our study can form the basis for further mechanistic studies and ultimately aid in patient-tailored selection of therapeutic strategies.
funding
Hellenic Cooperative Oncology Group (HE R_5G research grant).
disclosure
The authors declare no conflicts of interest.
Supplementary Material
acknowledgements
Microarray studies were carried out by the Molecular Genetics Core Facility at Children's Hospital Boston supported by NIH-P50-NS40828 and NIH-P30-HD18655.
references
- 1.Carvalho AL, Nishimoto IN, Califano JA, Kowalski LP. Trends in incidence and prognosis for head and neck cancer in the United States: a site-specific analysis of the SEER database. Int J Cancer. 2005;114:806–816. doi: 10.1002/ijc.20740. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 3.Hoffman HT, Porter K, Karnell LH, et al. Laryngeal cancer in the United States: changes in demographics, patterns of care, and survival. Laryngoscope. 2006;116:1–13. doi: 10.1097/01.mlg.0000236095.97947.26. [DOI] [PubMed] [Google Scholar]
- 4.Pfister DG, Ang K, Brockstein B, et al. NCCN Practice Guidelines for Head and Neck Cancers. Oncology (Williston Park) 2000;14:163–194. [PubMed] [Google Scholar]
- 5.Mendenhall WM, Werning JW, Hinerman RW, et al. Management of T1-T2 glottic carcinomas. Cancer. 2004;100:1786–1792. doi: 10.1002/cncr.20181. [DOI] [PubMed] [Google Scholar]
- 6.Yuce I, Cagli S, Bayram A, Guney E. Occult metastases from T1-T2 supraglottic carcinoma: role of primary tumor localization. Eur Arch Otorhinolaryngol. 2009;266:1301–1304. doi: 10.1007/s00405-008-0859-6. [DOI] [PubMed] [Google Scholar]
- 7.Wiernik G, Millard PR, Haybittle JL. The predictive value of histological classification into degrees of differentiation of squamous cell carcinoma of the larynx and hypopharynx compared with the survival of patients. Histopathology. 1991;19:411–417. doi: 10.1111/j.1365-2559.1991.tb00230.x. [DOI] [PubMed] [Google Scholar]
- 8.Bryne M, Jenssen N, Boysen M. Histological grading in the deep invasive front of T1 and T2 glottic squamous cell carcinomas has high prognostic value. Virchows Arch. 1995;427:277–281. doi: 10.1007/BF00203395. [DOI] [PubMed] [Google Scholar]
- 9.Karatzanis AD, Waldfahrer F, Psychogios G, et al. Resection margins and other prognostic factors regarding surgically treated glottic carcinomas. J Surg Oncol. 2010;101:131–136. doi: 10.1002/jso.21449. [DOI] [PubMed] [Google Scholar]
- 10.Chirila M, Bolboaca SD, Cosgarea M, et al. Perineural invasion of the major and minor nerves in laryngeal and hypopharyngeal cancer. Otolaryngol Head Neck Surg. 2009;140:65–69. doi: 10.1016/j.otohns.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 11.Yilmaz T, Hosal AS, Gedikoglu G, et al. Prognostic significance of vascular and perineural invasion in cancer of the larynx. Am J Otolaryngol. 1998;19:83–88. doi: 10.1016/s0196-0709(98)90100-4. [DOI] [PubMed] [Google Scholar]
- 12.Ma XJ, Wang Z, Ryan PD, et al. A two-gene expression ratio predicts clinical outcome in breast cancer patients treated with tamoxifen. Cancer Cell. 2004;5:607–616. doi: 10.1016/j.ccr.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 13.Boutros PC, Lau SK, Pintilie M, et al. Prognostic gene signatures for non-small-cell lung cancer. Proc Natl Acad Sci U S A. 2009;106:2824–2828. doi: 10.1073/pnas.0809444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buyse M, Loi S, van't Veer L, et al. Validation and clinical utility of a 70-gene prognostic signature for women with node-negative breast cancer. J Natl Cancer Inst. 2006;98:1183–1192. doi: 10.1093/jnci/djj329. [DOI] [PubMed] [Google Scholar]
- 15.Garman KS, Acharya CR, Edelman E, et al. A genomic approach to colon cancer risk stratification yields biologic insights into therapeutic opportunities. Proc Natl Acad Sci U S A. 2008;105:19432–19437. doi: 10.1073/pnas.0806674105. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Markowski J, Oczko-Wojciechowska M, Gierek T, et al. Gene expression profile analysis in laryngeal cancer by high-density oligonucleotide microarrays. J Physiol Pharmacol. 2009;60(Suppl 1):57–63. [PubMed] [Google Scholar]
- 17.Markowski J, Tyszkiewicz T, Jarzab M, et al. Metal-proteinase ADAM12, kinesin 14 and checkpoint suppressor 1 as new molecular markers of laryngeal carcinoma. Eur Arch Otorhinolaryngol. 2009;266:1501–1507. doi: 10.1007/s00405-009-1019-3. [DOI] [PubMed] [Google Scholar]
- 18.Ma LJ, Li W, Zhang X, et al. Differential gene expression profiling of laryngeal squamous cell carcinoma by laser capture microdissection and complementary DNA microarrays. Arch Med Res. 2009;40:114–123. doi: 10.1016/j.arcmed.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 19.Colombo J, Fachel AA, De Freitas Calmon M, et al. Gene expression profiling reveals molecular marker candidates of laryngeal squamous cell carcinoma. Oncol Rep. 2009;21:649–663. [PubMed] [Google Scholar]
- 20.Carinci F, Arcelli D, Lo Muzio L, et al. Molecular classification of nodal metastasis in primary larynx squamous cell carcinoma. Transl Res. 2007;150:233–245. doi: 10.1016/j.trsl.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 21.Mittempergher L, de Ronde JJ, Nieuwland M, et al. Gene expression profiles from formalin fixed paraffin embedded breast cancer tissue are largely comparable to fresh frozen matched tissue. PLoS One. 2011;6:e17163. doi: 10.1371/journal.pone.0017163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.April C, Klotzle B, Royce T, et al. Whole-genome gene expression profiling of formalin-fixed, paraffin-embedded tissue samples. PLoS One. 2009;4:e8162. doi: 10.1371/journal.pone.0008162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan JB, Yeakley JM, Bibikova M, et al. A versatile assay for high-throughput gene expression profiling on universal array matrices. Genome Res. 2004;14:878–885. doi: 10.1101/gr.2167504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bibikova M, Talantov D, Chudin E, et al. Quantitative gene expression profiling in formalin-fixed, paraffin-embedded tissues using universal bead arrays. Am J Pathol. 2004;165:1799–1807. doi: 10.1016/S0002-9440(10)63435-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bibikova M, Yeakley JM, Wang-Rodriguez J, Fan JB. Quantitative expression profiling of RNA from formalin-fixed, paraffin-embedded tissues using randomly assembled bead arrays. Methods Mol Biol. 2008;439:159–177. doi: 10.1007/978-1-59745-188-8_11. [DOI] [PubMed] [Google Scholar]
- 26.Bair E, Tibshirani R. Semi-supervised methods to predict patient survival from gene expression data. PLoS Biol. 2004;2:E108. doi: 10.1371/journal.pbio.0020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoshida Y, Brunet JP, Tamayo P, et al. Subclass mapping: identifying common subtypes in independent disease data sets. PLoS One. 2007;2:e1195. doi: 10.1371/journal.pone.0001195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Effron B, Tibshirani R. On testing the significance of sets of genes. Ann Appl Stat. 2007;1(1):107–129. [Google Scholar]
- 29.Bild AH, Yao G, Chang JT, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:353–357. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- 30.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 31.Chung CH, Parker JS, Karaca G, et al. Molecular classification of head and neck squamous cell carcinomas using patterns of gene expression. Cancer Cell. 2004;5:489–500. doi: 10.1016/s1535-6108(04)00112-6. [DOI] [PubMed] [Google Scholar]
- 32.Anguiano A, Tuchman SA, Acharya C, et al. Gene expression profiles of tumor biology provide a novel approach to prognosis and may guide the selection of therapeutic targets in multiple myeloma. J Clin Oncol. 2009;27:4197–4203. doi: 10.1200/JCO.2008.19.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davicioni E, Anderson JR, Buckley JD, et al. Gene expression profiling for survival prediction in pediatric rhabdomyosarcomas: a report from the children's oncology group. J Clin Oncol. 2010;28:1240–1246. doi: 10.1200/JCO.2008.21.1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoshida Y, Villanueva A, Kobayashi M, et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med. 2008;359:1995–2004. doi: 10.1056/NEJMoa0804525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jonsson G, Busch C, Knappskog S, et al. Gene expression profiling-based identification of molecular subtypes in stage IV melanomas with different clinical outcome. Clin Cancer Res. 2010;16:3356–3367. doi: 10.1158/1078-0432.CCR-09-2509. [DOI] [PubMed] [Google Scholar]
- 36.Korkola JE, Houldsworth J, Feldman DR, et al. Identification and validation of a gene expression signature that predicts outcome in adult men with germ cell tumors. J Clin Oncol. 2009;27:5240–5247. doi: 10.1200/JCO.2008.20.0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sotiriou C, Pusztai L. Gene-expression signatures in breast cancer. N Engl J Med. 2009;360:790–800. doi: 10.1056/NEJMra0801289. [DOI] [PubMed] [Google Scholar]
- 38.Simon R, Radmacher MD, Dobbin K, McShane LM. Pitfalls in the use of DNA microarray data for diagnostic and prognostic classification. J Natl Cancer Inst. 2003;95:14–18. doi: 10.1093/jnci/95.1.14. [DOI] [PubMed] [Google Scholar]
- 39.Parikh RR, Yang Q, Haffty BG. Prognostic significance of vascular endothelial growth factor protein levels in T1-2 N0 laryngeal cancer treated with primary radiation therapy. Cancer. 2007;109:566–573. doi: 10.1002/cncr.22432. [DOI] [PubMed] [Google Scholar]
- 40.Schneider S, Thurnher D, Kloimstein P, et al. Expression of the Sonic hedgehog pathway in squamous cell carcinoma of the skin and the mucosa of the head and neck. Head Neck. 2011;33(2):244–250. doi: 10.1002/hed.21437. [DOI] [PubMed] [Google Scholar]
- 41.Mincione G, Di Marcantonio MC, Artese L, et al. Loss of expression of TGF-beta1, TbetaRI, and TbetaRII correlates with differentiation in human oral squamous cell carcinomas. Int J Oncol. 2008;32:323–331. [PubMed] [Google Scholar]
- 42.Fukai Y, Fukuchi M, Masuda N, et al. Reduced expression of transforming growth factor-beta receptors is an unfavorable prognostic factor in human esophageal squamous cell carcinoma. Int J Cancer. 2003;104:161–166. doi: 10.1002/ijc.10929. [DOI] [PubMed] [Google Scholar]
- 43.Bertagnolo V, Benedusi M, Querzoli P, et al. PLC-beta2 is highly expressed in breast cancer and is associated with a poor outcome: a study on tissue microarrays. Int J Oncol. 2006;28:863–872. [PubMed] [Google Scholar]
- 44.Duiker EW, van der Zee AG, de Graeff P, et al. The extrinsic apoptosis pathway and its prognostic impact in ovarian cancer. Gynecol Oncol. 2010;116:549–555. doi: 10.1016/j.ygyno.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 45.Shimada H, Nakagawa A, Peters J, et al. TrkA expression in peripheral neuroblastic tumors: prognostic significance and biological relevance. Cancer. 2004;101:1873–1881. doi: 10.1002/cncr.20557. [DOI] [PubMed] [Google Scholar]
- 46.Ishikawa K, Miyamoto M, Yoshioka T, et al. Up-regulation of CD40 with juxtacrine activity in human nonsmall lung cancer cells correlates with poor prognosis. Cancer. 2008;113:530–541. doi: 10.1002/cncr.23618. [DOI] [PubMed] [Google Scholar]
- 47.Abramovitz M, Ordanic-Kodani M, Wang Y, et al. Optimization of RNA extraction from FFPE tissues for expression profiling in the DASL assay. Biotechniques. 2008;44:417–423. doi: 10.2144/000112703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Antonov J, Goldstein DR, Oberli A, et al. Reliable gene expression measurements from degraded RNA by quantitative real-time PCR depend on short amplicons and a proper normalization. Lab Invest. 2005;85:1040–1050. doi: 10.1038/labinvest.3700303. [DOI] [PubMed] [Google Scholar]
- 49.Conway C, Mitra A, Jewell R, et al. Gene expression profiling of paraffin-embedded primary melanoma using the DASL assay identifies increased osteopontin expression as predictive of reduced relapse-free survival. Clin Cancer Res. 2009;15:6939–6946. doi: 10.1158/1078-0432.CCR-09-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Waddell N, Cocciardi S, Johnson J, et al. Gene expression profiling of formalin-fixed, paraffin-embedded familial breast tumours using the whole genome-DASL assay. J Pathol. 2010;221:452–461. doi: 10.1002/path.2728. [DOI] [PubMed] [Google Scholar]
- 51.Ginos MA, Page GP, Michalowicz BS, et al. Identification of a gene expression signature associated with recurrent disease in squamous cell carcinoma of the head and neck. Cancer Res. 2004;64:55–63. doi: 10.1158/0008-5472.can-03-2144. [DOI] [PubMed] [Google Scholar]
- 52.Kuriakose MA, Chen WT, He ZM, et al. Selection and validation of differentially expressed genes in head and neck cancer. Cell Mol Life Sci. 2004;61:1372–1383. doi: 10.1007/s00018-004-4069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pyeon D, Newton MA, Lambert PF, et al. Fundamental differences in cell cycle deregulation in human papillomavirus-positive and human papillomavirus-negative head/neck and cervical cancers. Cancer Res. 2007;67:4605–4619. doi: 10.1158/0008-5472.CAN-06-3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Slebos RJ, Yi Y, Ely K, et al. Gene expression differences associated with human papillomavirus status in head and neck squamous cell carcinoma. Clin Cancer Res. 2006;12:701–709. doi: 10.1158/1078-0432.CCR-05-2017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.