

Abstract
The relationship between bats and coronaviruses (CoVs) has received considerable attention since the severe acute respiratory syndrome (SARS)-like CoV was identified in the Chinese horseshoe bat (Rhinolophidae) in 2005. Since then, several bats throughout the world have been shown to shed CoV sequences, and presumably CoVs, in the feces; however, no bat CoVs have been isolated from nature. Moreover, there are very few bat cell lines or reagents available for investigating CoV replication in bat cells or for isolating bat CoVs adapted to specific bat species. Here, we show by molecular clock analysis that alphacoronavirus (α-CoV) sequences derived from the North American tricolored bat (Perimyotis subflavus) are predicted to share common ancestry with human CoV (HCoV)-NL63, with the most recent common ancestor between these viruses occurring approximately 563 to 822 years ago. Further, we developed immortalized bat cell lines from the lungs of this bat species to determine if these cells were capable of supporting infection with HCoVs. While SARS-CoV, mouse-adapted SARS-CoV (MA15), and chimeric SARS-CoVs bearing the spike genes of early human strains replicated inefficiently, HCoV-NL63 replicated for multiple passages in the immortalized lung cells from this bat species. These observations support the hypothesis that human CoVs are capable of establishing zoonotic-reverse zoonotic transmission cycles that may allow some CoVs to readily circulate and exchange genetic material between strains found in bats and other mammals, including humans.
INTRODUCTION
Bats are known to be reservoir hosts for several human viruses, including rabies, Marburg, Nipah, Hendra, and the severe acute respiratory syndrome coronavirus (SARS-CoV) (5). In addition, virome studies have shown unprecedented numbers of viruses present in the fecal samples of this ancient mammalian species (11, 24). However, little is known about the genetic architecture of most bat species, the virus variation and gene flow that occur through different species, the potential of different bat species to support human virus replication, the differences between the bat and human immune systems, or the potential of bat viruses to undergo zoonotic transmission to humans and other mammals.
Coronaviruses are the largest known RNA viruses; these viruses contain single-stranded plus sense genomes and are classified in the family Coronaviridae, which is divided into three genera, including Alphacoronavirus (α-CoV), Betacoronavirus (β-CoV), and Gammacoronavirus (γ-CoV). Five CoVs are known to cause human disease, including the β-CoVs SARS-CoV, human CoV (HCoV)-OC43, and HCoV-HKU1 and the α-CoVs HCoV-229E and HCoV-NL63 (35). Three of these HCoVs have been shown to or have been predicted to have spilled over from zoonotic reservoirs, including SARS-CoV, which likely emerged from the Chinese horseshoe bat (Rhinolophidae) (26), HCoV-OC43, which probably emerged from bovine CoV (BCoV) (50), and HCoV-229E (36), which was predicted by molecular clock analysis to share a most recent common ancestor (MRCA) just over 200 hundred years ago with a bat CoV found in the leaf-nosed bat (Hipposideros caffer ruber) bat in Ghana (36). The close link between bat and human CoVs has led to the speculation that all human, and perhaps mammalian, CoVs may have originated in bats (19, 36, 49).
HCoV-NL63 was first discovered in 2004 as a new HCoV isolated from a 7-month-old baby suffering from bronchiolitis (48). A similar virus was isolated around the same time in samples derived from an 8-month-old baby with pneumonia (16). Since 2004, this HCoV has been detected in 1.0 to 9.3% of respiratory tract samples collected from several different countries (15), which indicates that HCoV-NL63 is distributed worldwide. There is no known reservoir for this virus, and little is known about its evolutionary history prior to 2004, although phylogenetic evidence suggests that HCoV-NL63 has infected humans for centuries, as it is predicted to have diverged from HCoV-229E approximately 1,000 years ago (38).
There are more than 1,100 species of bats, with bat populations inhabiting every continent except Antarctica. Bats belong to the order Chiroptera and are further divided into the suborders of Yinpterochiroptera, which include mostly the megabats (formerly known as the megachiroptera) and Yangochiroptera, which includes most of the microbat (formerly known as the microchiroptera) families (46). In the United States, most bat species are small nocturnal bats that belong to the family Vespertilionidae, and all are insectivores. In Maryland, previous surveys have yielded fecal samples from the big brown bat (Eptesicus fuscus), the little brown myotis (Myotis lucifugus), the northern long-eared myotis (Myotis septentrionalis), the eastern small-footed myotis (Myotis leibii), the hoary bat (Lasiurus cinereus), the red bat (Lasiurus borealis), and the tricolored bat (Perimyotis subflavus). In our previous work, we have shown that five of these species shed unique α-CoV sequences (11); however, the reagents necessary to isolate these viruses and determine if North American bat CoVs pose a threat to public health are currently lacking.
In this study, we identified nucleic acid sequences that potentially indicate the presence of a novel α-CoV that is predicted to share an RCA with HCoV-NL63 in the tricolored bat. We developed an immortalized lung cell line for this U.S. bat species and show that human CoVs grow in these bat cells. This observation suggests that human CoVs are capable of infecting multiple mammalian hosts, potentially establishing zoonotic-reverse zoonotic cycles that allow CoVs to maintain viral populations in multiple hosts, evolve novel recombinant viruses with viral genes derived from human and animal CoVs, and traffic back into human populations at a later date. In addition, these results suggest that HCoV-NL63 may have originated in bats and crossed the species barrier to infect humans roughly 563 to 822 years ago. Supporting a growing body of literature, our data support the hypothesis that cross-species transmission events and the emergence and colonization of new species represent common features of the Coronaviridae.
MATERIALS AND METHODS
Bat sequences for determining bat phylogeny.
All of the available cytochrome b genes (1) from the North American bats found in Maryland and other bats of interest were downloaded from GenBank along with the same genes from several other common mammals. The nucleotide gene sequences were then aligned by ClustalX, a maximum likelihood tree was generated using PhyML with 100 bootstraps, and the tree image was edited and exported using the bioinformatics tools available in the Geneious software suite version 5.4.3 (13).
Identification of novel α-CoVs in North American bats.
In our previous studies, we demonstrated that α-CoV sequences are present in the fecal samples of eastern North American bat species (11). Using the exact procedure as previously described (11), we used Roche 454 sequencing to determine the viral sequences present in bat fecal samples from big brown bats captured in the Saratoga National Historical park in New York (New England CoV [NECoV]) and tricolored bats from the Chesapeake and Ohio Canal National Historical Park in Maryland (Appalachian Ridge CoV strain 2 [ARCoV.2]). Then, we used previously reported primers and protocols (11) to amplify a >2,200-nucleotide (nt) fragment in the replicase region of these viruses, encompassing a portion of nsp13, all of nsp14, and a portion of nsp15. The amplified fragments were electrophoresed on a 1% agarose gel, and the >2,200-nt band was excised, purified, and subjected to Sanger sequencing as previously described (11). These sequences were deposited into GenBank (see below).
Phylogenetic and molecular clock analyses of α-CoVs found in North American bats. (i) Phylogenetic analysis.
The sequences of the >2,200-nt fragments of ARCoV.1, ARCoV.2, and NECoV were compared to the same region of several known CoV sequences downloaded from GenBank. The sequences were aligned using ClustalX as implemented in Geneious 5.4.3 (13), and the alignment was manually trimmed and corrected to generate a 2,321-nucleotide alignment. A maximum likelihood tree was generated using PhyML with 100 bootstraps, and the tree image was edited and exported using the bioinformatics tools available in the Geneious software suite version 5.4.3 (13). This was the largest fragment available for all three genomes, and that was the basis for generating the tree using these sequences.
(ii) Molecular clock analysis.
Molecular clock analysis was conducted using BEAST version 1.7.1 (14), following the same protocol as that used by Pfefferle et al. (2009) (36) and using the same 650- to 800-nt fragment of the replicase region of several known CoVs to estimate the date of the most recent common ancestor for ARCoV.1 and ARCoV.2. The replicase sequences for ARCoV.1 (11), NECoV, and ARCoV.2 were derived from sequence reads obtained by 454 sequencing, and because NECoV and ARCoV.1 were nearly identical, only the ARCoV.1 sequence was used in the analysis. Of note, this sequence is a portion of the viral replicase gene (nsp12), which is arguably the most conserved region of the CoV genome, making it the most appropriate target for molecular clock analysis. These replicase fragment sequences were deposited in GenBank (see below). Most of the sequences were dated in years before present, which was 2011 when this study was conducted. Using the date found by Vijgen et al. (2005) (50) for the HCoV-OC43 and bovine CoV sequences, and following the method of Pfefferle et al. (2009) (36), a normal probabilistic prior with a mean of 121 years before the present time and a standard deviation of 13 years was used to calibrate the analysis (36, 50). Both the GTR+Gamma 4 + I and the SRD06 models were tested under the assumption of an uncorrelated lognormal clock and a constant population size. In addition, an exponential population size assumption was tested using the SRD06 model. Markov chain Monte Carlo chains were set to 200,000,000 iterations with sampling every 20,000 generations. Bayes factor analysis, which was used to compare the models, was conducted using the program Tracer version 1.5 (http://tree.bio.ed.ac.uk/software/tracer). The SRD06 model was found to be superior to the GTR model by a log10 Bayes factor of 240.
Bat tissue samples.
Bats for this study were obtained from the Save Lucy Campaign, an education and bat rehabilitation center in Virginia. This organization specializes in rehabilitating bats that are sick or injured, and all of the bats in the rescue were observed for symptoms of rabies virus on intake. Only nonrabid bats that were badly injured beyond recovery were euthanized for this study. These included two big brown bats (EpFu) and one tricolored bat (PeSu). The bats were euthanized at the rescue center, and the bat organs were surgically removed and placed into processing medium (calcium- and magnesium-free PBS supplemented with 100 mg disodium EDTA, 1% penicillin/streptomycin, and 1% gentamicin) (7). Organs harvested from each bat included heart, lung, intestine, brain, kidney, liver, and stomach. The organs in medium were delivered on ice within 8 h to our laboratory for processing.
Biosafety considerations.
All of the bat organs harvested at the bat rescue were transported to our lab at the University of North Carolina (UNC). The tissues were further processed in a separate and locked biosafety level 2+ (BSL2+) room that required all personnel to have received a prophylactic series of rabies vaccines. All work was performed under certified biosafety cabinets, and all personnel wore gloves and Tyvek aprons (Fisher Scientific) when working with the fresh tissue.
Generation of primary bat cell lines from North American bats.
The organs were removed from transport buffer, washed with cold processing media, placed in specimen tubes with fresh processing media, and then placed on ice. To prepare the tissues, we followed a published protocol by Crameri et al. (2009) (7). The tissues from each organ were finely dissected using a sterile scalpel and washed with cold processing media. The dissected tissues were then transferred to a 50-ml conical tube, covered with cold 0.25% trypsin (Gibco), and incubated at 4°C overnight. The following day, the samples were centrifuged at 37°C on a benchtop shaker set at 200 rpm for 1 h. Supernatants were filtered through a 40-μm cell strainer (Fisher) into a 50-ml conical tube containing 10 ml of fetal calf serum (FCS) (HiClone). The cell strainer was then rinsed with phosphate-buffered saline (PBS) to ensure that all of the individual cells were delivered through the strainer. Next, the larger pieces of dissected tissues were incubated with 0.25% trypsin (Gibco) at 37°C for 30 min on the shaker, and then the supernatants were add to the FCS in the 50-ml conical tube. This was repeated until most of the cells were removed from the tissue scaffolds. Next, the cells were pelleted at 800 × g for 5 min in a Beckman benchtop centrifuge. The cells were then resuspended in primary cell culture medium (Dulbecco's modified Eagle medium [DMEM]/F12-Ham's media supplemented with 15% Fetal Clone II, 1% penicillin/streptomycin, 1% nonessential amino acids, and 1% gentamicin), transferred to T25 flasks, and incubated at 37°C with 5% CO2. Robust cultures of primary heart, lung, brain, and kidney cells were established. The cells were fed by removing the medium and replacing it with fresh primary medium with 15% FCS every 3 to 5 days until the cells reached confluence. The primary cells were then maintained for 2 to 5 passages to generate cell stocks that were cryopreserved and stored at −140°C.
Generation of immortalized bat cell lines from North American bats.
Primary cells that propagated well enough to reach sufficient numbers in up to three passages were selected as candidates for immortalization. Cells were prepared in 6-well plates and grown to 70 to 80% confluence at 37°C with 5% CO2 in primary cell culture media. The cells were then infected, as previously described (18), with lentivirus vectors expressing the human telomerase reverse transcriptase (hTERT) gene and the murine Bmi-1 gene (18), an oncogene that promotes efficient self-renewing cell divisions. A mixture of hTERT and Bmi-1 was created by placing 1.5 ml of each vector into a 15-ml conical tube at vector concentrations of >106 infectious units/ml. Next, we added 6 μl of Polybrene at a concentration of 8 μg/ml to increase the infectivity of the lentivirus vectors, and the mixture was vortexed gently and stored on ice. Next, we removed the medium from the cells in the 6-well plate and washed the cells twice with PBS (Gibco). For the cells to be immortalized, we added 1 ml of the hTERT–Bmi-1–Polybrene mixture and incubated it for 3 h at 37°C with 5% CO2. For the control cells, 1 ml of medium was added prior to incubation under the same conditions. After the incubation, the supernatants were removed from the cells and discarded into a 10% bleach solution. Primary cell culture medium was then added to all of the wells, and the cultures were incubated overnight at 37°C with 5% CO2. On the following day, the cells were transfected a second time using the same protocol. The cells were then monitored daily to look for morphological changes in the cells and to determine if the immortalization process was successful. Passages were conducted when the cells reached confluence, which varied by cell type, with the lung cells requiring 3 to 4 days between passages. Immortalization was determined to be successful in those cells that continued to thrive beyond the number of passages required for the primary cells to reach senescence and eventual cell death. To determine the extent of the immortalization, the infected cells were propagated for several additional passages, with the immortalized lung cells from the tricolored bat showing the best postsenescence growth phenotype. Several stocks of primary and immortalized cells were cryopreserved at each passage and then stored at −140°C for future use.
Infection of PESU-B5L cells with HCoV-NL63 and SARS-CoV.
Immortalized bat lung cells from the tricolored bat (Perimyotis subflavus) were seeded onto a 6-well plate and grown in primary cell culture medium at 37°C with 5% CO2 until they reached 70 to 80% confluence and a concentration of ∼3 × 105 cells per well. The supernatants were then removed, and the cells were infected with 1 ml of wild-type HCoV-NL63 (wtNL63) or HCoV-NL63 expressing green fluorescent protein (GFP) (NL63gfp) (12) or PBS (mock) at a multiplicity of infection (MOI) of 1. For the adsorption period, the cells were incubated at 32°C with 5% CO2 for 1 h with agitation every 15 min. At the end of the adsorption period, an additional 2 ml of culture medium was added to each well, and the infected cells were maintained at 32°C with 5% CO2 and monitored for 8 days. For SARS-CoVgfp (44) and several related strains, the experimental infections were conducted under biosafety level 3 containment following the same parameters as described above, using mouse-adapted SARS-CoV (MA15), a SARS-CoV strain adapted to be lethal in mice, GD03, a recombinant chimeric SARS-CoV clone bearing the spike sequence of a 2003 human clinical isolate, and HC/SZ/61/03, a recombinant chimeric SARS-CoV clone bearing the spike sequence of an early human isolate (8, 39, 41, 42, 44). These infected cells were maintained at 37°C with 5% CO2 and monitored for 5 days.
Verification of SARS-CoV and HCoV-NL63 replication in PESU-B5L.
To verify SARS-CoV and its related strains and HCoV-NL63 replication in the PESU-B5L lung cells, we used primers designed to amplify the subgenomic leader containing mRNA transcripts of each virus by reverse transcriptase PCR (RT-PCR). The amplified leader-containing cDNAs are specific to each HCoV and would be detected only if the virus was undergoing active transcription and replication. For HCoV-NL63, we used the primer sets and the exact RT-PCR conditions to amplify the subgenomic mRNA encoding the nucleocapsid (N) gene as previously described (12). The resulting bands were also excised, purified, and subjected to Sanger sequencing at the UNC Genomic Analysis Center to verify the origins and sequence of the leader-containing cDNAs. For SARS-CoV and its related strains, we used primers designed to detect subgenomic transcription of multiple viral proteins using primers and reaction conditions that have been published elsewhere (53). The resulting band for the SARS-CoV membrane gene was excised, purified, and sequenced in the same way as described for the HCoV-NL63 N gene.
Detection of HCoV-NL63 in PESU-B5L by immunofluorescence assay.
PESU-B5L cells were infected with HCoV-NL63 at an MOI of 0.7 or mock infected. Seventy-two hours postinfection, monolayers were washed twice with cold phosphate-buffered saline and fixed with a 50% methanol-50% acetone solution. Cells were then permeabilized with 0.1% Triton X-100–5% bovine serum albumin (BSA) in PBS and blocked with 5% BSA in PBS. Rabbit anti-NL63 nucleocapsid sera (the kind gift of Lia van der Hoek) were used as the primary antibody. Unbound primary antibody was removed by washing three times with 1% BSA–0.05% Nonidet P-40 in PBS, followed by addition of an Alexa 546-conjugated goat anti-rabbit IgG (Invitrogen). After washing off unbound secondary antibody, infected cells were photographed using an inverted microscope with a mercury lamp light source and a tetramethyl rhodamine isothiocyanate (TRITC) filter.
Detection of HCoV-NL63 in PESU-B5L cells by Western blotting.
PESU-B5L cells were infected with HCoV-NL63 at a multiplicity of infection of 0.7 or mock infected. Seventy-two hours postinfection, cells were washed with cold PBS followed by lysis in AV lysis buffer (20 mM Tris-HCl [pH 7.6], 150 mM NaCl, 0.5% deoxycholine, 1.0% Nonidet P-40, and 0.1% sodium dodecyl sulfate [SDS]). The lysates were spun down at 13,000 rpm for 10 min to pellet the nuclei. Equal volumes of 0.9% SDS and 10 mM EDTA were added to the postnuclear supernatants (to raise the concentration of SDS to 0.5% for virus inactivation), and the mixture was incubated at room temperature for 15 min to ensure complete inactivation of HCoV-NL63. Equal amounts of cell lysates were then loaded onto a 4 to 12% Bis-Tris Nupage gel (Invitrogen), and postelectrophoresis, the proteins were transferred onto polyvinylidene difluoride (PVDF) membranes using an Xcell blot module (Invitrogen) according to the manufacturer's instructions. Membranes were blocked with 5% nonfat dry milk in TBS-T buffer (0.1% Tween 20 in Tris-buffered saline) for 1 h and incubated overnight with rabbit anti-NL63 nucleocapsid sera. Unbound antibody was removed by washing in TBS-T followed by incubation with an antirabbit horseradish peroxidase (HRP) secondary antibody. Proteins were visualized using a Pierce Super Signal Western blot chemiluminescence kit. For the loading control, a purified mouse monoclonal antibody to beta actin (catalog no. 612656; BD Transduction Technologies) was used, followed by staining with secondary antimouse HRP antibody (GE Amersham Biosciences).
Nucleotide sequence accession numbers.
The sequences determined in this study were deposited into GenBank under accession numbers JX537911 through JX537914.
RESULTS
Evolutionary relatedness of North American bats.
The mitochondrial cytochrome b genes of several bat and other mammalian species were compared to show the evolutionary relatedness of North American bats frequently found in Maryland, including the big brown bat (EpFu), the little brown myotis (MyLu), the eastern small-footed myotis (MyLe), the northern long-eared myotis (MySe), and the tricolored bat (PeSu) (Fig. 1). The North American bat species group in the same cluster with other bat species, showing a closer relationship between these species. EpFu and PeSu were more closely related to each other than to other myotine species found in Maryland (Fig. 1).
Fig 1.
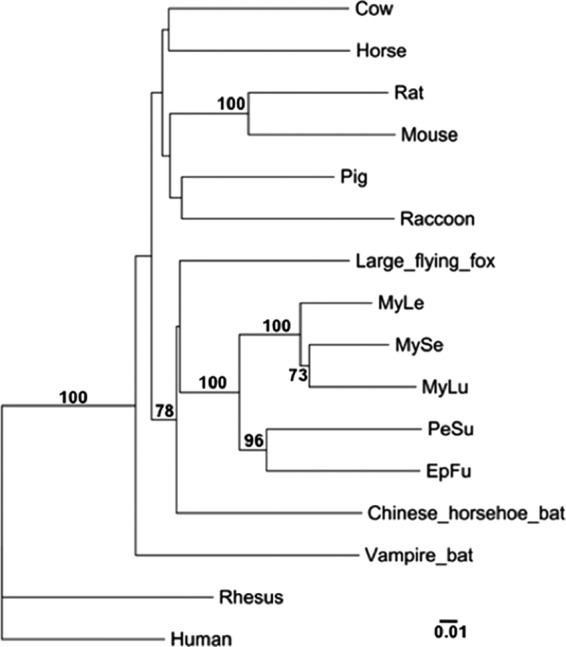
Phylogeny of the mitochondrial cytochrome b gene from several mammalian species. This maximum likelihood tree shows that North American bats of the family Vespertilionidae are more closely related to themselves than to other bat or common mammalian species. EpFu, big brown bat; PeSu, tricolored bat; MyLu, little brown myotis; MyLe, eastern small-footed myotis; MySe, northern long-eared myotis. The scale bar represents nucleotide substitutions. Only nodes with bootstrap support above 70% are labeled.
Identification and characterization of novel α-CoV sequences in big brown bats and the tricolored bat.
Roche 454 sequencing was used to identify α-CoV sequences in two additional bat populations, including a big brown bat population from Saratoga National Historical Park in New York (NECoV) and a tricolored bat population that was sampled at the Chesapeake and Ohio National Historical Park in Maryland. RT-PCR was used to amplify a >2,200-nt region of these viruses (comprising a portion of nsp13, all of nsp14, and a portion of nsp15), and these were sequenced and compared to the same fragment that had been amplified previously from a big brown bat population sampled in Maryland and was known as ARCoV.1 (11). Other regions of these genomes have been determined by 454 sequencing, but this was the largest fragment available in common between all three sequences. Interestingly, the two >2,200-nt sequences derived from big brown bats were nearly identical (approximately 97% identical at the nucleotide level), suggesting that big brown bats harbor similar α-CoVs regardless of geographic location (Fig. 2A and B). In contrast, the sequence from ARCoV.2 was approximately 74% identical at the nucleotide level, suggesting that different U.S. bat species harbor unique α-CoV strains (Fig. 2B). Together, these fragments of North American bat CoVs suggest a new clade in the Alphacoronavirus genus (Fig. 2B).
Fig 2.

A novel α-CoV in the tricolored bat is closely related to HCoV-NL63. Novel α-CoV sequences have been found in the fecal samples of several North American bat species. (A) Schematic showing where the two fragments (used for the trees in panels B and C) occur in the NL63 genome. (B) A >2.2-kb fragment of the replicase region (starting at position 16480 in the schematic) was sequenced from three samples, including two from the big brown bat (NECoV and ARCoV.1) and one from the tricolored bat (ARCoV.2). A maximum likelihood tree comparing the nucleotide sequences of these bat CoVs to other known CoVs indicated that NECoV and ARCoV.1 are very closely related while ARCoV.2 is significantly different from NECoV and ARCoV.1. The three novel α-CoV sequences form a novel cluster in the α-CoV group. (C) Molecular clock analysis using a 650- to 800-nt portion of the highly conserved replicase region (starting at position 13810 in panel A) predicted that the MRCA of bat CoVs from the Hipposideros caffer ruber bats and HCoV-229E was likely to have existed 212 to 350 years ago (in agreement with Pfefferle et al., 2009 [36]). The MRCA for HCoV-NL63 and ARCoV.2 was predicted to have existed 563 to 822 years ago. NECOV and ARCoV.1 were identical in this region, so only ARCoV.1 is shown, and it clustered with other bat α-CoVs.
Molecular clock analysis.
During the annotation stage, many of the CoV-like sequences from tricolored bat samples were most closely related, albeit distantly, to HCoV-NL63 (Fig. 2B). Therefore, molecular clock analysis was conducted to estimate an MRCA between ARCoV.2 and HCoV-NL63, using a similar data set and the same parameters as those employed by Pfefferle et al. (2009) (36). This analysis was performed using sequence fragments obtained from the highly conserved viral polymerase (nsp12), which is arguably the most conserved region of the CoV genome (Fig. 2A). Briefly, both the GTR+Gamma 4 + I and SRD06 models were tested under the assumption of an uncorrelated lognormal clock and constant population size. In addition, an exponential population size assumption was tested using the SRD06 model. Markov chain Monte Carlo chains were run for 200,000,000 iterations with sampling every 20,000 generations. Bayes factor analysis, used to compare the models, was conducted using the program Tracer. The SRD06 model was found to be superior to the GTR model by a log10 Bayes factor of 240. The exponential population assumption was found to be superior to the constant assumption, but not significantly so (log10 Bayes factor of 2). The results of the analyses are shown with highest posterior densities (HPD) in Table 1. Interestingly, the analysis recapitulated the results obtained by Pfefferle et al. (2009) (36) and provided an estimated MCRA between ARCoV.2 and HCoV-NL63 of approximately 1449 CE (range, 1190 to 1449 CE) (Table 1 and Fig. 2C). This observation suggests that ARCoV.2 and HCoV-NL63 originated from the same ancestor, predicting that a potential cross-species transmission event occurred about the time that Columbus arrived in North America. Because ARCoV.1 and NECoV were nearly identical, only ARCoV.1 was used in this analysis, and this sequence clustered with other α-CoVs of bats (Fig. 2C).
Table 1.
Models used and highest posterior densities
| Model | Root (range)a | Mean substitution rateb (range) |
|---|---|---|
| GTR + G + I, constant | 3469 (1237–5923) | 1.629 × 10−4 (1.034 × 10−4–2.3499 × 10−4) |
| SRD06, constant | 5940 (2424–9921) | 1.76 × 10−4 (1.073 × 10−4–2.570 × 10−4) |
| SRD06, exponential | 5450 (2143–9358) | 1.847 × 10−4 (1.078 × 10−4–2.668 × 10−4) |
Values are years BCE (before the common era).
Mean substitution rate is the number of substitutions per site per year.
Generating primary cells from sacrificed bats.
Bat tissues used for this study were obtained from The Save Lucy Campaign, which operates as a bat rescue in Virginia. Bats that were too badly damaged to be rescued were euthanized, and their organs were harvested and transported to UNC, where the tissues were surgically dissected and trypsinized, and the filtered primary cells were plated and grown. Once a sufficient number of cultured primary cells were available, these were tested for rabies virus by RT-PCR. All of the tested bat tissues were negative for rabies virus (data not shown).
A female tricolored bat that arrived in the rescue in mid-July of 2011, suffering injuries sustained from being hit by a car, produced the best results. Brain, heart, liver, lung, intestine, stomach, and kidney tissues were obtained from this bat, and primary cells were successfully generated for the brain, lung, stomach, and heart. The other tissue types grew initially, and either they reached senescence before reaching confluence or there were too few cells to establish long-term growth. The tricolored bat lung cells grew most efficiently, and these cells were passaged three times to generate a large stock of primary lung cells for this bat (Fig. 3A). Stocks of these cells were cryopreserved at −140°C for future use.
Fig 3.

Immortalization of tricolored bat lung cells, PESU-B5L. Primary tricolored bat lung cells were grown in enriched growth medium and passaged four times to generate a suitable quantity for immortalization. Primary cells were immortalized with lentiviral vectors containing hTERT and the Bmi-1 proto-oncogene. (A) Primary tricolored bat lung cells. (B) Primary PESU-B5L lung cells reached senescence by passage 10. (C) Immortalized cells at passage 10, including 4 passages as primary cells and 6 passages postimmortalization. The immortalized PESU-B5L cells were still going strong after more than 30 passages (over 100 doublings).
Generating immortalized lungs cells from the tricolored bat.
Primary tricolored bat lung cells that had been passaged 3 times were immortalized by two concurrent infections with two lentivirus vectors, one containing hTERT and one containing Bmi-1 (18). Immortalized and primary control cells were then passaged at 3- to 4-day intervals to determine if the immortalization process extended the viability of those cells infected with the immortalization vectors. By passage 10, the primary cells had reached senescence and had stopped growing completely, while the cells that were immortalized continued to grow and divide efficiently (Fig. 3B and C). The immortalized cells were named PESU-B5L cells, and these cells have continued to show prolific growth. At the time of writing, we have PESU-B5L cells that have undergone over 30 passages and over 100 doublings. There were no observable morphological changes in these cells over passage, although early primary cultures had heterogenous mixtures of cell types. However, the immortalization process appears to have selected for a single fibroblast-resembling cell population (Fig. 3C). Total RNA was extracted from the immortalized cells, and RT-PCR and sequencing were used to determine that the murine Bmi-1 gene was expressed in the immortalized cells (data not shown).
Experimental infection of PESU-B5L lung cells with human CoVs.
To assess whether or not bat cells could support growth of HCoVs, we infected the PESU-B5L cells with HCoV-NL63, a human α-CoV, and SARS-CoV, a human β-CoV, using engineered versions of these viruses that express GFP during active replication (12, 17, 44, 53), so that we could monitor the presence of a productive infection by fluorescence microscopy. In the case of HCoV-NL63, we detected GFP fluorescence at day 2 postinfection and it continued until day 8, when the cells were harvested due to overconfluence. While a cytopathic effect was observed in some cells by day 5 postinfection (Fig. 4), the cultures remained viable, suggesting that many cells were resistant to infection. The progeny virions harvested from the supernatants of these infections were viable upon passage to fresh cells and formed plaques on LLC-MK2 cells; however, the titer was low (<103 PFU/ml), suggesting a potential block in viral egress. Attempts to generate HCoV-NL63 plaques in the PESU-B5L cells were unsuccessful.
Fig 4.
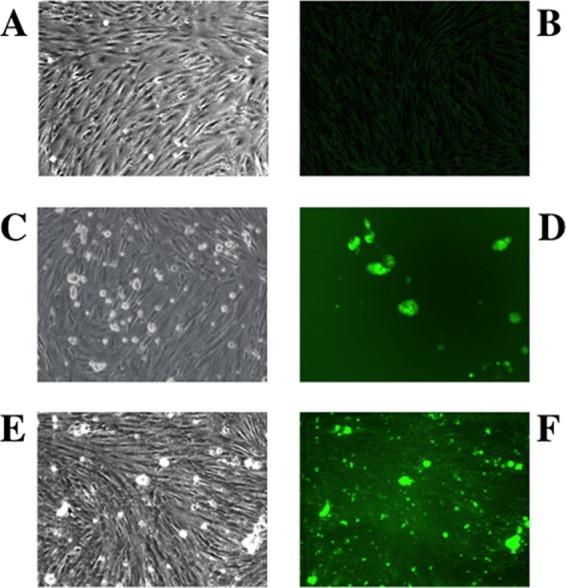
Infection of PESU-B5L cells with human coronavirus NL63 expressing GFP. PESU-B5L cells were infected with mock or NL63gfp at an MOI of 0.25 and monitored by bright-field and fluorescence microscopy. (A and B) Mock-infected cells by bright-field (A) and fluorescence (B) microscopy. (C through F) Cells infected with NL63gfp visualized at day 3 postinfection by bright-field microscopy (C) and by fluorescence microscopy (D) and at day 5 postinfection by bright-field microscopy (E) and by fluorescence microscopy (F).
In the case of SARS-CoVgfp, no GFP expression was detected in PESU-B5L cells over 5 days of infection, but GFP fluorescence was readily detected in Vero E6 cells infected with the same virus as a positive control by 24 h postinfection (data not shown), indicating that the stock of SARS-CoVgfp virus was viable.
Detection and verification of replication of HCoV-NL63 in PESU-B5L cells.
To verify that HCoV-NL63 replication had occurred in the PESU-B5L lung cells, we used RT-PCR to amplify leader-containing transcripts of the subgenomic N gene. The RT-PCR resulted in a ladder of amplicons representing subgenomic transcripts that had been generated during the course of efficient virus replication (Fig. 5A), and these were sequenced to verify the presence of leader-containing transcripts during HCoV-NL63 replication (Fig. 5B). In addition, infection of PESU-B5L cells by HCoV-NL63 was detected by immunofluorescence assay (IFA) (Fig. 6A) and Western blotting (Fig. 6B), both directed against the nucleocapsid protein. For the IFA, cells expressing HCoV-NL63 nucleocapsid appeared bright green under fluorescent conditions, while uninfected cells were not stained. Western blotting of cell lysates using the anti-NL63 N antibody revealed that the infected cells strongly expressed a protein of approximately 45 kDa, which was consistent with the size of the HCoV-NL63 nucleocapsid protein.
Fig 5.
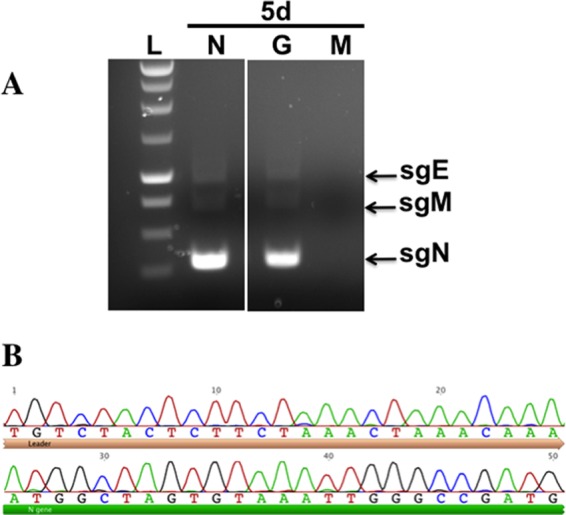
Verification of NL63 replication in PESU-B5L cells. Total RNA was harvested from cells on day 5 postinfection and subjected to RT-PCR using primers designed to detect leader-containing transcripts of the N gene, which would be present only if NL63 replicated. (A) Subgenomic transcripts were detected in the PESU-B5L cells infected with both wtNL63 and NL63gfp. (B) The sgN band for wtNL63 was excised, and the DNA was purified and sequenced to verify that the HCoV-NL63 leader sequence and the 5′ end of the N gene were present. L, ladder; N, wt-NL63; G, NL63gfp; M, mock. The leader sequence has a beige bar beneath it, and the 5′ end of the N gene has a green bar beneath it.
Fig 6.
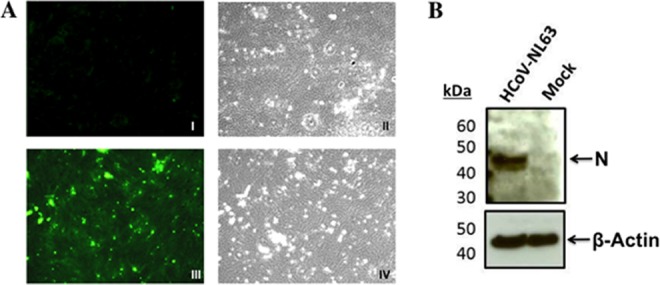
Evidence of HCoV-NL63 infection in PESU-B5L cells. PESU-B5L cells were infected with HCoV-NL63 at an MOI of 0.8. At 72 h postinfection, cells were probed for the nucleocapsid protein of HCoV-NL63 by immunofluorescence assay and Western blotting. (A) I, mock-infected cells, fluorescence microscopy; II, mock-infected cells, bright-field microscopy; III, HCoV-NL63-infected cells, fluorescence microscopy; IV, HCoV-NL63-infected cells, bright-field microscopy. (B) Western blot of HCoV-NL63 and mock-infected PESU-B5L cell lysates with β-actin as a loading control. Nucleocapsid (N) protein appears as a distinct band at ∼45 kDa.
Detection of replication of SARS-CoV in PESU-B5L cells.
Despite the fact that SARS-CoVgfp was not detected by fluorescence in the infected cultures of PESU-B5L cells, RT-PCR was conducted on RNA harvested from these cells at 24 and 48 h using primers designed to detect the subgenomic GFP transcript, and this resulted in the detection of a band in the 24-h culture, indicating that subgenomic transcription was ongoing in these cells (Fig. 7). No viruses or replication was detected upon passage of supernatants from these cultures.
Fig 7.
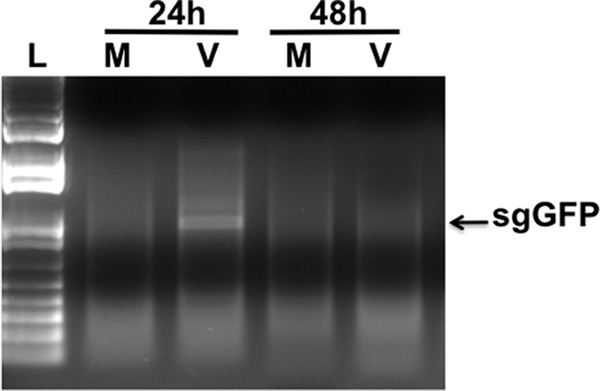
Evidence of SARS-CoV replication in PESU-B5L cells. PESU-B5L cells were infected with SARS-CoV at an MOI of ∼1, and total RNA was harvested from the cells at 24-h intervals and subjected to RT-PCR using primers designed to detect leader-containing transcripts of the subgenomic GFP (sgGFP), indicative of SARS-CoV replication. Subgenomic transcripts were detected at 24 h postinfection but disappeared by 48 h postinfection, suggesting that SARS-CoV replicates in these cells at a low level. L, Promega 1-kb ladder; M, mock-infected cells; V, SARS-CoV-infected cells.
Detection of replication of MA15 and chimeric SARS-CoV strains in PESU-B5L cells.
In addition to performing infections with SARS-CoV expressing GFP, we also conducted experimental infections with MA15 and two chimeric SARS-CoV viruses that encoded the spike proteins from two early human isolates known as GD03 and HC/SZ/61/03. All three of these viruses replicated subgenomic RNAs in the PESU-B5L lung cells (Fig. 8), as detected by RT-PCR using the total RNA of cells harvested at 48 and 72 h postinfection and primers designed to detect subgenomic transcription of viral proteins. Subgenomic transcripts were detected at both time points for all three strains (Fig. 8A). The band representing the M gene was excised and sequenced by Sanger sequencing for all three of these strains, and all contained the appropriate sequence to verify the presence of the leader sequence combined with the M gene (Fig. 8B and data not shown). Interestingly, despite the fact that the PESU-B5L cells infected with MA15 and the two chimeric viruses showed moderate amounts of cytopathology that occurred prior to cell senescence (data not shown), we were unable to detect any signs of viral replication when the supernatants from these infections were passed onto fresh PESU-B5L cells.
Fig 8.
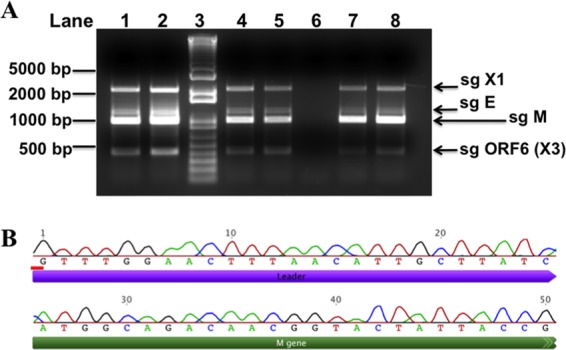
Evidence of SARS-CoV replication in PESU-B5L cells. PESU-B5L cells were infected with the mouse-adapted strain of SARS-CoV, MA15, and with chimeric SARS-CoV viruses bearing the spike genes of GD03 and HC/SZ/61/03 at an MOI of ∼1. Total RNA was harvested from the cells at 48 and 72 h postinfection and subjected to RT-PCR using primers designed to detect leader-containing subgenomic transcripts indicative of replication. (A) Subgenomic (sg) transcripts for X1, envelope (E), membrane (M), and open reading frame 6 [ORF6 (X3)] were detected at both time points for all three strains. Lane 1, MA15 at 48 h; lane 2, MA15 at 72 h; lane 3, Invitrogen 1-kb Plus ladder; lane 4, SARS+GD03 at 48 h; lane 5, SARS+GD03 at 72 h; lane 6, mock-infected cells; lane 7, SARS+HC/SZ/61/03 at 48 h; and lane 8, SARS+HC/SZ/61/03 at 72 h. (B) The sg M bands for all three strains were excised, and the DNA was purified and sequenced to verify that the SARS-CoV leader sequence and the 5′ end of the M gene were present. Shown here is sg M from SARS-CoV strain GD03.
DISCUSSION
Bats are important reservoir hosts for a large number of emerging viruses that cause human disease. However, only a small number of species (∼36 of 1,100) (5, 11, 24) have been studied and shown to harbor RNA viruses, and many of these viruses share sequence similarity to human strains. The fact that CoVs have been found in multiple Old World and New World bat species suggests that bats may be particularly suited for maintaining CoVs in nature. Including the findings in this study, at least three human CoVs have been linked to bat CoVs (26, 36), and there is mounting evidence that all mammalian CoVs may have originated from bats (19, 36, 49), particularly human coronaviruses.
Although the exact number of infections is unknown, HCoV-NL63 has been shown to be distributed worldwide; it infects a significant number of children and elderly persons each year, and HCoV-OC43 and -229E account for nearly one-third of all colds. Moreover, SARS-CoV infected over 8,000 people worldwide, with a mortality rate approaching 10% (34). These observations indicate that CoVs are important human pathogens that cause significant morbidity and frequent mortality. The fact that over 1,000 bat species have not been assessed for the presence of CoVs or other RNA and DNA viruses may have enormous public health consequences in an outbreak setting. Given that there are more than 1,100 species of bats and nearly one-half of the 36 bat species that have been studied for viruses have shown evidence of CoV sequences, there may be hundreds of novel coronaviruses in bats throughout the world. If this is the case, then the next CoV spillover from bats to humans is just a matter of ecological opportunity.
North American bats have been shown to harbor α-CoV sequences (10, 11, 24, 31, 33); however, no α-CoVs have been isolated from these bats, and in fact, no full-length CoV sequences have been obtained or reported from North American bats. Of note, in our metagenomics study of North American bats in the Eastern United States, we identified α-CoV sequences in multiple bat species, including the big brown bat and the tricolored bat (11). In this study, we detected α-CoV sequences in a big brown bat population in New York and from a tricolored bat population sampled in Maryland (Fig. 2). Interestingly, the two sequences derived from big brown bats (ARCoV.1 and NECoV) were nearly identical in a >2,200-nt fragment of the replicase gene, even though the two populations were from different regions (New York and Maryland) (Fig. 2B). In contrast, the same sequence fragment obtained from tricolored bats was significantly different (74% identity at the nucleotide level), suggesting that different bat species harbor CoVs that have adapted specifically to that species (Fig. 2B). Whether or not a compartmentalized CoV from one species is more likely to infect humans, as occurs with the rabies virus (6, 32, 40), is yet to be determined. However, it is clear that specific bat species harbor bat SARS-like viruses that are more closely related to SARS-CoV (26).
The fact that some of the Roche 454 sequences derived from the tricolored bat were more closely related to HCoV-NL63 than any other α-CoV suggested that this novel North American α-CoV may be related to HCoVs. Therefore, we conducted molecular clock analysis using BEAST to determine the relatedness of this α-CoV to other CoVs, by adding it to previous data sets used to determine ancestry for other CoVs (36, 50). Of note, ARCoV.2 and HCoV-NL63 appear to have an MRCA that occurred just over 560 years ago, based on a 650- to 800-nt portion of the highly conserved replicase gene. This analysis was limited to this fragment so that we could use the data obtained from previous studies to calibrate the clock and validate our results. If these predictions are correct, this observation suggests that HCoV-NL63 may have originated from bats between 1190 and 1449 CE. However, it is possible that recombination between bat and human CoVs or other mammalian strains may have occurred in this region, and therefore, more sequence information is necessary to work out the evolutionary history of these CoVs. Comparing the two trees in Fig. 2 suggests that recombination has likely occurred, as the tree topologies are slightly different even in the nonstructural gene sequences that are known to be highly conserved among CoVs.
Immortalized cell lines are important reagents for investigating the similarities and differences between viruses that grow in bats and other mammals. To date, only a few immortalized bat cell lines have been developed, and the majority of these are for fruit bats, which allows for further studies with the Nipah and Hendra viruses (4, 7, 22). In this study, we successfully immortalized bat cells (Fig. 3) from a bat species known to harbor a unique α-CoV that appears to share common ancestry with HCoV-NL63. The facts that SARS-CoV and related strains replicate (Fig. 7 and 8) and that HCoV-NL63 grows in the PESU-B5L immortalized lung cell line indicate the importance of developing robust reagents to a number of different bat species. To our knowledge, this is the first bat cell line that supports growth of HCoVs. We are currently in the process of characterizing the innate immune response in these cells, and we have primary cells available for three additional New World bat species that will be targeted for immortalization. These cell lines will be used to generate reagents for directly studying CoVs in bats and for assessing the potential of these viruses to infect humans. Our data suggest that the lentivirus-based immortalization system reported herein would also be successful for multiple North American bat species and likely represents a robust tool for rapidly establishing continuous cell lines from bats and other zoonotic species.
Only a small number of immortalized bat cell lines currently exist, and these were derived both from Old World bats, including the Egyptian fruit bat (Rousettus aegyptiacus) (22), the straw-colored fruit bat (Eidolon helvum) (4), and the black flying fox (Pteropus alecto) (7), and from New World bats, such as the Brazilian free-tailed bat (Tadarida brasiliensis) (ATCC CCL-88). In these cases, the cell lines were immortalized using three different methods, including the simian virus 40 (SV40) large tumor antigen (4, 7), the adenovirus serotype 5 E1A and E1B genes driven from the promoters for human phosphoglycerate kinase and thymidine kinase of herpes simplex virus (22), and hTERT (7). Initially, we attempted to immortalize the various primary bat cells using hTERT, but these attempts did not result in immortalized cells (data not shown). Interestingly, it has been shown that hTERT alone cannot immortalize primary human bronchial epithelial (hBE) cells (30), possibly due to suboptimal culture conditions. Moreover, while hTERT in combination with viral oncogenes has been used to immortalize mammalian cell lines, these immortalizations have often resulted in immortalized cell lines that were limited in differentiation capacity and that frequently exhibited other morphological and genetic abnormalities (18). To circumvent these limitations, we used a viral-oncogene-independent approach that was successfully used to immortalize hBE cells for the study of cystic fibrosis (18). The primary difference in this approach was that it used the expression of the murine Bmi-1 gene (18), which is a proto-oncogene that normally maintains stem cell populations but has also been shown to recapitulate normal cell structure and function (18). The PESU-B5L cells immortalized by this approach have shown no morphological modifications over time and appear to be a homogenous population of lung epithelial cells.
The isolation and growth of novel α-CoVs from the North American and other global bat populations continue to be a difficult problem. We have attempted to isolate α-CoVs of several bat species from fecal samples that were positive by PCR for an α-CoV by inoculating freshly filtered fecal supernatants from these samples onto a variety of cell types, including PESU-B5L lung cells. To date, we have been unsuccessful in isolating a CoV using this approach. Therefore, we have been using high-throughput and Sanger sequencing approaches to identify and fill in genomic sequences that can be engineered into infectious clones that will allow us to study these viruses in the laboratory. We previously used this synthetic approach to resurrect Bat CoV HKU3, the SARS-like CoV identified in the Chinese horseshoe bat (26), and showed that the only block to replication of this bat virus in primate cells was the 180-amino-acid portion of the spike glycoprotein known as the receptor-binding domain (RBD) (3).
In this paper, we report the first evidence that traditional human CoVs are capable of growth in bat cells. Interestingly, both HCoV-NL63 and SARS-CoV and its related viruses replicated in these cells; however, replication occurred over multiple passages with HCoV-NL63. Recent studies have demonstrated that SARS-CoV and HCoV-NL63 use angiotensin-1 converting enzyme-2 (ACE2) as a receptor for entry into permissive cells (25, 28, 37, 45). However, crystallography studies have shown that these HCoVs interact with ACE2 using different receptor binding domains in the spike glycoproteins of each virus to interact with different regions of the ACE2 molecule (23, 27, 52). In fact, we would predict that it is differences in these interactions that impact the ability of SARS-CoV to efficiently replicate upon passage in these cells. Interestingly, while SARS-CoVgfp replicated leader-containing transcripts at 24 h, transcription was not detected at 48 h and no GFP fluorescence was observed in these cultures. However, MA15 and the two chimeric human SARS-CoVs bearing spike genes from the early stage of the SARS epidemic appeared to replicate leader-containing transcripts more efficiently and exhibited more cytopathology, indicating that these viruses grew better in these cells than did SARS-CoVgfp. This observation suggests that the spike protein may be the primary determinant for infectivity in these cells and that spike proteins that are more adapted to human ACE2, such as SARS-CoV, are less capable of establishing infection in these bat cells. However, HCoV-NL63 appeared to replicate more efficiently than any of the SARS-CoV variants, suggesting that the HCoV-NL63 interaction site on the tricolored bat ACE2 may be the primary determinant of infection. The interactions between PESU-B5L cells and various spike proteins are currently being explored in more detail. Interestingly, a recent study has shown that signatures of recurrent positive selection in the bat ACE2 gene map almost perfectly to known SARS-CoV interaction surfaces, suggesting that ACE2 utilization preceded the emergence of SARS-CoV-like viruses from bats (9).
In addition, the sequence of the orthologous ACE2 receptor of the tricolor bat has not been determined but likely presents the determinants of cross-species transmission, allowing HCoVs to infect bats. Supporting this hypothesis, several bat ACE2 receptors were recently shown to function as receptors for SARS-CoV docking and entry, and notably, HCoV-NL63 receptor usage was not evaluated in these studies (21). Of note, most α-CoVs use aminopeptidase N (APN) molecules as the cellular receptor, and the exogenous expression of feline APN in cells that are refractory to infection has rendered these cells susceptible to most α-CoVs (47). The fact the HCoV-NL63, which is an α-CoV, uses ACE2 is an unusual feature of this HCoV, although it is not know which receptors the bat α-CoVs use. We are currently in the process of cloning and sequencing all of the known CoV receptor orthologues for this bat species to determine which receptor(s) is used by SARS-CoV and HCoV-NL63 and to assess the risk of reverse zoonotic transmission from humans to bats. We also plan to evaluate PESU-B5L infectivity and receptor usage by other coronaviruses such as HCoV-229E and HKU3 in future experiments. Attempts to obtain these viruses for this study were unsuccessful.
CoVs have a long history of cross-species transmission (35). BCoV and HCoV-OC43 are closely related, with an MRCA predicted to have occurred ∼100 years ago (2, 50, 51). BCoV strains have also spread to alpaca and wild ruminants. These observations suggest that HCoV-OC43 arose from a cross-species transmission of BCoV into humans, although it is equally likely that the transmission happened in reverse (2, 50, 51). In the α-CoV genus, canine CoV (CCoV), feline CoV (FCoV), and the porcine CoV transmissible gastroenteritis virus (TGEV) carry genetic evidence of recombination with each other, suggesting that these CoVs likely originated from or infected the same host prior to becoming host range restricted. In fact, early strains CCoV-1 and FCoV-1 are thought to have diverged from a common ancestor, and multiple recombination events between these strains and an unknown CoV (in an unknown host) likely resulted in the novel CoVs CCoV-II and FCoV-II (20a). Sequence analysis looking at the similarities between CCoV-II and TGEV suggests that TGEV emerged from a cross-species transmission of CCoV-II from an infected canine to pigs (29).
SARS-CoV emerged from the Chinese wet markets, where the virus is thought to have crossed the species barriers from Chinese horseshoe bats to masked palm civets (Paguma larvata) and raccoon dogs (Nyctereutes procyonoides) to humans (20). However, given that the human and civet SARS-CoV genomes were 96% identical and that the primary restriction to host range occurred at a small number of amino acid positions in the RBD, SARS-CoV may have been present in humans first and then been transmitted to civets and other animals in the markets (20). This is particularly intriguing, as the SARS-CoV takes a more generalist approach to host range, whereby it can infect cells expressing human, civet, and bat ACE2 (42, 43). In contrast, the civet SARS-CoV is restricted to infecting cells expressing civet ACE2 (42, 43). This study provides evidence that HCoV-NL63 is also a generalist, as it can infect bat cells as well as primate (48) and human (12) cells. Interestingly, HCoV-NL63 does not grow in mice.
These are important observations because they indicate that some human and animal CoVs are generalists that can grow in a variety of different mammals, which is in contrast to many other mammalian CoVs that appear to be restricted to a single host. Recognizing that few mammalian coronaviruses have been tested in bat cell lines, these data suggest that humans and perhaps other select mammals may serve as the gateway species for the evolutionary expansion of mammalian CoVs. In addition, this observation suggests that human and/or other mammalian CoVs could potentially circulate back and forth between bats and humans, establishing a zoonotic-reverse zoonotic cycle that may allow the virus to maintain viral populations in multiple hosts, exchange genetic information to alter pathogenesis or transmission characteristics, and potentially evolve variants that are capable of efficiently infecting humans or other mammals. Further work is necessary to determine the risks of such a cycle, evaluate the impact that this could have on public health, and determine whether or not this represents a general feature of the mammalian Coronaviridae, which are predicted to have originated from bats.
ACKNOWLEDGMENTS
This project was funded by an ARRA grant through SERCEB no. U54-AI057157PI awarded to E.F.D., J.E.G., and M.B.F.
We gratefully acknowledge the work of Aimee Haskew, Lisa Smith, Jenny Saville, and Angela Sjollema, all of whom participated in collecting bat samples for this work.
Footnotes
Published ahead of print 19 September 2012
REFERENCES
- 1. Agnarsson I, Zambrana-Torrelio CM, Flores-Saldana NP, May-Collado LJ. 2011. A time-calibrated species-level phylogeny of bats (Chiroptera, Mammalia). PLoS Curr. 3:RRN1212 doi:10.1371/currents.RRN1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alekseev KP, et al. 2008. Bovine-like coronaviruses isolated from four species of captive wild ruminants are homologous to bovine coronaviruses, based on complete genomic sequences. J. Virol. 82:12422–12431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Becker MM, et al. 2008. Synthetic recombinant bat SARS-like coronavirus is infectious in cultured cells and in mice. Proc. Natl. Acad. Sci. U. S. A. 105:19944–19949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Biesold SE, et al. 2011. Type I interferon reaction to viral infection in interferon-competent, immortalized cell lines from the African fruit bat Eidolon helvum. PLoS One 6:e28131 doi:10.1371/journal.pone.0028131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T. 2006. Bats: important reservoir hosts of emerging viruses. Clin. Microbiol. Rev. 19:531–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Childs JE, Trimarchi CV, Krebs JW. 1994. The epidemiology of bat rabies in New York State, 1988-92. Epidemiol. Infect. 113:501–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Crameri G, et al. 2009. Establishment, immortalisation and characterisation of pteropid bat cell lines. PLoS One 4:e8266 doi:10.1371/journal.pone.0008266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Deming D, et al. 2006. Vaccine efficacy in senescent mice challenged with recombinant SARS-CoV bearing epidemic and zoonotic spike variants. PLoS Med. 3:e525 doi:10.1371/journal.pmed.0030525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Demogines A, Farzan M, Sawyer SL. 2012. Evidence for ACE2-utilizing coronaviruses (CoVs) related to severe acute respiratory syndrome CoV in bats. J. Virol. 86:6350–6353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dominguez SR, O'Shea TJ, Oko LM, Holmes KV. 2007. Detection of group 1 coronaviruses in bats in North America. Emerg. Infect. Dis. 13:1295–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Donaldson EF, et al. 2010. Metagenomic analysis of the viromes of three North American bat species: viral diversity among different bat species that share a common habitat. J. Virol. 84:13004–13018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Donaldson EF, et al. 2008. Systematic assembly of a full-length infectious clone of human coronavirus NL63. J. Virol. 82:11948–11957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Drummond AJ, et al. 2010. Geneious v5.5, v5.5 ed BioMatters, Ltd., Auckland, New Zealand [Google Scholar]
- 14. Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214 doi:10.1186/1471-2148-7-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fielding BC. 2011. Human coronavirus NL63: a clinically important virus? Future Microbiol. 6:153–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fouchier RA, et al. 2004. A previously undescribed coronavirus associated with respiratory disease in humans. Proc. Natl. Acad. Sci. U. S. A. 101:6212–6216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Frieman M, et al. 2007. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J. Virol. 81:9812–9824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fulcher ML, et al. 2009. Novel human bronchial epithelial cell lines for cystic fibrosis research. Am. J. Physiol. Lung Cell. Mol. Physiol. 296:L82–L91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gloza-Rausch F, et al. 2008. Detection and prevalence patterns of group I coronaviruses in bats, northern Germany. Emerg. Infect. Dis. 14:626–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Graham RL, Baric RS. 2010. Recombination, reservoirs, and the modular spike: mechanisms of coronavirus cross-species transmission. J. Virol. 84:3134–3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20a. Herrewegh AA, Smeenk I, Horzinek MC, Rottier PJ, de Groot RJ. 1998. Feline coronavirus type II strains 79-1683 and 79-1146 originate from a double recombination between feline coronavirus type I and canine coronavirus. J. Virol. 72:4508–4514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hou Y, et al. 2010. Angiotensin-converting enzyme 2 (ACE2) proteins of different bat species confer variable susceptibility to SARS-CoV entry. Arch. Virol. 155:1563–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jordan I, Horn D, Oehmke S, Leendertz FH, Sandig V. 2009. Cell lines from the Egyptian fruit bat are permissive for modified vaccinia Ankara. Virus Res. 145:54–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li F, Li W, Farzan M, Harrison SC. 2005. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 309:1864–1868 [DOI] [PubMed] [Google Scholar]
- 24. Li L, et al. 2010. Bat guano virome: predominance of dietary viruses from insects and plants plus novel mammalian viruses. J. Virol. 84:6955–6965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li W, Choe H, Farzan M. 2006. Insights from the association of SARS-CoV S-protein with its receptor, ACE2. Adv. Exp. Med. Biol. 581:209–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li W, et al. 2005. Bats are natural reservoirs of SARS-like coronaviruses. Science 310:676–679 [DOI] [PubMed] [Google Scholar]
- 27. Li W, et al. 2007. The S proteins of human coronavirus NL63 and severe acute respiratory syndrome coronavirus bind overlapping regions of ACE2. Virology 367:367–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li W, et al. 2005. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 24:1634–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lorusso A, et al. 2008. Gain, preservation, and loss of a group 1a coronavirus accessory glycoprotein. J. Virol. 82:10312–10317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lundberg AS, et al. 2002. Immortalization and transformation of primary human airway epithelial cells by gene transfer. Oncogene 21:4577–4586 [DOI] [PubMed] [Google Scholar]
- 31. Misra V, et al. 2009. Detection of polyoma and corona viruses in bats of Canada. J. Gen. Virol. 90:2015–2022 [DOI] [PubMed] [Google Scholar]
- 32. Morimoto K, et al. 1996. Characterization of a unique variant of bat rabies virus responsible for newly emerging human cases in North America. Proc. Natl. Acad. Sci. U. S. A. 93:5653–5658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Osborne C, et al. 2011. Alphacoronaviruses in new world bats: prevalence, persistence, phylogeny, and potential for interaction with humans. PLoS One 6:e19156 doi:10.1371/journal.pone.0019156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Peiris JS, Guan Y, Yuen KY. 2004. Severe acute respiratory syndrome. Nat. Med. 10:S88–S97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Perlman S, Netland J. 2009. Coronaviruses post-SARS: update on replication and pathogenesis. Nat. Rev. Microbiol. 7:439–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pfefferle S, et al. 2009. Distant relatives of severe acute respiratory syndrome coronavirus and close relatives of human coronavirus 229E in bats, Ghana. Emerg. Infect. Dis. 15:1377–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pohlmann S, et al. 2006. Interaction between the spike protein of human coronavirus NL63 and its cellular receptor ACE2. Adv. Exp. Med. Biol. 581:281–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pyrc K, et al. 2006. Mosaic structure of human coronavirus NL63, one thousand years of evolution. J. Mol. Biol. 364:964–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Roberts A, et al. 2007. A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice. PLoS Pathog. 3:e5 doi:10.1371/journal.ppat.0030005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shankar V, et al. 2005. Genetic divergence of rabies viruses from bat species of Colorado, U. S. A. Vector Borne Zoonotic Dis. 5:330–341 [DOI] [PubMed] [Google Scholar]
- 41. Sheahan T, Deming D, Donaldson E, Pickles R, Baric R. 2006. Resurrection of an “extinct” SARS-CoV isolate GD03 from late 2003. Adv. Exp. Med. Biol. 581:547–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sheahan T, Rockx B, Donaldson E, Corti D, Baric R. 2008. Pathways of cross-species transmission of synthetically reconstructed zoonotic severe acute respiratory syndrome coronavirus. J. Virol. 82:8721–8732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sheahan T, et al. 2008. Mechanisms of zoonotic severe acute respiratory syndrome coronavirus host range expansion in human airway epithelium. J. Virol. 82:2274–2285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sims AC, et al. 2005. Severe acute respiratory syndrome coronavirus infection of human ciliated airway epithelia: role of ciliated cells in viral spread in the conducting airways of the lungs. J. Virol. 79:15511–15524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Smith MK, et al. 2006. Human angiotensin-converting enzyme 2 (ACE2) is a receptor for human respiratory coronavirus NL63. Adv. Exp. Med. Biol. 581:285–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Teeling EC, et al. 2005. A molecular phylogeny for bats illuminates biogeography and the fossil record. Science 307:580–584 [DOI] [PubMed] [Google Scholar]
- 47. Tresnan DB, Levis R, Holmes KV. 1996. Feline aminopeptidase N serves as a receptor for feline, canine, porcine, and human coronaviruses in serogroup I. J. Virol. 70:8669–8674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. van der Hoek L, et al. 2004. Identification of a new human coronavirus. Nat. Med. 10:368–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vijaykrishna D, et al. 2007. Evolutionary insights into the ecology of coronaviruses. J. Virol. 81:4012–4020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vijgen L, et al. 2005. Complete genomic sequence of human coronavirus OC43: molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J. Virol. 79:1595–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vijgen L, Lemey P, Keyaerts E, Van Ranst M. 2005. Genetic variability of human respiratory coronavirus OC43. J. Virol. 79:3223–3224, 3224–3225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wu K, Li W, Peng G, Li F. 2009. Crystal structure of NL63 respiratory coronavirus receptor-binding domain complexed with its human receptor. Proc. Natl. Acad. Sci. U. S. A. 106:19970–19974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yount B, et al. 2003. Reverse genetics with a full-length infectious cDNA of severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. U. S. A. 100:12995–13000 [DOI] [PMC free article] [PubMed] [Google Scholar]