

Abstract
Previously we reported that the sesquiterpene lactone parthenolide induces oxidative stress in cardiac myocytes, which blocks Janus kinase (JAK) activation by the interleukin 6 (IL-6)-type cytokines. One implication suggested by this finding is that IL-6 signaling is dependent upon cellular anti-oxidant defenses or redox status. Therefore, the present study was undertaken to directly test the hypothesis that JAK1 signaling by the IL-6-type cytokines in cardiac myocytes is impaired by glutathione (GSH) depletion, since this tripeptide is one of the major anti-oxidant molecules and redox-buffers in cells. Cardiac myocytes were pretreated for 6 h with L-buthionine-sulfoximine (BSO) to inhibit GSH synthesis. After 24 h, cells were dosed with the IL-6-like cytokine, leukemia inhibitory factor (LIF). BSO treatment decreased GSH levels and dose-dependently attenuated activation of JAK1, Signal Transducer and Activator of Transcription 3 (STAT3), and extracellular signal regulated kinases 1 and 2 (ERK1/2). Addition of glutathione monoethyl ester, which is cleaved intracellularly to GSH, prevented attenuation of LIF-induced JAK1 and STAT3 activation, as did the reductant N-acetyl-cysteine. Unexpectedly, LIF-induced STAT1 activation was unaffected by GSH depletion. Evidence was found that STAT3 is more resistant than STAT1 to intermolecular disulfide bond formation under oxidizing conditions and more likely to retain the monomeric form, suggesting that conformational differences explain the differential effect of GSH depletion on STAT1 and STAT3. Overall, our findings indicate that activation of both JAK1 and STAT3 are redox-sensitive and the character of IL-6 type cytokine signaling in cardiac myocytes is sensitive to changes in the cellular redox status. In cardiac myocytes, activation of STAT1 may be favored over STAT3 under oxidizing conditions due to GSH depletion and/or augmented reactive oxygen species production, such as in ischemia-reperfusion and heart failure.
Keywords: Leukemia inhibitory factor, Oxidative stress, JAK1 kinase, STAT3 transcription factor, Redox
1. Introduction
The interleukin 6 (IL-6)-family of cytokines are important mediators of the acute-phase response to injury and infection (Heinrich et al., 2003). They require the transmembrane protein glycoprotein 130 (gp130) for signal transduction and include interleukin-6 (IL-6), leukemia inhibitory factor (LIF), and cardiotrophin-1 (CT-1). These 3 cytokines especially play a crucial role in cardiac pathophysiology, and have been implicated in cardiac protection from ischemia-reperfusion (I/R) injury (Kurdi and Booz, 2007a; Nian et al., 2004; Terrell et al., 2006). The IL-6-family cytokines are induced and released in the heart by I/R injury and infarction (Aoyama et al., 2000; Chandrasekar et al., 1999; Gritman et al., 2006; Gwechenberger et al., 1999; Kukielka et al., 1995), and are produced by cardiac myocytes in response to hypoxia and I/R (Gwechenberger et al., 1999; Hishinuma et al., 1999; Yamauchi-Takihara et al., 1995). Evidence from knockout mice indicates that IL-6 is critical for the late phase of ischemic preconditioning in the heart (Dawn et al., 2004). IL-6 is required for upregulating the co-mediators of late preconditioning, nitric oxide synthase-2 (NOS2) and cyclooxygenase-2 (COX-2) through activation of the transcription factor Signal Transducer and Activator of Transcription 3 (STAT3). Other genes linked to STAT3 activation in cardiac myocytes that are important to protection or repair from I/R injury are the angiogenic factor vascular endothelial growth factor (VEGF) and antioxidant defense molecules manganese superoxide dismutase (MnSOD), and metallothioneins (MT1 & MT2) (Kurdi and Booz, 2007a). Although recognized as important in cardiac pathophysiology, surprisingly little is known about the factors regulating IL-6-type cytokine signaling in cardiac myocytes, under either normal or stressed conditions (Kurdi and Booz, 2007a).
Intracellular signaling for the IL-6-type cytokines is initiated by ligand binding-induced gp130 homodimerization or gp130 heterodimerization with a homologous protein. This in turn leads to trans-autophosphorylation on two tandem tyrosine (Y) residues of the JAK1 proteins constitutively associated with the cytoplasmic tails of the receptors, leading to increased catalytic activity of JAK1 (Heinrich et al., 2003; Kurdi and Booz, 2007a). The activated JAK1 proteins phosphorylate tyrosine residues on gp130 or the homologous protein that participate in recruiting signaling molecules or scaffolding proteins linked to several signaling cascades. Most prominent among the signaling molecules recruited is STAT3, which following recruitment is phosphorylated by JAK1 on Y705 resulting in dimerization and translocation to the nucleus. By virtue of its constitutive association with gp130 and the gp130-homologous proteins, JAK1 is obligatory for receptor signaling by the IL-6-type cytokines, although these cytokines may activate other JAK kinase family members (Kurdi and Booz, 2007a; Rodig et al., 1998). Knockout of the JAK1 gene in mice showed that IL-6- and LIF-induced STAT3 activation was reduced >95% in cardiac myocytes (Rodig et al., 1998).
The impact of reactive oxygen species (ROS) on JAK-STAT signaling is poorly understood (Kurdi and Booz, 2009). There are multiple reports that oxidative stress, by hydrogen peroxide (H2O2) in particular, activates JAK-STAT3 signaling; yet no clear mechanism has been defined. Since activation of JAK2 by H2O2 is reported to be cell line-dependent, the effect of ROS on JAK activity is likely indirect through inhibition of a tyrosine phosphatase (Kurdi and Booz, 2009). We recently reported that the sesquiterpene lactone parthenolide, which occurs naturally in the feverfew plant, induces oxidative stress in cardiac myocytes, which in turn leads to blockade of JAK1 activation by the IL-6-type cytokines (Kurdi and Booz, 2007b). Thus, for this reason and because cardiac myocytes heavily rely on oxidative metabolism, these cells are an ideal model for studying the redox-sensitivity of gp130 cytokine signaling.
Here we directly tested the hypothesis that JAK-STAT activation by the IL-6-type cytokines in cardiac myocytes is adversely affected by glutathione (GSH) depletion, since the tripeptide GSH is one of the major anti-oxidant molecules in cells (Jones, 2002). Moreover, in both patients and animal models, reduced cardiac GSH levels occur in I/R (Akila et al., 2007; Morihira et al., 2006) and heart failure (Damy et al., 2009; Lombardi et al., 2009). Recently, GSH oxidation in cardiac myocytes was implicated in mitochondrial dysfunction and arrhythmias, ostensibly through enhanced oxidative stress (Brown et al., 2010; Slodzinski et al., 2008).
2. Materials and Methods
All tissue culture supplies including DMEM/F-12 and horse serum were from Invitrogen-Gibco (Carlsbad, CA). LIF was from EMD Millipore (Billerica, MA). L-buthionine-(S,R)-sulfoximine (BSO), N-acetyl-L-cysteine (NAC), sodium ortho-iodosobenzoate (o-IBZ), H2O2, dimedone, diamide, and protease inhibitor cocktail for use with mammalian cell and tissue extracts were purchased from Sigma-Aldrich (St. Louis, MO). Dithiothreitol (DTT) was from Thermo Fisher Scientific (Waltham, MA). Glutathione monoethyl ester (GME) was from EMD Chemicals USA (Gibbstown, NJ). Antibodies to phospho-STAT3 (Y705), phospho-STAT3 (S727), STAT3, phospho-STAT1 (Y701), and STAT1 were from Cell Signaling Technology (Beverly, MA). Antibodies to ERK1/2 and anti-ACTIVE MAPK were from Promega (Fitchburg, WI). Santa Cruz Biotechnology (Santa Cruz, CA) was the source for protein A/G PLUS-agarose, normal rabbit IgG, and antibodies for JAK1 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The anti-phosphotyrosine antibody was from Upstate Biotechnology (Lake Placid, NY), horseradish peroxidase-conjugated secondary antibodies from Bio-Rad (Hercules, CA), and chemiluminescence reagents from PerkinElmer Life Sciences (Waltham, MA). RIPA-based kinase extraction buffer and activated vanadate were from Boston Bioproducts (Ashland, MA). Sources for additional reagents used for sulfenic acid detection were as follows: Odyssey blocking buffer, nitrocellulose membranes, molecular weight marker, and IRDye secondary antibodies were from LI-COR Biosciences (Lincoln, NE); human recombinant JAK1 was from OriGene (Rockville, MD); anti-human JAK1 (anti-hJAK1) antibody was from BD Biosciences (Bedford, MA); and anti-cysteine-sulfenic acid antibody was from Millipore. 1-Nitrosocyclohexyl pivalate (NCP) was a gift from Dr. S. Bruce King (Department of Chemistry, Wake Forest University, Winston-Salem, NC, USA)
2.1. Isolation and treatment of neonatal rat ventricular myocytes
The study protocol was approved by the Institutional Animal Care and Use Committee (#1192). Ventricular myocytes were isolated from 1-2-day-old Sprague-Dawley rat pups and maintained as described (Kurdi and Booz, 2007b). Experiments were performed 3-4 days later on confluent cultures. Cardiac myocytes were treated for 6 h with various concentrations of BSO (30, 60, 120, and 200 μM), washed 2× with DMEM-F12 medium, and incubated for 24 h in serum-free medium containing vehicle, GME (2 mM), or NAC (10 mM). BSO solutions were prepared fresh from desiccated and refrigerated powder. Cells were then treated for 10 min with vehicle or LIF (2 ng/mL). Since BSO inhibits GSH synthesis, an extended period between BSO treatment and cytokine stimulation was selected to allow sufficient time for intracellular GSH to be depleted by normal cellular processes.
2.2. Glutathione assay
Reduced glutathione (GSH) was measured using a colorimetric assay kit from OXIS International (Foster City, CA) according to the manufacturer’s protocol and as described (Kurdi et al., 2007). Briefly, cells were placed on ice and washed 2× with cold phosphate-buffered saline (PBS). Cells were scraped into OXIS assay buffer and lysates prepared by sonication followed by centrifugation (20,000 g, 15 min, 4°C). An aliquot of the supernatant was taken for determining protein. GSH levels in the supernatant were measured after protein removal by acid precipitation and centrifugation. Cellular GSH content was normalized to protein levels and expressed as a percentage of the control.
2.3. Immunoprecipitations and Western analysis
Cell lysates were prepared as described (Kurdi and Booz, 2007b). Lysate proteins (30 – 50 μg) were resolved by SDS-PAGE and transferred to nitrocellulose membranes. Immunoreactive bands were visualized by enhanced chemiluminescence (ECL). Immunoprecipitations were carried out using 0.5 to 1 mg of protein from cell lysates (Kurdi and Booz, 2007b). Briefly, aliquots containing equal amounts of protein were pre-cleared for 30 min with protein A/G PLUS-agarose and normal rabbit IgG, and then incubated for 4 h at 4o C under constant rotation with anti-JAK1 antibody. Protein A/G PLUS-agarose beads were added and the incubation continued for 16 h. Immunoprecipitates were collected by centrifugation and washed 3× with ice-cold PBS. The agarose beads were suspended in 30 μl of Laemmli reducing buffer and heated for 5 min at 100o C. Samples were analyzed by SDS-PAGE following centrifugation. Loading controls are provided for each immunoblot to confirm equal protein loading. Immunoreactive bands were visualized by ECL and film. Signal intensity was quantified using Bio Rad Gel Documentation System and Quantity 1 software.
2.4. Determination of superoxide levels by electron paramagnetic resonance spectroscopy
Cardiac myocytes were isolated and treated with BSO for 6 h as described above. Samples were prepared 24 h after BSO treatment and treated for electron paramagnetic resonance (EPR) spectroscopy using 1 mM 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethyl pyrrolidine (CMH, Enzo Life Sciences) for 10 minutes. Stock solutions (10 mM) of CMH were made in nitrogen purged 0.9% (w/v) NaCl, 25 g/L Chelex 100 (Bio-Rad) and 0.1 mM DTPA (Dikalov et al., 2011). Samples were assayed in 50 μL glass capillary tubes at room temperature using the Bruker e-scan EPR spectrometer. Spectrometer settings were as follows: sweep width, 100 G; microwave frequency, 9.75 GHz; modulation amplitude, 1 G; conversion time, 5.12 ms; time constant 5.12 ms; receiver gain, 2 × 102; number of scans, 4. Relative superoxide levels were assessed by measuring the signal amplitude. In order to deplete ROS signals, samples were incubated with shaking using 2.1 units/μL poly-ethylene glycolated superoxide dismutase (PEG-SOD) and 0.4 units/μL Cu,Zn-SOD for 3 h.
2.5. JAK1 protein-sulfenic acid detection
Sulfenic acid was detected as described (Seo and Carroll, 2009). Human recombinant JAK1 (~25 μg/mL) in RIPA-based extraction buffer with vanadate and protease inhibitors was incubated at 4o C under constant rotation with anti-JAK1 (HR-785) antibody (Santa Cruz Biotechnology). Protein A/G PLUS-agarose beads were added after 4 h and the incubation continued for 16 h. Immunoprecipitates were collected by centrifugation at 3000 g for 5 min and washed 3× for 5 min at room temperature with redox neutral wash buffer (1% Triton X-100, 0.1 mM sodium vanadate, 50 mM NaCl, 10 mM HEPES, pH 7.4). After the third wash, immunoprecipitate complexes were pre-treated with 10 mM DTT, 2.5 mM o-IBZ, or 60 μM H2O2 for 1 hr at 4° C, washed 3x, and then incubated for 2 h with 10 mM dimedone at 4° C. The recovered immunoprecipitation complex was resolved by SDS-PAGE and transferred to LI-COR nitrocellulose membranes. The ratio of sulfenic acid to JAK1 was determined with the LI-COR Odyssey Detection System using anti-hJAK1 and anti-cysteine-sulfenic acid antibodies. The sulfenic acid signal intensity was normalized to JAK1 signal intensity.
2.6. Difference between STAT1 and STAT3 in response to oxidative stress
Ventricles from hearts of 2-3 month old male C57BL/6 mice (~27 g) were homogenized in 1.2 mL TSE buffer (20 mM Tris, pH 7.4, 250 mM sucrose, and 1 mM EDTA) with protease inhibitors and vanadate. The homogenates were cleared by centrifugation at 17,000 g for 15 min at 4°C. Four aliquots were incubated under constant rotation with vehicle, 500 μM NCP, 1 mM diamide, or 500 μM NCP + 1 mM diamide for 30 min at 22°C. The reaction was stopped by adding twice the volume of ice-cold TSE buffer. An aliquot was added to nonreducing Laemmli’s sample buffer and another aliquot to reducing Laemmli’s sample buffer with 2-mercaptoethanol. Both sets were boiled at 102°C for 5 min. Proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes, which were immunoblotted for STAT1 and STAT3 simultaneously using the Li-COR Odyssey detection system.
2.7. Statistics
All experiments with cardiac myocytes were performed using cultures from different heart dispersions. Results are expressed as mean ± SEM of n independent experiments. Statistical analysis involving multiple comparisons was carried out by an ANOVA and Newman– Keuls or Dunnett’s multiple comparison post-test.
3. Results
3.1. BSO decreases GSH levels and leads to an increase in oxidative stress
To deplete intracellular stores of GSH, neonatal rat cardiac myocytes were pretreated for 6 h with BSO (30, 60, 120 and 200 μM), a specific and irreversible inhibitor of the rate limiting enzyme of GSH synthesis, γ-glutamylcysteine synthetase. Changes in intracellular GSH levels 24 h after BSO treatment are shown in Figure 1A. Treatment with BSO decreased GSH levels in a dose-dependent manner. Significant reduction of intracellular GSH content was observed at concentrations equal to or greater than 30 μM BSO. We next sought to assess whether GSH depletion was associated with increased superoxide formation. To deplete intracellular stores of GSH, neonatal rat cardiac myocytes were pretreated for 6 h with 30 or 60 μM BSO. Changes in superoxide levels were tested 24 h after BSO treatment and are shown in Figure 1B. Treatment with BSO increased superoxide levels in a dose-dependent manner. A significant increase in ROS levels was seen at concentrations equal to 60 μM BSO, which reduced cellular GSH levels by > 50%. EPR signals were ablated with the use of SOD, indicating the signal was due to a superoxide reaction with the EPR spin probe (Fig. 1C).
Figure 1.
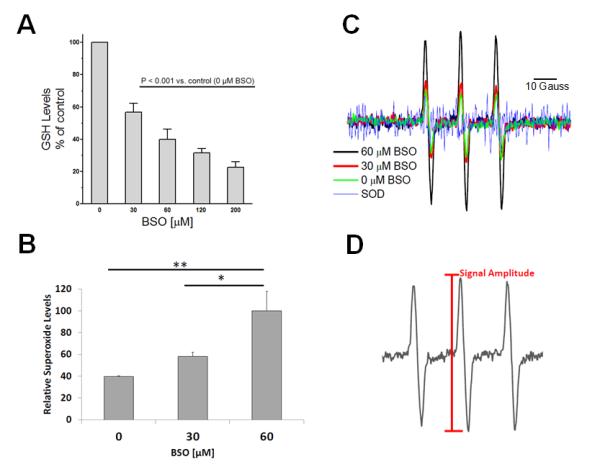
Depletion of cellular GSH by treatment with L-buthionine-(S,R)-sulfoximine (BSO) induces oxidative stress. (A) Cardiac myocytes were treated for 6 h with various concentrations of BSO, a specific inhibitor of the first enzyme in glutathione biosynthesis.. Cells were then washed 2× with medium and incubated in serum-free medium. After 24 h, cell lysates were prepared and equal protein amounts of the lysates analyzed for GSH. Data were expressed as a percentage of the control and represent the mean ± SE of ≥ 4 independent experiments. All values are significantly different from each other except for 120 vs. 60 and 200 vs. 120. (B) ROS detection in cardiac myocytes. Neonatal rat ventricular myocytes were treated for 6 h with vehicle or with 30 μM or 60 μM BSO to deplete cellular GSH then washed 2× with medium and incubated in serum-free medium for 24 h. To detect ROS production, cells were isolated and incubated for 10 min with 1 mM CMH. Samples were normalized to 60 μM BSO to reflect the relative abundance of superoxide and expressed as mean ± SEM of 3 independent experiments (**P < 0.01 vs. control or * P< 0.05 vs. 30 μM BSO). (C) Representative superoxide EPR spectra for BSO and SOD-treated samples. (D) Representative EPR spectrum illustrating how signal amplitude was measured.
3.2. GSH depletion inhibits LIF-induced STAT3 activation
We determined the effect of GSH depletion on STAT3 activation in cardiac myocytes. Cells were pretreated for 6 h with various concentrations of BSO followed by incubation for 24 h in serum-free medium before treatment with LIF (2 ng/mL) for 10 min. As Figure 2A shows, LIF induced robust STAT3 Y705 phosphorylation (lane 2 vs. 1), which was decreased by increasing BSO concentrations (lanes 3 – 6). BSO also inhibited in a dose-dependent manner STAT3 S727 phosphorylation induced by LIF. The effect of BSO on LIF-induced STAT3 Y705 and S727 phosphorylation is summarized in Figures 2B and 2C, respectively.
Figure 2.
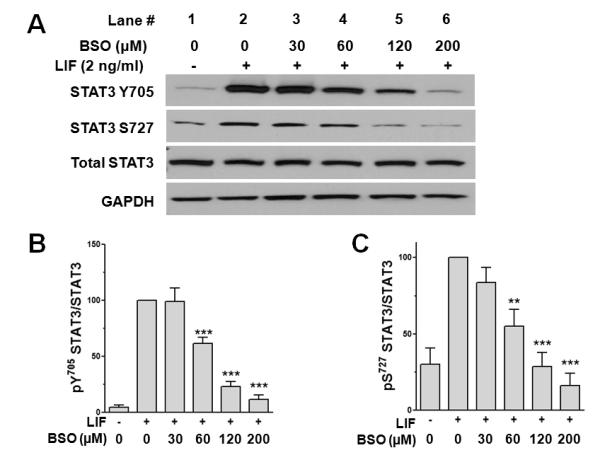
GSH depletion inhibits LIF-induced activation of STAT3. Cardiac myocytes were treated for 6 h with various concentrations of BSO to deplete cellular GSH (lanes 3-6), then washed 2× with medium and incubated in serum-free medium for 24 h. (A) Following treatment with vehicle (lane 1) or 2 ng/mL LIF (lanes 2-6) for 10 min, cell lysates were prepared and analyzed by Western immunoblotting for STAT3 Y705 phosphorylation or STAT3 S727 phosphorylation. Membranes were stripped and reprobed for total STAT3 and GAPDH to confirm equal loading. Densitometric analysis for STAT3 Y705 phosphorylation (B) and STAT3 S727 phosphorylation (C) normalized to total STAT3. Values shown are mean ± SE of 5 independent experiments. **P < 0.01 and ***P < 0.001 vs. LIF alone.
3.3. GSH depletion decreases LIF–induced JAK1 and ERK1/2 activation, but not that of STAT1
We assessed whether GSH depletion inhibits JAK1 activation induced by LIF, which is the initiating step in IL-6-type cytokine signaling. Cardiac myocytes were pretreated with various concentrations of BSO. After 24 h in serum-free medium, the cells were stimulated with LIF (2 ng/mL) for 10 min. As Figures 3A shows, LIF stimulation induced JAK1 phosphorylation while BSO prevented phosphorylation in a dose-dependent manner, although the impact of GSH depletion on JAK1 appeared to be less than on STAT3 activation (Fig. 2B).
Figure 3.
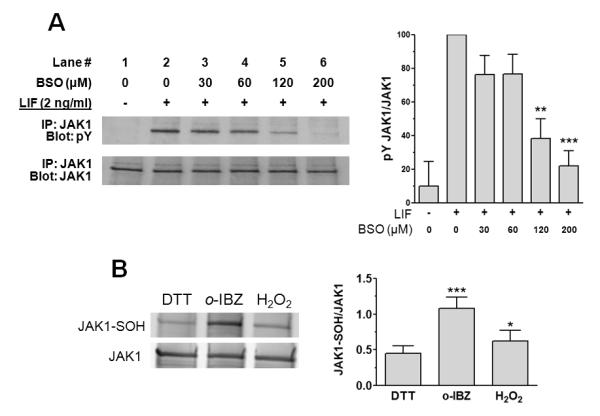
GSH depletion inhibits LIF-induced activation of JAK1 and evidence JAK1 is redox-sensitive. (A) Cardiac myocytes were treated 6 h with various concentrations of BSO (lanes 3-6), washed 2× with medium, and incubated 24 h in serum-free medium. Following treatment for 10 min with vehicle (lane 1) or 2 ng/mL LIF (lanes 2-6), cell lysates were prepared. JAK1 in lysates was immunoprecipitated and analyzed for tyrosine phosphorylation (pY) by Western immunoblotting. Membranes were stripped and reprobed for total JAK1 as a loading control. Graph shows densitometric analysis of immunoreactive bands. Values are mean ± SE of 4 independent experiments. **P < 0.01 and ***P < 0.001 vs. LIF alone. (B) Oxidant exposure increases sulfenic acid formation in purified recombinant JAK1. Purified recombinant JAK1 was immunoprecipitated and treated with either 10 mM DTT, 2.5 mM o-IBZ or 60 μM H2O2 for 1 hr at 4°C, then processed as described under “Materials and Methods” to determine the sulfenic acid content within JAK1. Levels of cysteine-sulfenic acid and JAK1 were quantified by the Li-COR Odyssey Detection System. Treatment with o-IBZ or H2O2 resulted in a significant increase in relative sulfenic acid content. *P < 0.05 and ***P < 0.001 vs. DTT, n = 4.
The above evidence shows that depleting intracellular GSH levels thereby creating a more oxidizing intracellular redox state, impairs LIF-JAK1-STAT3 signal propagation. Relatively stable protein-sulfenic acid formation occurs in select proteins that have redox-sensitive cysteines (Poole et al., 2004). Treatment with the oxidant o-IBZ, resulted in a substantial increase in the relative sulfenic acid content of purified recombinant JAK1, as compared to that of JAK1 which was treated with the reducing agent DTT (Fig. 3B). A milder oxidizing protocol (treatment with 60 μM H2O2) also increased JAK1 sulfenic acid formation (Fig. 3B). These observations are consistent with the presence of redox-reactive cysteines in JAK1. GSH depletion also attenuated, but did not totally block, LIF-induced ERK1/2 activation (Fig. 4A); however, unexpectedly, LIF-induced STAT1 activation was not affected by GSH depletion (Fig. 4B).
Figure 4.
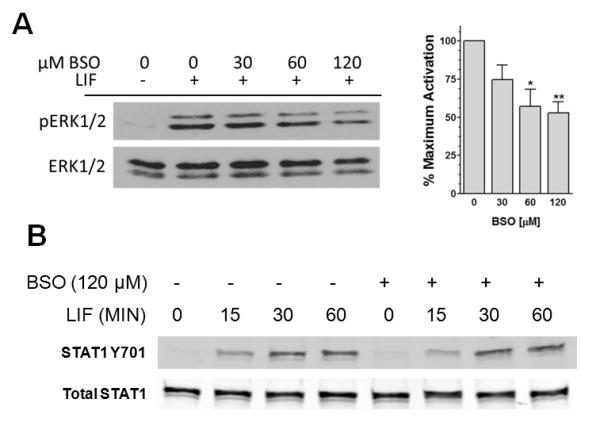
GSH depletion attenuates LIF-induced ERK1/2, but not STAT1 activation. (A) Cardiac myocytes that were treated with various concentrations of BSO were dosed with 2 ng/mL LIF for 10 min. Cell extracts were prepared and analyzed for activated ERK1/2 (pERK1/2) and total ERK1/2 as a loading control. Relative levels of inhibition were determined. *P < 0.05 and **P < 0.001 vs.LIF alone, ANOVA and Dunnett’s. (B) Cardiac myocytes were pretreated with 120 μM BSO and dosed with 2 ng/mL LIF for various times. Cell extracts were analyzed for pY701 STAT1 and total STAT1. Blot shown is representative of 3 independent experiments.
3.4. Replenishment of intracellular GSH reverses the inhibitory effect of BSO
We asked whether addition of the cell-permeable GME (2 mM), which is cleaved intracellularly to GSH, would prevent the impairment of LIF-induced JAK1 and STAT3 activation produced by GSH depletion. As Figures 5A and 5C show, the inhibitory effect of BSO on LIF-induced STAT3 activation was reversed by treatment of cells with GME (lanes 7 and 8 vs. 3 and 4). The inhibitory effect on LIF-induced JAK1 activation was reversed as well (Figs. 5B and 5C).
Figure 5.
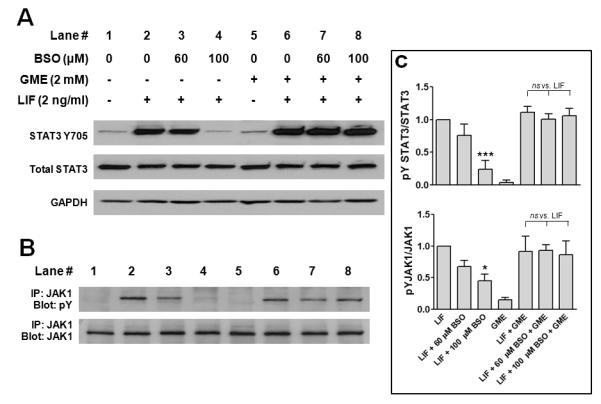
Replenishment of intracellular GSH reverses the inhibitory effect of GSH depletion on LIF-induced STAT3 and JAK1 activation. Cardiac myocytes were treated for 6 h with different concentrations of BSO (lanes 3,4,7,8), washed 2× with medium, and incubated for 24 h in serum-free medium containing vehicle or 2 mM glutathione monoethyl ester (GME) (lanes 5-8). Cells were dosed with vehicle (lanes 1 & 5) or 2 ng/mL LIF (lanes 2-4 & 6-8) for 10 min. (A) Cell lysates were prepared and analyzed by Western immunoblotting for STAT3 Y705 phosphorylation. Membranes were stripped and reprobed for total STAT3 and GAPDH to confirm equal loading. (B) Immunoprecipitates of JAK1 from cell lysates were immunoblotted for tyrosine phosphorylated JAK1 (upper) and total JAK1 (lower) to ensure equal loading. Blots shown are representative of 5 independent experiments. (C) Densitometric analysis of immunoreactive bands. *P < 0.05 and ***P < 0.001 vs. maximal activation with LIF, ANOVA and Dunnett’s multiple comparison test.
3.5. N-Acetyl-L-cysteine prevents the inhibitory effect of GSH depletion
To establish a role for oxidative stress in the effect of GSH depletion on LIF signaling, thereby confirming that JAK1-STAT3 signaling is redox-sensitive, we post-treated cardiac myocytes with NAC, a thiol-donating compound. As Figure 6 shows, post-treatment with NAC (10 mM) prevented the inhibitory effect of BSO on LIF-induced STAT3 (Fig. 6A, lanes 6 & 7 vs. lanes 4 & 5, and Fig. 6C) and JAK1 activation (Fig. 6B, lanes 6 & 7 vs. lanes 4 & 5, and Fig. 6C). Pretreatment with NAC alone produced a significant enhancement in LIF-induced JAK1 activation (Figs. 6B and 6C).
Figure 6.
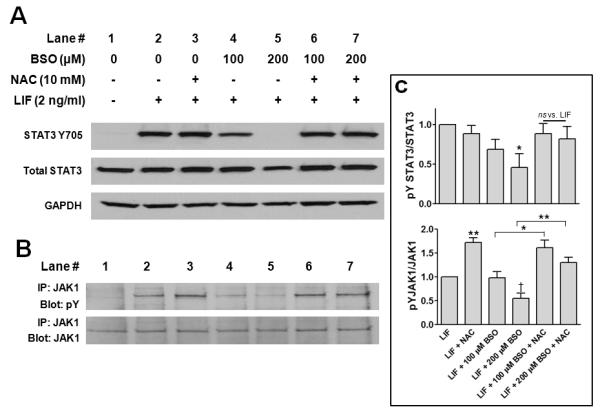
N-Acetylcysteine (NAC) prevents the inhibitory effect of GSH depletion on LIF-induced STAT3 and JAK1 activation. Cardiac myocytes were treated for 6 h with different concentrations of BSO, washed 2× with medium, and incubated for 24 h in serum-free medium containing vehicle or NAC (10 mM). Cells were dosed for 10 min with vehicle (lane 1) or 2 ng/mL LIF (lanes 2 - 7). (A) Western immunoblots of cell lysates were sequentially probed for STAT3 Y705 phosphorylation, STAT3, and GAPDH (equal loading control). (B) Immunoprecipitated JAK1 from cell lysates was resolved by SDS-PAGE and the blots sequentially probed for phosphotyrosine and total JAK1 protein. The blots shown are representative of 4 and 3 independent experiments for A and B, respectively. (C) Densitometric analysis of immunoreactive bands. *P < 0.05 vs. maximal activation with LIF, ANOVA and Dunnett’s multiple comparison test (top panel). *P < 0.05 and **P < 0.01 noted comparisons and †P < 0.05 vs. LIF, ANOVA and Newman–Keuls post-test (bottom panel).
3.6 Differential effect of crosslinking agents on STAT1 and STAT3
Our evidence indicates that JAK1, which is upstream of STAT3, is redox-sensitive and is targeted by oxidative stress. However, the finding that STAT3 but not STAT1 activation by LIF was affected by GSH depletion suggests that STAT3 is directly affected by the cellular redox status. One possible scenario is that formation of an intramolecular disulfide bond under conditions of oxidative stress induces a conformational change in STAT3 that impedes interaction with other proteins. To assess this possibility, we incubated heart homogenates with two different crosslinking reagents under conditions favorable for protein-protein interactions: NCP, which selectively oxidizes thiolate (reactive cysteine) residues (Shoman et al., 2011) and diamide, a broader based cysteine crosslinking agent. The STAT1 pool was more sensitive to depletion by the crosslinking procedure using either NCP or diamide than was the STAT3 pool (Fig. 7). Conversely, a higher percentage of the monomeric form was retained under oxidizing conditions for STAT3 than STAT1.
Figure 7.
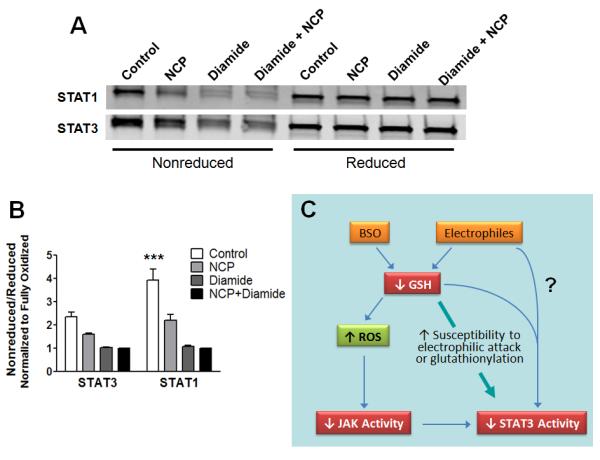
Differential response of STAT1 and STAT3 to crosslinking agents. (A & B) Aliquots of mouse heart homogenates were incubated with vehicle, 500 μM NCP, 1 mM diamide, or 500 μM NCP + 1 mM diamide for 30 min. Samples were processed for SDS-PAGE and Western analysis in nonreducing or reducing sample buffer. (A) Membranes were probed for STAT3 and STAT1 using the Li-COR Odyssey detection system. (B) For STAT1 and STAT3, the intensity of the band in the nonreduced sample was normalized to the intensity of the band after reduction and expressed relative to the total oxidized sample (NCP + diamide). ***P < 0.001 vs. Control STAT3, 2-way ANOVA and Bonferroni post-test (n = 3 mouse hearts). (C) Proposed scheme based on our findings and the literature to explain the accumulating evidence that oxidative stress attenuates gp130 cytokine signaling by targeting JAK1/2 and STAT3.
4. Discussion
Here we report that JAK1-STAT3 activation in cardiac myocytes is inhibited by GSH depletion, which was associated with increased superoxide formation. The attenuation of LIF-induced JAK1 and STAT3 activation observed with GSH depletion could be reversed by the addition of either GME, which is cleaved intracellularly to GSH, or the reductant NAC. Moreover, we also found that chemical oxidation of recombinant JAK1 causes cysteine-sulfenic acid formation, a reversible protein modification that is a hallmark of redox-sensitive proteins (Winterbourn and Hampton, 2008). However, our observation of a differential action of GSH depletion on STAT3 and STAT1 highlights the point that oxidative stress targets STAT3 as well. These findings are novel and support the conclusion that gp130 JAK1-STAT3 cytokine signaling in cardiac myocytes is redox-sensitive.
The JAK family of non-receptor tyrosine kinases consists of 4 members, JAK1, JAK2, JAK3, and TYK2. JAK1 and JAK2 are widely expressed, and their catalytic activity is regulated by the autophosphorylation of 2 conserved tandem tyrosine residues found in their activation loop (Liu et al., 1997; Feng et al., 1997). Recently, we reported site-directed mutagenesis experiments demonstrating the presence of 2 cysteine residues (C866 and C917) in the catalytic domain of rat JAK2 that function as a redox switch to control its catalytic activity (Smith et al., 2012). Among JAK family members these cysteine residues are highly conserved across species and in rat JAK1 are C891 and C943. Cysteine-sulfenic acid formation is one mechanism that conveys redox sensitivity to proteins and was recently demonstrated in recombinant JAK2 (Smith et al., 2012). Here, we report the formation of protein-sulfenic acid in JAK1 under oxidizing conditions imposed with o-IBZ (Fig. 3B). There was also enhanced sulfenic acid formation in JAK1 treated with 60 μM H2O2 but not to the same extent as seen with o-IBZ; however, H2O2 may require the presence of some catalyst to effectively form protein-sulfenic acids (Roos and Messens, 2011).
Although GSH depletion reduced LIF-induced STAT3 activation, STAT1 activation was unaffected (Fig. 4B). Evidence reported by others (Li and Shaw, 2004) indicates that the conformational response of STAT3 in cells treated with peroxide is different from that of STAT1. In our study, residual JAK1 activity in GSH depleted cardiac myocytes (Fig. 3A) may have been sufficient to activate STAT1. Alternatively, there is a recent report showing that LIF may couple to STAT activation independent of JAKs (Sun et al., 2011). Our finding that GSH depletion affected LIF-induced STAT3 activation, but not STAT1 activation, may have clinical importance. In cardiac myocytes, STAT3 has been linked to a protective program, while STAT1 has been linked to apoptosis (Zgheib et al., 2012). Therefore, IL-6 family cytokine signaling in cardiac myocytes could be skewed towards an adverse outcome under conditions of oxidative stress. Interestingly, others have recently reported evidence of impaired angiotensin II-induced STAT3 activation in failing human myocytes (Cambi et al., 2012). Moreover, both expression and phosphorylation of STAT3 are reported to be reduced in patients with heart failure (Podewski et al., 2003).
Our findings are consistent with accumulating evidence that gp130 cytokine signaling is redox-sensitive at the level of both JAK1 and STAT3 (Fig. 7C). Two naturally occurring terpenes that possess an α,β-unsaturated carbonyl group and are electrophiles were reported to decrease intracellular GSH content in the human acute monocytic leukemia cell line THP-1 and inhibit IL-6 signaling (Butturini et al., 2011). In that study, S-glutathionylation of STAT3 was observed, which others reported renders STAT3 a poor substrate for recombinant JAK2 (Xie et al., 2009); however, IL-6-induced JAK1 activation was inhibited as well. The α,β-unsaturated flavonoid chalcone was reported to inhibit IL-6-induced STAT3 activation in bovine aortic endothelial cells (BAEC) and depletion of GSH with BSO enhanced the effects of chalcone (Liu et al., 2007). In this study, BSO by itself did not affect IL-6-induced STAT3 activation, perhaps because in these cells endogenous levels of ROS generated by GSH depletion are less than in more metabolically-active cardiac myocytes. In mouse embryonic stem cells, the electrophile 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) inhibited LIF-induced JAK1 and STAT3 activation (Rajasingh and Bright, 2006), while in BAEC this arachidonic acid metabolite targeted only STAT3 (Wung et al., 2006).
Others reported that agents that induce oxidative stress by reducing cellular GSH levels cause S-glutathionylation of STAT3 and inhibition of IL-6 signaling in human HepG2 hepatocarcinoma cells (Xie et al., 2009). S-Glutathionylation of proteins is often seen with oxidative stress and may be triggered by GSH depletion (Franco and Cidlowski, 2009). What role, if any, this post-translational modification has in inhibiting JAK1 activation in GSH-depleted cardiac myocytes is an area of future investigation. Based on the differential response of STAT1 and STAT3 to crosslinking agents (Fig. 7), we propose however that STAT3 has a greater propensity to form intramolecular disulfide bonds that preclude interactions with other proteins.
Although the issue of apoptosis was not specifically addressed in our study, we saw no gross evidence for cell death with GSH depletion during the course of our experiments (e.g., membrane blebbing, cell lift-off, and loss of cellular protein or contractility). Relevant to this issue is a study reporting that BSO treatment of rats in vivo selectively reduced the cytoplasmic GSH pool in cardiac myocytes, but not the mitochondrial pool (Ghosh et al, 2005). Notably, apoptosis was only seen if the mitochondrial pool of GSH was also depleted by the addition of diethyl maleate along with BSO to deplete both cellular pools of GSH. The results of the GSH measurements in our study are suggestive of a pool of GSH that is resistant to BSO treatment (Fig. 1A)
Because there are both direct and indirect redox regulatory mechanisms affecting cytokine/JAK/STAT pathways, and because there are cellular differences in oxidative defense mechanisms and redox buffering capacities, one cannot extrapolate results from one cell (or tissue) type to another. For example, there are reports that BSO may increase basal STAT3 signaling in some cases. Enhanced STAT3-specific binding of nuclear extracts was detected following treatment of rat-1 fibroblasts and epidermally derived A-431 cells with BSO (Simon et al., 1998). Increased STAT3 tyrosine phosphorylation was seen in livers from rats treated with BSO (Torres L et al., 2009), although activation was short term (3-6 hours) and over by 24 h. In these two cases, ROS may have activated JAK2 indirectly through inhibition of a protein tyrosine phosphatase or may have activated a Src kinase family member, a JAK-alternative means of activating STAT3 (Kurdi and Booz, 2009). Notably, in our study we did not observe enhanced basal STAT3 activation with BSO treatment.
The gp130 cytokines have been shown to exert protective effects on the heart and/or cardiac myocytes in the context of I/R injury in part by upregulating cellular anti-oxidant defenses and by stimulating the production of angiogenic factors (Fischer and Hilfiker-Kleiner, 2007; Kusano et al., 2007; Lipsic et al., 2006; Ueda et al., 2006). The possibility of therapeutically exploiting gp130 cytokine signaling in the heart has been proposed (Kurdi and Booz 2007a; Fujio et al., 2011). Paradoxically, the results of the present study suggest that a normal intracellular redox environment is necessary for the IL-6 family cytokines to activate anti-oxidant defenses. This observation has implications for understanding events underlying the progression of post-infarct remodeling to heart failure, as increased cardiac generation of ROS leading to oxidative stress plays a major part in the pathogenesis of heart failure (Grieve et al., 2004; Nediani et al., 2007; Nojiri et al., 2006; Tsutsui et al., 2006). Recently, GSH deficiency in heart failure patients was correlated to their functional status and structural cardiac abnormalities (Damy et al., 2009). Notably, GSH depletion in wild-type Friend virus B (FVB) mice was found to result in cardiomyopathy at 2 weeks (Ren et al., 2008). Furthermore, treatment with NAC, a precursor of glutathione, reversed cardiac remodeling and systolic dysfunction in a rabbit genetic model of human cardiomyopathy (Lombardi et al., 2009).
In summary, we have found that LIF-induced JAK1-STAT3 signaling in cardiac myocytes is attenuated by depletion of the anti-oxidant GSH. These findings suggest that the STAT3-mediated protective effects of the IL-6-type cytokines may be attenuated during heart failure and other acute or chronic cardiac diseased conditions in which an overall oxidizing environment is present.
ACKNOWLEDGEMENTS
The authors wish to thank Barak Gunter, Carlos Zgheib, and Fouad Zouein for their expert assistance with some of the experiments. The authors are grateful to Dr. Vabren L. Watts for obtaining neonatal rat ventricular myocytes for EPR studies. This work was supported by grants to MK from The Lebanese University (MK-02-2011), The Lebanese National Council for Scientific Research (CNRS;05-10-09), and The COMSTECH-TWAS (09-122 RG/PHA/AF/AC_C); grants from the National Heart, Lung, and Blood Institute to GWB (R01HL088101-06), MAA & NP (R01HL091923-01), and VD (T32HL007227); a grant to VS from the Fondation Leducq Transatlantic Network of Excellence (09 CVD 01); and a grant from the National Institute of Diabetes and Digestive and Kidney Diseases to RJD (1R56DK082781-01).
Footnotes
DISCLOSURES None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Akila, D’souza B, Vishwanath P, D’souza V. Oxidative injury and antioxidants in coronary artery bypass graft surgery: off-pump CABG significantly reduces oxidative stress. Clin Chim Acta. 2007;375:147–52. doi: 10.1016/j.cca.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Aoyama T, Takimoto Y, Pennica D, Inoue R, Shinoda E, Hattori R, Yui Y, Sasayama S. Augmented expression of cardiotrophin-1 and its receptor component, gp130, in both left and right ventricles after myocardial infarction in the rat. J Mol Cell Cardiol. 2000;32:1821–30. doi: 10.1006/jmcc.2000.1218. [DOI] [PubMed] [Google Scholar]
- Brown DA, Aon MA, Frasier CR, Sloan RC, Maloney AH, Anderson EJ, O’Rourke B. Cardiac arrhythmias induced by glutathione oxidation can be inhibited by preventing mitochondrial depolarization. J Mol Cell Cardiol. 2010;48:673–9. doi: 10.1016/j.yjmcc.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butturini E, Cavalieri E, de Prati AC, Darra E, Rigo A, Shoji K, Murayama N, Yamazaki H, Watanabe Y, Suzuki H, Mariotto S. Two naturally occurring terpenes, dehydrocostuslactone and costunolide, decrease intracellular GSH content and inhibit STAT3 activation. PLoS One. 2011;6:e20174. doi: 10.1371/journal.pone.0020174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambi GE, Lucchese G, Djeokeng MM, Modesti A, Fiaschi T, Faggian G, Sani G, Modesti PA. Impaired JAK2-induced activation of STAT3 in failing human myocytes. Mol Biosyst. 2012 Jun 26; doi: 10.1039/c2mb25120e. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Chandrasekar B, Mitchell DH, Colston JT, Freeman GL. Regulation of CCAAT/Enhancer binding protein, interleukin-6, interleukin-6 receptor, and gp130 expression during myocardial ischemia/reperfusion. Circulation. 1999;99:427–33. doi: 10.1161/01.cir.99.3.427. [DOI] [PubMed] [Google Scholar]
- Damy T, Kirsch M, Khouzami L, Caramelle P, Le Corvoisier P, Roudot-Thoraval F, Dubois-Randé JL, Hittinger L, Pavoine C, Pecker F. Glutathione deficiency in cardiac patients is related to the functional status and structural cardiac abnormalities. PLoS ONE. 2009;4:e4871. doi: 10.1371/journal.pone.0004871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawn B, Xuan YT, Guo Y, Rezazadeh A, Stein AB, Hunt G, Wu WJ, Tan W, Bolli R. IL-6 plays an obligatory role in late preconditioning via JAK-STAT signaling and upregulation of iNOS and COX-2. Cardiovasc Res. 2004;64:61–71. doi: 10.1016/j.cardiores.2004.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikalov SI, Kirilyuk IA, Voinov M, Grigor’ev IA. EPR detection of cellular and mitochondrial superoxide using cyclic hydroxylamines. Free Radic Res. 2011;45:417–30. doi: 10.3109/10715762.2010.540242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Witthuhn BA, Matsuda T, Kohlhuber F, Kerr IM, Ihle JN. Activation of JAK2 catalytic activity requires phosphorylation of Y1007 in the kinase activation loop. Mol Cell Biol. 1997;17:2497–501. doi: 10.1128/mcb.17.5.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer P, Hilfiker-Kleiner D. Survival pathways in hypertrophy and heart failure: The gp130-STAT3 axis. Basic Res Cardiol. 2007;102:279–97. doi: 10.1007/s00395-007-0658-z. [DOI] [PubMed] [Google Scholar]
- Franco R, Cidlowski JA. Apoptosis and glutathione: beyond an antioxidant. Cell Death Differ. 2009;16:1303–14. doi: 10.1038/cdd.2009.107. [DOI] [PubMed] [Google Scholar]
- Fujio Y, Maeda M, Mohri T, Obana M, Iwakura T, Hayama A, Yamashita T, Nakayama H, Azuma J. Glycoprotein 130 cytokine signal as a therapeutic target against cardiovascular diseases. J Pharmacol Sci. 2011;117:213–22. doi: 10.1254/jphs.11r05cr. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Pulinilkunnil T, Yuen G, Kewalramani G, An D, Qi D, Abrahani A, Rodrigues B. Cardiomyocyte apoptosis induced by short-term diabetes requires mitochondrial GSH depletion. Am J Physiol Heart Circ Physiol. 2005;289:H768–76. doi: 10.1152/ajpheart.00038.2005. [DOI] [PubMed] [Google Scholar]
- Grieve DJ, Byrne JA, Cave AC, Shah AM. Role of oxidative stress in cardiac remodelling after myocardial infarction. Heart Lung Circ. 2004;13:132–8. doi: 10.1016/j.hlc.2004.02.008. [DOI] [PubMed] [Google Scholar]
- Gritman K, Van Winkle DM, Lorentz CU, Pennica D, Habecker BA. The lack of cardiotrophin-1 alters expression of interleukin-6 and leukemia inhibitory factor mRNA but does not impair cardiac injury response. Cytokine. 2006;36:9–16. doi: 10.1016/j.cyto.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwechenberger M, Mendoza LH, Youker KA, Frangogiannis NG, Smith CW, Michael LH, Entman ML. Cardiac myocytes produce interleukin-6 in culture and in viable border zone of reperfused infarctions. Circulation. 1999;99:546–51. doi: 10.1161/01.cir.99.4.546. [DOI] [PubMed] [Google Scholar]
- Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hishinuma S, Funamoto M, Fujio Y, Kunisada K, Yamauchi-Takihara K. Hypoxic stress induces cardiotrophin-1 expression in cardiac myocytes. Biochem Biophys Res Commun. 1999;264:436–40. doi: 10.1006/bbrc.1999.1535. [DOI] [PubMed] [Google Scholar]
- Jones DP. Redox potential of GSH/GSSG couple: assay and biological significance. Methods Enzymol. 2002;348:93–112. doi: 10.1016/s0076-6879(02)48630-2. [DOI] [PubMed] [Google Scholar]
- Kukielka GL, Smith CW, Manning AM, Youker KA, Michael LH, Entman ML. Induction of interleukin-6 synthesis in the myocardium. Potential role in postreperfusion inflammatory injury. Circulation. 1995;92:1866–75. doi: 10.1161/01.cir.92.7.1866. [DOI] [PubMed] [Google Scholar]
- Kurdi M, Booz GW. Can the protective actions of JAK-STAT in the heart be exploited therapeutically? Parsing the regulation of IL-6-type cytokine signaling. J Cardiovasc Pharmacol. 2007a;50:126–41. doi: 10.1097/FJC.0b013e318068dd49. [DOI] [PubMed] [Google Scholar]
- Kurdi M, Booz GW. Evidence that IL-6-type cytokine signaling in cardiomyocytes is inhibited by oxidative stress: Parthenolide targets JAK1 activation by generating ROS. J Cell Physiol. 2007b;212:424–31. doi: 10.1002/jcp.21033. [DOI] [PubMed] [Google Scholar]
- Kurdi M, Booz GW. JAK redux: a second look at the regulation and role of JAKs in the heart. Am J Physiol Heart Circ Physiol. 2009;297:H1545–56. doi: 10.1152/ajpheart.00032.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurdi M, Bowers MC, Dado J, Booz GW. Parthenolide induces a distinct pattern of oxidative stress in cardiac myocytes. Free Radic Biol Med. 2007;42:474–81. doi: 10.1016/j.freeradbiomed.2006.11.012. [DOI] [PubMed] [Google Scholar]
- Kusano K, Tsutsumi Y, Dean J, Gavin M, Ma H, Silver M, Thorne T, Zhu Y, Losordo DW, Aikawa R. Long-term stable expression of human growth hormone by rAAV promotes myocardial protection post-myocardial infarction. J Mol Cell Cardiol. 2007;42:390–9. doi: 10.1016/j.yjmcc.2006.10.016. [DOI] [PubMed] [Google Scholar]
- Li L, Shaw PE. A STAT3 dimer formed by inter-chain disulphide bridging during oxidative stress. Biochem Biophys Res Commun. 2004;322:1005–11. doi: 10.1016/j.bbrc.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Lipsic E, Schoemaker RG, van der Meer P, Voors AA, van Veldhuisen DJ, van Gilst WH. Protective effects of erythropoietin in cardiac ischemia: from bench to bedside. J Am Coll Cardiol. 2006;48:2161–7. doi: 10.1016/j.jacc.2006.08.031. [DOI] [PubMed] [Google Scholar]
- Liu KD, Gaffen SL, Goldsmith MA, Greene WC. Janus kinases in interleukin-2 mediated signaling: JAK1 and JAK3 are differentially regulated by tyrosine phosphorylation. Curr Biol. 1997;7:817–26. doi: 10.1016/s0960-9822(06)00369-1. [DOI] [PubMed] [Google Scholar]
- Liu YC, Hsieh CW, Wu CC, Wung BS. Chalcone inhibits the activation of NF-κB and STAT3 in endothelial cells via endogenous electrophile. Life Sci. 2007;80:1420–30. doi: 10.1016/j.lfs.2006.12.040. [DOI] [PubMed] [Google Scholar]
- Lombardi R, Rodriguez G, Chen SN, Ripplinger CM, Li W, Chen J, Willerson JT, Betocchi S, Wickline SA, Efimov IR, Marian AJ. Resolution of established cardiac hypertrophy and fibrosis and prevention of systolic dysfunction in a transgenic rabbit model of human cardiomyopathy through thiol-sensitive mechanisms. Circulation. 2009;119:1398–407. doi: 10.1161/CIRCULATIONAHA.108.790501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morihira M, Hasebe N, Baljinnyam E, Sumitomo K, Matsusaka T, Izawa K, Fujino T, Fukuzawa J, Kikuchi K. Ischemic preconditioning enhances scavenging activity of reactive oxygen species and diminishes transmural difference of infarct size. Am J Physiol Heart Circ Physiol. 2006;290:H577–83. doi: 10.1152/ajpheart.00817.2004. [DOI] [PubMed] [Google Scholar]
- Nediani C, Borchi E, Giordano C, Baruzzo S, Ponziani V, Sebastiani M, Nassi P, Mugelli A, d’Amati G, Cerbai E. NADPH oxidase-dependent redox signaling in human heart failure: relationship between the left and right ventricle. J Mol Cell Cardiol. 2007;42:826–34. doi: 10.1016/j.yjmcc.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Nian M, Lee P, Khaper N, Liu P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res. 2004;94:1543–53. doi: 10.1161/01.RES.0000130526.20854.fa. [DOI] [PubMed] [Google Scholar]
- Nojiri H, Shimizu T, Funakoshi M, Yamaguchi O, Zhou H, Kawakami S, Ohta Y, Sami M, Tachibana T, Ishikawa H, Kurosawa H, Kahn RC, Otsu K, Shirasawa T. Oxidative stress causes heart failure with impaired mitochondrial respiration. J Biol Chem. 2006;281:33789–801. doi: 10.1074/jbc.M602118200. [DOI] [PubMed] [Google Scholar]
- Podewski EK, Hilfiker-Kleiner D, Hilfiker A, Morawietz H, Lichtenberg A, Wollert KC, Drexler H. Alterations in Janus kinase (JAK)-signal transducers and activators of transcription (STAT) signaling in patients with end-stage dilated cardiomyopathy. Circulation. 2003;107:798–802. doi: 10.1161/01.cir.0000057545.82749.ff. [DOI] [PubMed] [Google Scholar]
- Poole LB, Karplus PA, Claiborne A. Protein-sulfenic acids in redox signaling. Annu Rev Pharmacol Toxicol. 2004;44:325–47. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- Rajasingh J, Bright JJ. 15-Deoxy-Δ12,14-prostaglandin J2 regulates leukemia inhibitory factor signaling through JAK-STAT pathway in mouse embryonic stem cells. Exp Cell Res. 2006;312:2538–46. doi: 10.1016/j.yexcr.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Ren J, Privratsky JR, Yang X, Dong F, Carlson EC. Metallothionein alleviates glutathione depletion-induced oxidative cardiomyopathy in murine hearts. Crit Care Med. 2008;36:2106–16. doi: 10.1097/CCM.0b013e31817bf925. [DOI] [PubMed] [Google Scholar]
- Rodig SJ, Meraz MA, White JM, Lampe PA, Riley JK, Arthur CD, King KL, Sheehan K, Yin L, Pennica D, Johnson EM, Jr, Schreiber RD. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell. 1998;93:373–83. doi: 10.1016/s0092-8674(00)81166-6. [DOI] [PubMed] [Google Scholar]
- Roos G, Messens J. Protein sulfenic acid formation: from cellular damage to redox regulation. Free Radic Biol Med. 2011;51:314–26. doi: 10.1016/j.freeradbiomed.2011.04.031. [DOI] [PubMed] [Google Scholar]
- Seo YH, Carroll KS. Profiling protein thiol oxidation in tumor cells using sulfenic acid-specific antibodies. Proc Natl Acad Sci USA. 2009;106:16163–8. doi: 10.1073/pnas.0903015106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoman ME, DuMond JF, Isbell TS, Crawford JH, Brandon A, Honovar J, Vitturi DA, White CR, Patel RP, King SB. Acyloxy nitroso compounds as nitroxyl (HNO) donors: kinetics, reactions with thiols, and vasodilation properties. J Med Chem. 2011;54:1059–1070. doi: 10.1021/jm101432z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slodzinski MK, Aon MA, O’Rourke B. Glutathione oxidation as a trigger of mitochondrial depolarization and oscillation in intact hearts. J Mol Cell Cardiol. 2008;45:650–60. doi: 10.1016/j.yjmcc.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon AR, Rai U, Fanburg BL, Cochran BH. Activation of the JAK-STAT pathway by reactive oxygen species. Am J Physiol. 1998;275:C1640–52. doi: 10.1152/ajpcell.1998.275.6.C1640. [DOI] [PubMed] [Google Scholar]
- Smith JK, Patil CN, Patlolla S, Gunter BW, Booz GW, Duhé RJ. Identification of a redox-sensitive switch within the JAK2 catalytic domain. Free Radic Biol Med. 2012;52:1101–10. doi: 10.1016/j.freeradbiomed.2011.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Wang J, Xiong J, Yang L, Liu H. Free LIF receptor α-chain distal cytoplasmic motifs enhance Jak2-independent STAT3 phosphorylation and induce differentiation in HL-60 cells. Oncol Rep. 2011;26:399–404. doi: 10.3892/or.2011.1289. [DOI] [PubMed] [Google Scholar]
- Terrell AM, Crisostomo PR, Wairiuko GM, Wang M, Morrell ED, Meldrum DR. Jak/STAT/SOCS signaling circuits and associated cytokine-mediated inflammation and hypertrophy in the heart. Shock. 2006;26:226–34. doi: 10.1097/01.shk.0000226341.32786.b9. [DOI] [PubMed] [Google Scholar]
- Torres L, Sandoval J, Penella E, Zaragozá R, García C, Rodríguez JL, Viña JR, García-Trevijano ER. In vivo GSH depletion induces c-myc expression by modulation of chromatin protein complexes. Free Radic Biol Med. 2009;46:1534–42. doi: 10.1016/j.freeradbiomed.2009.03.005. [DOI] [PubMed] [Google Scholar]
- Tsutsui H, Ide T, Kinugawa S. Mitochondrial oxidative stress, DNA damage, and heart failure. Antioxid Redox Signal. 2006;8:1737–44. doi: 10.1089/ars.2006.8.1737. [DOI] [PubMed] [Google Scholar]
- Ueda K, Takano H, Hasegawa H, Niitsuma Y, Qin Y, Ohtsuka M, Komuro I. Granulocyte colony stimulating factor directly inhibits myocardial ischemia-reperfusion injury through Akt-endothelial NO synthase pathway. Arterioscler Thromb Vasc Biol. 2006;26:e108–13. doi: 10.1161/01.ATV.0000219697.99134.10. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008;45:549–61. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- Wung BS, Wu CC, Hsu MC, Hsieh CW. 15-Deoxy-Δ12,14-prostaglandin J2 suppresses IL-6-induced STAT3 phosphorylation via electrophilic reactivity in endothelial cells. Life Sci. 2006;78:3035–42. doi: 10.1016/j.lfs.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Yamauchi-Takihara K, Ihara Y, Ogata A, Yoshizaki K, Azuma J, Kishimoto T. Hypoxic stress induces cardiac myocyte-derived interleukin-6. Circulation. 1995;91:1520–4. doi: 10.1161/01.cir.91.5.1520. [DOI] [PubMed] [Google Scholar]
- Xie Y, Kole S, Precht P, Pazin MJ, Bernier M. S-glutathionylation impairs signal transducer and activator of transcription 3 activation and signaling. Endocrinology. 2009;150:1122–31. doi: 10.1210/en.2008-1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zgheib C, Zouein FA, Kurdi M, Booz GW. Differential STAT3 signaling in the heart: Impact of concurrent signals and oxidative stress. JAK-STAT. 2012;1:102–111. doi: 10.4161/jkst.19776. [DOI] [PMC free article] [PubMed] [Google Scholar]