

Abstract
One stereocenter makes all the difference
The synthesis and biological evaluation of 17-epi-cortistatin A is reported from a common intermediate used to procure natural cortistatin A. The synthesis features a unique stereocontrolled Raney-Ni reduction process that can be employed to reliably produce both α- and β-configured D-ring aryl steroids. The scope, generality, and mechanism of this process is discussed along with the first reported biological evaluations of synthetic “cortalogs”.
Keywords: natural products, angiogenesis, asymmetric synthesis
Natural products have always been a successful pool of molecules from which the pharmaceutical industry can find novel medicinal agents.[1] Steroids, in particular, continue to be the subject of medicinal investigations for two reasons: they are “privileged”[2] pharmacophores and their scrutiny for over half a century has resulted in a vast body of knowledge regarding their reactivity.[3] Recently, Kobayashi and his collaborators elucidated the structures of cortistatins A-L [see Figure 1 for the structure of cortistatin A (1)] from the sponge Corticium simplex.[4] These marine derived steroidal alkaloids exhibit potent anti-angiogenic activity against HUVECs (human umbilical vein endothelial cells) by a unique mechanism.[4] While the Kobayashi group has already delineated a preliminary SAR picture of the family (see Figure 1), their scarce natural supply renders chemical synthesis as the only means to decipher their medicinal potential. Particularly intriguing is the impact of the isoquinoline moiety on biological activity since its absence significantly lowers activity. This communication illustrates the dramatic influence of D-ring stereochemistry on biological activity with the synthesis of 17-epi-cortistatin A (4). Specifically, we have found that the C-17 stereochemistry may be removed all together as Δ16-cortistatin A (2) retains much of the potency of 1. This line of chemical inquiry has also led to the first useful method for the stereocontrolled preparation of other α-aryl-substituted D-ring steroids.
Figure 1.
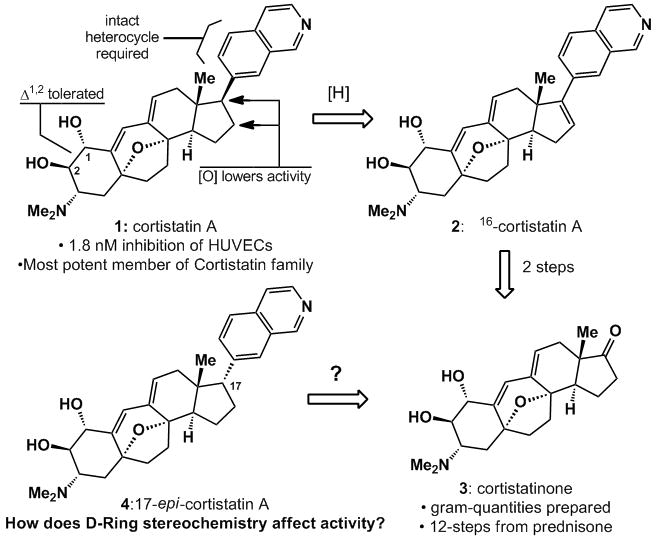
Cortistatin A (1), its known SAR,[4a, 4d] and relationship to its 17-epi relative (4).
Several approaches have been reported for the preparation of the core structure of cortistatins5 and two elegant total syntheses of the most potent member of this natural product class, cortistatin A (1), have appeared from the Nicolaou-Chen[6a] and Shair groups.[6b]
Our own synthetic plan[7] was profoundly affected by the strategic choice to use prednisone – a synthetic corticosteroid produced annually on multi-ton scale by microbial oxidations of naturally occurring steroids – as a starting material. Aside from economical considerations, this choice was made with the knowledge that semi-synthetic approaches have enjoyed decades of success in the pharmaceutical industry. Thus, a twelve-step sequence was utilized from this starting point to arrive at cortistatinone (3) in gram quantities.[7] The synthesis concluded with a highly chemo- and stereoselective Raney nickel (Ra-Ni) mediated hydrogenation of Δ16-cortistatin A (2). In order to evaluate the importance of a β-oriented isoquinoline moiety, an estrone-derived model (5, Scheme 1) was employed as a testbed for a strategy that would deliver both epimers from a common intermediate.
Scheme 1.
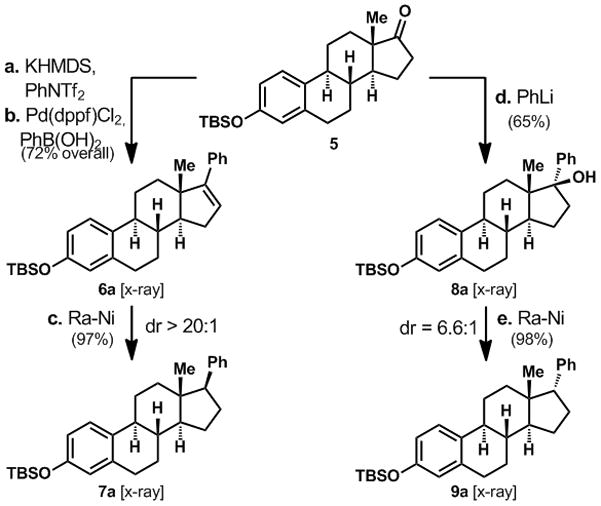
Divergent access to α- and β-configured C-17 aryl estrone derivatives.
By analogy to the synthesis of 2, estrone model 5 was converted to the D-ring styrene 6a as depicted in Scheme 1. Regardless of the reducing conditions, the only observed product was the expected β-aryl substituted product 7a. Ra-Ni mediated reduction led to a 97% isolated yield of 7a. This is not surprising given the fact that an overwhelming majority of nucleophilic, electrophilic, and radical substitution reactions at C-17 occur from the α-face.[8] Attention was therefore turned to an alternative approach that began with tertiary alcohol 8a, derived from addition of PhLi to 5. Based on preliminary evidence gathered in-house,[9] and a report that Ra-Ni reductions of benzylic alcohols occurred with retention of configuration,[10] alcohol 8a was subjected to Ra-Ni in toluene at reflux. To our delight, a diastereomeric pair of compounds was isolated in a 6.6:1 ratio, the major isomer of which bore the desired α-stereochemistry. The structures of 6a–9a were all verified by X-ray crystallography.[12]
The generality of this reagent system, a synthesis of 17-epi-cortistatin A, mechanistic analysis of these reductive processes, and biological evaluation of cortistatin analogs are presented below.
As shown in Table 1, both pathways (6 to 7 and 8 to 9) are amenable to the incorporation of electron rich, neutral, and withdrawn arenes, as demonstrated by the successful deoxygenations and hydrogenations of C-17 phenyl, p-tolyl, p-anisyl, and 3-pyridyl steroids. Hydrogenation of Δ16-17-arylsteroids 6 was carried out in 10% toluene in isopropanol with Ra-Ni at 60°C for 2 h, providing 7 with yields varying between 68% and 98%. The diastereoselectivity of this transformation is generally over 20 : 1 (17-β : 17-α). Alternatively, deoxygenation of 17-β-hydroxy-17-α-arylsteroids 8 was carried out in toluene with Ra-Ni at 110°C for 5 h. Yields varied between 68% and 98% with moderate to good diastereoselectivities.
Table 1.
Deoxygenation and hydrogenation mediated by Ra-Ni.
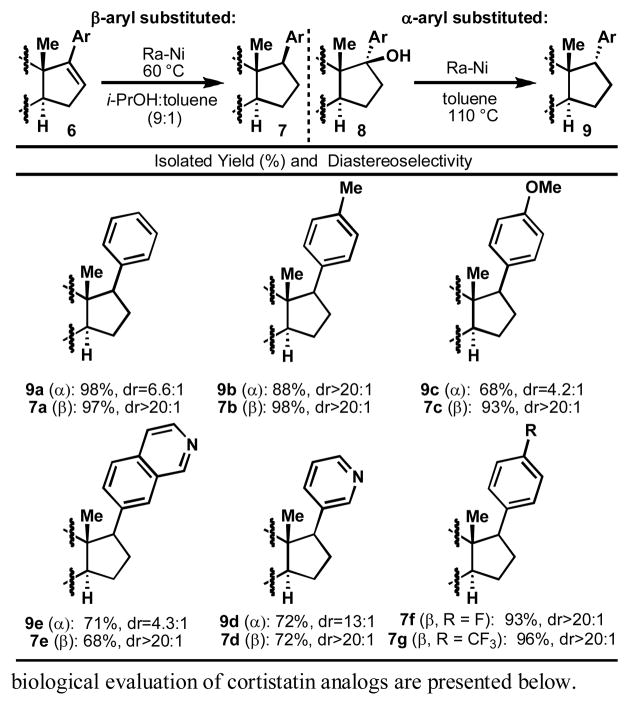
|
To determine the source of hydrogen in the reductions, deuterium labelling experiments were carried out as shown in Scheme 2. For Ra-Ni mediated transfer hydrogenation, the reaction of 6a was conducted in deuterated isopropanol and toluene (toluene or toluene-d8 gave identical results) with D2O-washed Ra-Ni, affording 7a-d2 with deuterium incorporation at C-16 and C-17. For Ra-Ni mediated deoxygenation, the reaction of 8a employed deuterated toluene with D2O-washed Ra-Ni. Surprisingly, 9a-d4 was obtained as the major product in 96% yield. However, 7a and 9a exhibited identical aromatic deuterium substitution when subjected to the same reduction conditions as 8a, demonstrating that this aromatic deuteration is independent of the deoxygenation process.
Scheme 2.
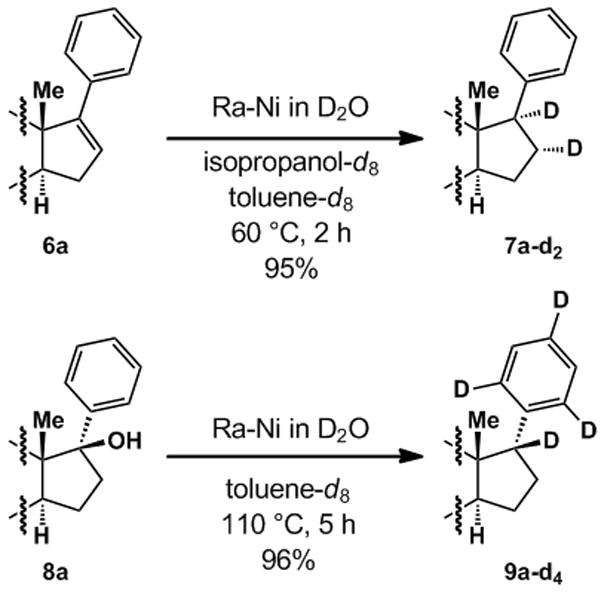
Deuterium labelling of hydrogenation and deoxygenation.
The dichotomy in observed stereochemical outcome between 7 and 9 seemingly excludes the intermediacy of free-radicals in deoxygenation (8→9; radical deoxygenation produces β-stereochemistry at C-17, vide supra). The differential stereoselectivity between 6 and 8 can be rationalized based on the facial selectivity of chemo-adsorption to the metal surface as depicted in Figure 2. Previous studies[11] have demonstrated that a high degree of stereoselectivity can be incurred in Ra-Ni mediated reductions based on stereoselective adsorption. In the case of 6, adsorption likely occurs most favorably on the relatively flat α-face, away from the angular methyl group (C-18). Hydrogens and/or electrons are then transfered from the metal surface to the preferentially adsorbed face.[11] In the case of 8, adsorption possibly takes place on the convex face, with interaction occuring between the surface and both the aromatic π-system and the benzylic hydroxyl, followed by hydrogen/deuterium delivery from the metal.
Figure 2.
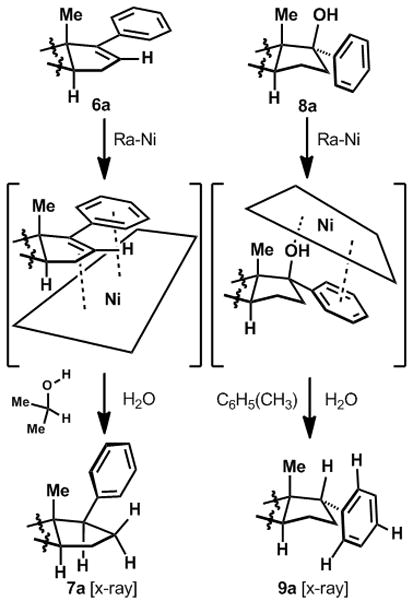
Possible explanation for observed stereochemical dichotomy.
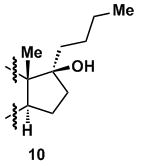
Finally, the mechanistic requirement of an aryl group at C-17 during deoxygenation is supported by the fact that 17-β-hydroxy-17-α-(n-butyl)-estrone (10) was inert to deoxygenation using Ra-Ni. In addition to submitting 10 to the reaction condition, a control experiment was carried out with an equimolar mixture of 10 and 8a premixed in the same reaction vessel and treated with Ra-Ni. While 8a was completely deoxygenated, 10 was quantitatively recovered.
The utility of the present invention is aptly demonstrated by the synthesis of 17-epi-cortistatin (4), as shown in Scheme 3. Thus, protection of the diol motif in cortistatinone (3) with TMS-imidazole, followed by treatment with an excess of 7-lithioisoquinoline in a THF/TMEDA solvent mixture at −78 °C generated an alkanolisoquinoline that was deoxygenated with Ra-Ni to deliver 17-epi-cortistatin A (4) in 16% yield over three steps.
Scheme 3.
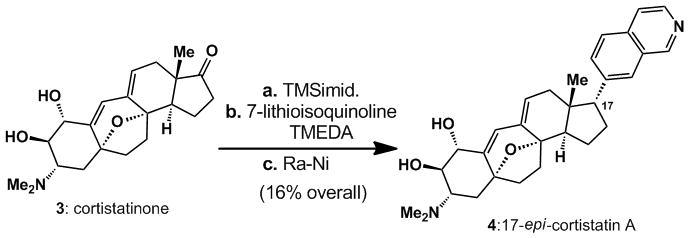
Synthesis of 17-epi-cortistatin A.
This substance proved crucial in testing the substrate scope/specificity of cortistatin A’s biological target. The importance of this substrate is clear since the greatest modulation of biological activity in the naturally occurring cortistatins stems from structure variations of the C-17 substituent.[4]
In an assay to determine activity against HUVECs (carried out by Pfizer Inc.), synthetic cortistatin A exhibited an IC50 value of 2.43 nM, which is in good agreement with the reported value.4a Remarkably, 2 still retains high potency against HUVECs, with an IC50 of 3.88 nM. This result is a significant step forward in the simplification of the overall cortistatin structure from a synthesis standpoint. However, 4 does not exhibit useful levels of activity (>1 μM). This profound difference of biological activity clearly indicates that the C-17 stereochemistry is essential for biological behavior. Modeling studies (shown in Figure 3) suggested that 1, 2, and 4 exhibit rigid architectures that differ only in the angle in which the isoquinoline moiety would be presented to the active site. Several estrone model compounds were also found to be inactive in the HUVEC screen.
Figure 3.
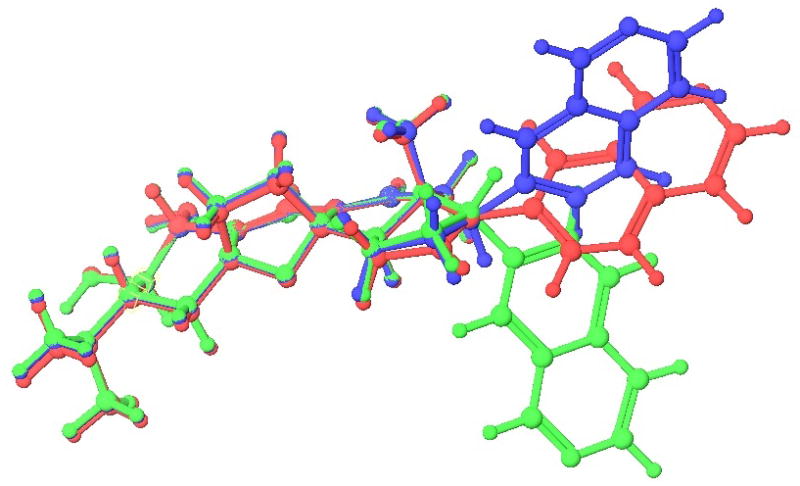
Superimposed structures of the lowest energy conformation of 1 (blue), 2 (red), and 4 (green) by Schrödinger software dihedral drive macromodel.
In summation, two processes were identified for the production of either α- or β-oriented C-17 aryl steroids, conveniently using the C-17 ketone as a common starting material. Because of this divergence, it is now possible to efficiently produce both epimers of C-17 aryl steroids, thus adding the α-epimers to the cadre of unnatural biomolecules available for biological and other studies. The relevance and utility of such a transformation has been demonstrated by the synthesis of 1, its epimer 2, and biological evaluations thereof.[12] Finally, the compelling finding that 2 retains much of the potency of 1 should considerably simplify SAR studies in this family.
Experimental Section
General Procedure for the Standardization of Raney Nickel (Ra-Ni)
Raney® 2800 Nickel (ca. 1 g of a 1g/mL slurry in H2O, pH = 9, Sigma-Aldrich) was placed in a vial. The water was removed by pipette, and the Ra-Ni was washed by 5 seconds of shaking, followed by removal of the supernatant: first H2O (2 × 2 mL), then sat. aq. Rochelle’s salt (2 × 2 mL), then H2O (10 × 2 mL). After all washes, the Ra-Ni aqueous solution (pH = 7) was stored under H2O (1 mL).
General Procedure for Hydrogenation
To a solution of styrene in i-PrOH/ toluene (9:1, 0.01 M), was added the suspension of Ra-Ni prepared above (the Ra-Ni suspension was removed by 5.75′ pipette from the thick bottom layer of the vial; 1 drop suspension per 0.1 mL solution). The reaction flask was immersed in an oil bath preheated to 60 °C and stirred vigorously for 120 minutes. After cooling to ambient temperature, the reaction mixture was passed through Celite, the Ra- Ni washed with CH2Cl2, and the combined filtrates were concentrated in vacuo. The product was purified by flash column chromatography.
General procedure for Deoxygenation
To a solution of alcohol in toluene (0.01M), was added the suspension of Ra-Ni prepared above (the Ra-Ni suspension was removed by 5.75’ pipette from the thick bottom layer of the vial; 1 drop suspension per 0.1 mL solution). The reaction flask was immersed in an oil bath preheated to 110°C and stirred vigorously for 5 hours. After cooling to ambient temperature, the reaction mixture was passed through Celite, the Ra-Ni washed with CH2Cl2, and the combined filtrates were concentrated in vacuo. The product was purified by flash column chromatography.
Supplementary Material
Table 2.
Selective growth inhibition of cortistatins against HUVECs.
Acknowledgments
We are grateful to Jennifer Lafontaine, Morena Cobbs, Sean Riley and Judy Goldman of Pfizer Inc. for helpful discussions and biological testing, Professor Jin-Quan Yu for mechanistic insight, Dr. Laura Pasternack for help with computations, Dr. Curtis Moore and Professor Arnold Reingold for X-ray crystallographic measurements, and Dr. Dee-Hua Huang for NMR assistance. Financial support for this work was provided by Pfizer, Bristol-Myers Squibb (predoctoral fellowship to J. S.), the Department of Defense (predoctoral fellowship to R.A.S.), the NIH (predoctoral fellowship to C.A.G.), and the Uehara Memorial Foundation (postdoctoral fellowship to H.S.).
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Jun Shi, Department of Chemistry, The Scripps Research Institute, 10650 North Torrey Pines Road, La Jolla, CA, 92037 (USA), Fax: (+1) 858-784-7375.
Dr. Hiroki Shigehisa, Department of Chemistry, The Scripps Research Institute, 10650 North Torrey Pines Road, La Jolla, CA, 92037 (USA), Fax: (+1) 858-784-7375
Carlos A. Guerrero, Department of Chemistry, The Scripps Research Institute, 10650 North Torrey Pines Road, La Jolla, CA, 92037 (USA), Fax: (+1) 858-784-7375
Ryan A. Shenvi, Department of Chemistry, The Scripps Research Institute, 10650 North Torrey Pines Road, La Jolla, CA, 92037 (USA), Fax: (+1) 858-784-7375
Dr. Chuang-Chuang Li, Department of Chemistry, The Scripps Research Institute, 10650 North Torrey Pines Road, La Jolla, CA, 92037 (USA), Fax: (+1) 858-784-7375
Prof. Phil S. Baran, Email: pbaran@scripps.edu, Department of Chemistry, The Scripps Research Institute, 10650 North Torrey Pines Road, La Jolla, CA, 92037 (USA), Fax: (+1) 858-784-7375.
References
- 1.Baker DD, Chu M, Oza U, Rajgarhia V. Nat Prod Rep. 2007;24:1225– 1244. doi: 10.1039/b602241n. [DOI] [PubMed] [Google Scholar]
- 2.Wolff ME, editor. Burger’s Medicinal Chemistry and Drug Discovery. Vol. 5. John Wiley & Sons, Inc; 1997. pp. 281–376. [Google Scholar]
- 3.Biellmann JF. Chem Rev. 2003;103:2019– 2033. doi: 10.1021/cr020071b. [DOI] [PubMed] [Google Scholar]
- 4.a) Aoki S, Watanabe Y, Sanagawa M, Setiawan A, Kotoku N, Kobayashi M. J Am Chem Soc. 2006;128:3148–3149. doi: 10.1021/ja057404h. [DOI] [PubMed] [Google Scholar]; b) Watanabe Y, Aoki S, Tanabe D, Setiawan A, Kobayashi M. Tetrahedron. 2007;63:4074–4079. [Google Scholar]; c) Aoki S, Watanabe Y, Tanabe D, Setiawan A, Arai M, Kobayashi M. Tetrahedron Lett. 2007;48:4485–4488. [Google Scholar]; d) Aoki S, Watanabe Y, Tanabe D, Arai M, Suna H, Miyamoto K, Tsujibo H, Tsujikawa K, Yamamoto H, Kobayashi M. Bioorg Med Chem. 2007;15:6758–6762. doi: 10.1016/j.bmc.2007.08.017. [DOI] [PubMed] [Google Scholar]; e) Sato Y, Kamiyama H, Usui T, Saito T, Osada H, Kuwahara S, Kiyota H. Biosci Biotechnol Biochem. 2008;72:2992– 2997. doi: 10.1271/bbb.80562. [DOI] [PubMed] [Google Scholar]
- 5.Dai M, Danishefsky SJ. Heterocycles. 2009;77:157–161.Simmons EM, Hardin AR, Guo X, Sarpong R. Angew Chem Int Ed. 2008;47:6650–6653. doi: 10.1002/anie.200802203.Yamashita S, Iso K, Hirama M. Org Lett. 2008;10:3413–3415. doi: 10.1021/ol8012099.Craft DT, Gung BW. Tetrahedron Lett. 2008;49:5931–5934. doi: 10.1016/j.tetlet.2008.07.155.Dai M, Danishefsky SJ. Tetrahedron Lett. 2008;49:6610–6612. doi: 10.1016/j.tetlet.2008.09.018.Dai M, Wang Z, Danishefsky SJ. Tetrahedron Lett. 2008;49:6613–6616. doi: 10.1016/j.tetlet.2008.09.019.Kotoku N, Sumii Y, Hayashi T, Kobayshi M. Tetrahedron Lett. 2008;49:7078–7081.Kürti L, Czakó B, Corey EJ. Org Lett. 2008;10:5247–5250. doi: 10.1021/ol802328n.Yamashita S, Kitajima K, Iso K, Hirama M. Tetrahedron Lett. ASAP.Gung BW, Craft DT. Tetrahedron Lett. ASAP.review, see: Nishing CF, Bräse S. Angew Chem Int Ed. 2008;47:9389–9391. doi: 10.1002/anie.200803720.
- 6.a) Nicolaou KC, Sun Y-P, Peng X-S, Polet D, Chen DY-K. Angew Chem Int Ed. 2008;47:7310–7313. doi: 10.1002/anie.200803550. [DOI] [PubMed] [Google Scholar]; b) Lee HM, Nieto-Oberhuber C, Shair MD. J Am Chem Soc. 2008;130:16864– 16866. doi: 10.1021/ja8071918. [DOI] [PubMed] [Google Scholar]
- 7.Shenvi RA, Guerrero CA, Shi J, Li C-C, Baran PS. J Am Chem Soc. 2008;130:7241– 7243. doi: 10.1021/ja8023466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fieser LF, Fieser M. Steroids. Reinhold; New York: 1959. [Google Scholar]
- 9.a) Guerrero CA. Ph D Thesis. The Scripps Research Institute; 2008. [Google Scholar]; b) Shenvi RA. Ph D Thesis. The Scripps Research Institute; 2008. [Google Scholar]
- 10.a) Mitsui S, Imaizumi S. Bull Chem Soc Jpn. 1961;34:774–778. [Google Scholar]; b) Krafft ME, Crooks WJ., III J Org Chem. 1988;53:432– 434. [Google Scholar]
- 11.Mitsui S, Imaizumi S, Esashi Y. Bull Chem Soc Jpn. 1970;43:2143–2152. and references cited therein. [Google Scholar]
- 12.See supporting information for detailed experimental procedures and copies of all spectra. CCDC-721528 (6a), -726038 (7a), -721529 (8a), -721530 (9a), and -721527 (10), contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.