

Abstract
The exquisite selectivity and unique transport properties of membrane proteins can be harnessed for a variety of engineering and biomedical applications if suitable membranes can be produced. Amphiphilic block copolymers (BCPs), developed as stable lipid analogs, form membranes that functionally incorporate membrane proteins and are ideal for such applications. While high protein density and planar membrane morphology are most desirable, BCP–membrane protein aggregates have so far been limited to low protein densities in either vesicular or bilayer morphologies. Here, we used dialysis to reproducibly form planar and vesicular BCP membranes with a high density of reconstituted aquaporin-0 (AQP0) water channels. We show that AQP0 retains its biological activity when incorporated at high density in BCP membranes, and that the morphology of the BCP–protein aggregates can be controlled by adjusting the amount of incorporated AQP0. We also show that BCPs can be used to form two-dimensional crystals of AQP0.
Introduction
Membrane proteins mediate specific and efficient transport of water, ions and solutes across cell membranes. They also serve the cell as sensors that detect environmental conditions, ranging from pH to specific signaling molecules and toxins. Incorporating such membrane proteins into stable membranes formed by lipid analogs could provide materials with targeted applications in sensors,1,2 enzymatic reactions,3 drug screening,4 and even water purification.5 Block copolymers (BCPs) form membranes that mimic the architecture of lipid bilayers and allow incorporation of functional membrane proteins.6 However, in contrast to lipids, BCPs can be tailored to have the stability and durability associated with polymeric materials7 and hence are more suitable for the production of membrane protein-based devices and other applications. Furthermore, BCPs can be designed to form membranes with specific physical properties and unique morphologies simply by the choice of the blocks and their lengths8 or their length ratio.9 Physical properties that can be engineered include the toughness and permeability of the membrane, as well as its morphology (e.g., micellar, vesicular, cylindrical or planar). BCP end groups can also be modified by molecules such as biotin and 4-formylbenzoate (for recognition and immobilization),10,11 methacrylate (for stabilization by cross-linking),12 fluorescent molecules (for imaging),10 and even drugs (for drug delivery).13
Recent efforts to insert membrane proteins into BCP membranes resulted in the incorporation of only a small number of proteins into either vesicles (reviewed in ref (14)) or painted, supported, or suspended bilayers.15,16 While vesicles are excellent vectors for drug delivery,17 other applications such as sensors, reactive surfaces, drug screening, and water purification would benefit greatly from a planar membrane morphology. Also, film rehydration, a technique commonly used to make polymer–protein vesicles, appears to limit the amount of membrane protein that can be incorporated into BCP vesicles, even if high protein concentrations are used.5 Painted bilayers are excellent tools for studying the function, in particular the conductance, of membrane proteins,18 but the number of membrane proteins that are incorporated is usually low, and the stability of the membrane can be limited due to the presence of residual solvent. Supported and suspended bilayers have also shown low reconstitution of membrane proteins. Dialysis of mixtures of lipids and membrane proteins dissolved in nonionic detergents is a method commonly used to reconstitute membrane proteins into lipid membranes for functional and structural studies. Here, we adapted the dialysis approach to BCPs, which, compared to lipids, are less soluble in detergents19,20 and can form aggregates even in the presence of high detergent concentrations.
We selected two BCPs each from the two systems most commonly used for polymer vesicle formation and membrane protein insertion: polybutadiene-polyethyleneoxide (PB-PEO)-based diblock copolymers and polymethyloxazoline-polydimethylsiloxane (PMOXA-PDMS)-based triblock copolymers. The architectures and block compositions of these polymers are provided in Table 1 and Supporting Information Figure S1. Table 1 also summarizes the molecular weight (MW) of the chosen BCPs as well as their calculated hydrophilic weight and volume ratios, the fractions of the polymers that are comprised of hydrophilic blocks by weight and volume, respectively. While PMOXA-PDMS polymers have previously been shown to incorporate membrane proteins (summarized in ref (21)), PB-PEO polymers have not yet been tested for their ability to incorporate membrane proteins. As test protein for incorporation into BCP membranes, we selected the lens-specific water channel aquaporin-0 (AQP0), because it forms regular arrays in the native lens membrane22,23 as well as upon reconstitution with various lipids.24,25 Lipid membranes with increasing concentrations of reconstituted AQP0 also show a morphological transition from vesicles to planar membranes, and finally to 2D crystals, thus providing an excellent basis for comparison with reconstitution of AQP0 into BCP membranes. Our studies show that all BCPs tested form vesicular and planar membranes in the presence of specific amounts of AQP0, and also that the amount of incorporated AQP0 strongly influences the morphology of the resulting BCP–AQP0 aggregates. At very high concentrations, AQP0 forms 2D arrays in two of the BCPs, similar to those seen with lipids. Most of the transitions between different membrane morphologies occur at similar volume fraction values of incorporated AQP0 (the fractional volume occupied by AQP0 in the polymer and calculated by considering the membrane-spanning part of AQP0 as a hydrophobic cylinder). We also characterized the function of AQP0 in one of the BCPs and show that its biological function is preserved in BCP membranes even at high packing densities.
Table 1. Block Copolymers Used in This Study.
| polymer ID | block composition | type | MW (g/mol) | fhydrophilic (w)/fhydrophilic (vol)a |
|---|---|---|---|---|
| PB12 | PEO10-PB12 | Diblock | 1089 | 0.40/0.34 |
| PB22 | PEO14-PB22 | Diblock | 1806 | 0.34/0.28 |
| ABA42 | PMOXA20-PDMS42-PMOXA20 | Triblock | 6508 | 0.52/0.51 |
| ABA55 | PMOXA12-PDMS55-PMOXA12 | Triblock | 6110 | 0.33/0.33 |
fhydrophilic (w), hydrophilic weight ratio, fhydrophilic (vol), hydrophilic volume ratio; the calculation of these values is described in Supporting Information.
Results
Optimization of AQP0 Incorporation into BCP Membranes
Complete initial polymer dissolution in detergent and a slow detergent removal rate were found to be critical for successful incorporation of AQP0 into BCP membranes. The polymers were initially dissolved in various concentrations of octyl-β,d-glucoside (OG) (2%, 4%, 10% and 40%) and dodecyl-β,d-maltoside (DDM) (2% and 10%). Examination by transmission electron microscopy (EM) revealed that all four polymers tested here required an OG concentration of 10% for complete dissolution. DDM was not used for further experiments as it has a low critical micellar concentration (cmc) and is thus not well suited for removal by dialysis. For detergent removal by dialysis, various parameters were tested, including ionic strength, pH, and divalent metal ion concentration (MgCl2) of the dialysis buffer, temperature, and detergent removal rate. Most efficient and reproducible incorporation of AQP0 into BCP membranes was obtained by performing the dialysis at 4 °C with dialysis buffer (10 mM MES, pH 6, 100 mM NaCl, 50 mM MgCl2, 0.01% NaN3) that initially contained 4% OG. In the course of the dialysis, the detergent concentration was gradually lowered (by doubling the dialysis buffer volume every 24 h with detergent-free buffer) until it reached 0.25% on the fourth day. On the fifth day the buffer was exchanged with detergent-free buffer three times every four hours, and the dialysis buttons were harvested. A contact angle-based approach26 was used to follow detergent removal during dialysis, which established that under the chosen conditions the rate was 5.1 (mg/mL)/day when the detergent concentration reached the cmc of OG, and that the residual OG concentration at the end of the dialysis was ∼0.0001%. Details of the methods used to vary the dialysis rate, and to determine detergent concentrations and detergent removal rates are provided in Supporting Information and Figures S2–S4.
Aggregate Morphology at Different Polymer-to-Protein Ratios
In a systematic study of the dependence of aggregate morphology on the amount of incorporated protein, we reconstituted AQP0 with the four BCPs using a wide range of polymer-to-protein-ratios (PoPRs). For easier comparison, PoPRs of triblock copolymer/AQP0 mixtures are reported as twice the value actually used, as a triblock polymer molecule is equivalent to two lipid or diblock copolymer molecules in a bilayer configuration. We observed that with increasing protein concentration the aggregates transitioned from the native structures, which the polymer forms in the absence of protein (network structures, vesicles with attached tubes, small vesicles), to vesicles (larger, more monodisperse), to mixtures of vesicles and planar membranes, and finally to only planar membranes. With two polymers, PB12 and ABA42, AQP0 organized into 2D crystals at low PoPR values. Because these transitions occurred at different PoPRs for the different BCPs, we chose for presentation in Figure 1 and Supporting Information Figure S5 PoPRs at which a particular morphology of the BCP–AQP0 aggregates was dominant. For two polymers, PB12 and ABA42, the transitions are described in the following paragraphs, and for the other two polymers, PB22 and ABA55, data are shown and described in Supporting Information.
Figure 1.
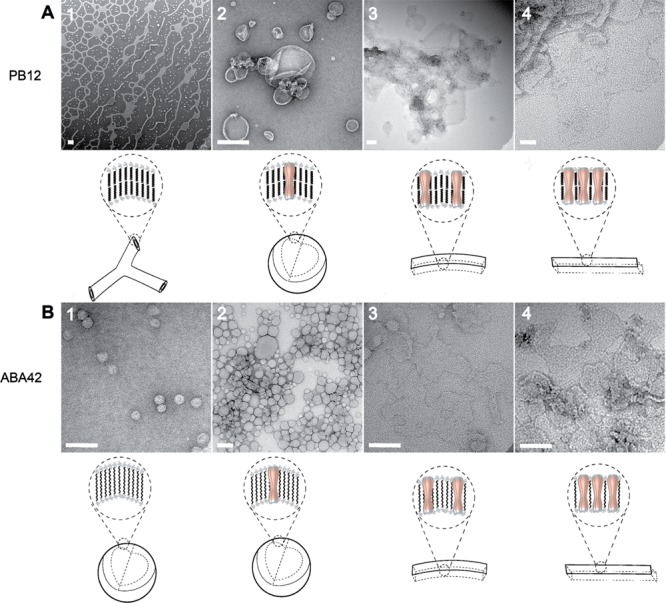
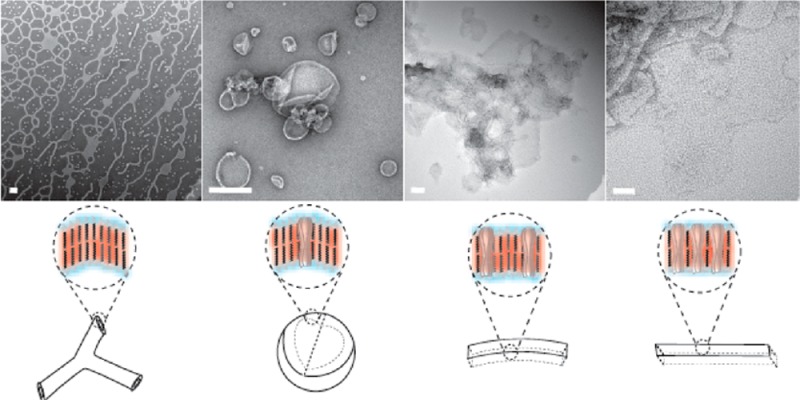
The membrane protein concentration has a large effect on the morphology of the resulting self-assembled membrane protein–block copolymer aggregate. The micrographs show PoPRs that are representative of the range in which a particular aggregate morphology is dominant. (A) Reconstitution of AQP0 with PB12 at molar PoPRs of (1) ∞ (no protein), (2) 15.5, (3) 3.9, and (4) 1.3. The increase in incorporated protein leads to a transition from the network structures formed by pure polymer as described before (ref (27)), to a mixture of network structures and vesicles (shown in Supporting Information Figure S5), to vesicles only, membranes, and finally to crystalline membrane patches. (B) Reconstitution of AQP0 with ABA42 at PoPRs of (1) ∞ (no protein), (2) 43.2, (3) 2.2, and (4) 0.6. The aggregates transition from vesicles only, to larger vesicles, to membranes only, and finally to crystalline membrane patches. Schematics below each panel show the presumed arrangement of polymer bilayers (for PB12) or monolayers (for ABA42) and the location of AQP0 in the formed membranes. Scale bars are 100 nm.
The PB12–AQP0 system showed the strongest transitions with changing PoPRs. In the absence of protein, Jain and Bates reported that several PB-PEO polymers self-assemble into similar network structures.27 Although it was hypothesized in the earlier study that PB-PEO polymers with MWs as low as that of the PB12 polymer studied here do not form such network structures, we found that PB12 also forms network structures, and in a similar weight fraction range (∼0.4) of the hydrophilic PEO block (Figure 1A, panel 1). The concentration of polymers used in our study is much lower (0.1% rather than 1%), and our procedure differs from that used in the Jain and Bates study, in which solid polymers were mixed in deionized water and equilibrated for several days to weeks. These experimental differences may explain why PB12 did form network structures in our experiments. Upon incorporation of protein, at a PoPR of first ∼250 and then ∼50, the native network structures evolved into a mixture of network structures and vesicles (Supporting Information Figure S5). An increase in protein concentration to a PoPR of 15.5 resulted in the formation of exclusively vesicles that were 200–300 nm in diameter (Figure 1A, panel 2). A further increase in protein concentration to a PoPR of 3.9 led to the formation of planar membrane sheets (Figure 1A, panel 3), and at a PoPR of 1.3 AQP0 began to form crystalline arrays in the PB12 membranes (Figure 1A, panel 4).
Dialysis at higher detergent removal rates reduced the efficiency of AQP0 incorporation into PB12 membranes and changed the morphology of the resulting PB12–AQP0 aggregates (see Supporting Information and Supporting Information Figure S6 for more information).
In the ABA42–AQP0 system, aggregates transitioned with decreasing PoPRs from vesicles only, to vesicles associated with planar membranes, to planar membranes, and finally to crystalline patches. Without protein, ABA42 formed small vesicles (Figure 1B, panel 1). With the incorporation of protein, at a PoPR of 43.2, larger vesicles formed, and at PoPRs of 8.6 and then 2.2 the native structures evolved into a mixture of vesicles and membrane patches (Supporting Information Figure S5) and then larger membrane areas (Figure 1B, panel 3). With a further increase in the incorporated protein, at a PoPR of 0.6, many membrane patches showed crystalline AQP0 arrays (Figure 1B, panel 4).
Figure 2 shows enlarged images of the crystalline AQP0 arrays that formed with PB12 at a PoPR of 1.3 and ABA42 at a PoPR of 0.6. Fourier transforms of images of such two-dimensional (2D) arrays provide information about the organization of the proteins in the array. After computational unbending of the crystal lattice with the 2dx software,28 calculated Fourier transforms of the BCP–AQP0 crystal images revealed clear diffraction spots (Figure 2, insets) that define a tetragonal unit cell of a = b = 6.5 nm. This unit cell is identical to those seen with AQP0 2D crystals produced with lipids,24,25 demonstrating that AQP0 tetramers in BCP membranes are organized in the same way as in lipid bilayers.
Figure 2.

2D crystals of AQP0 formed in BCPs. (A) AQP0 2D crystals in ABA42; (B) AQP0 2D crystals in PB12. The diffraction spots seen in the Fourier transforms of these images after unbending (insets) demonstrate the high degree of protein incorporation. The scale bar in the insets is (5 nm)−1. The unit cell dimensions of a = b = 6.5 nm are the same as those of AQP0 2D crystals formed with lipids.
Functional Characterization of Densely Packed AQP0 in Polymer Vesicles
We used stopped-flow measurements to assess the function of densely packed AQP0 in PB12 membranes. In stopped-flow studies, light scattering or fluorescence quenching is used to monitor the rapid change in vesicle size when vesicles are subjected to an osmotic gradient. This approach has been used extensively to determine the permeability of water channels reconstituted into liposomes.29−33 A prerequisite for the use of this method is that the protein is reconstituted into vesicles, and vesicular morphology was therefore confirmed by EM for all samples used in these studies.
With the dialysis procedure described above, all BCPs tested could be used to incorporate AQP0 at a high density. To determine whether the incorporated water channels were functional, we reconstituted AQP0 with PB12 at a PoPR of 15, which yields large vesicles densely packed with AQP0 (Figure 1A, panel 2). Proteoliposomes are often used to determine the transmembrane transport characteristics of reconstituted membrane proteins (reviewed in ref.34), but not in the case of AQP0. Instead, AQP0 function has been characterized extensively using native vesicles35 and expression in Xenopus laevis oocytes.32,36 Water permeability of AQP0 is low (2.5 × 10–15 cm3/s per molecule; ref (37)) compared to that of classical water channels such as AQP1 (1.17 × 10–13 cm3/s; ref (31)) and aquaporin Z (AqpZ) (∼1 × 10–13 cm3/s; ref (30)). The low water permeability necessitated a high density of AQPs in order to distinguish its permeability over the background permeability of the BCP membranes.
Since dialysis in the absence of AQP0 causes PB12 to form network structures (Figure 1A, panel 1), this method could not be used to obtain pure PB12 vesicles needed as control to measure the function of AQP0 reconstituted into PB12 vesicles. We therefore used the sucrose rehydration method7 to form pure PB12 vesicles, which were confirmed by EM and dynamic light scattering.
At pH 6.5, the water permeability of PB12 vesicles was 189.7 ± 61.3 μm/s (Figure 3), which is high compared to the measured permeability of other BCP vesicles, 2.5 μm/s for polyethylethylene-polyethylene oxide (PEE37-PEO40)7 and 0.7 μm/s for PMOXA15-PDMS110-PMOXA155.5 The high water permeability of PB12 vesicles, which is in the range of those seen for lipid vesicles (10–150 μm/s; ref (34)), is likely due to its hydrophobic block consisting of only 12 butadiene units, which is small compared to those of the BCPs analyzed before (30–110 hydrophobic repeat units).
Figure 3.

Comparison of the water permeability of PB12 and PB12–AQP0 vesicles. (A) Normalized light scattering traces of PB12 vesicles with (upper curve) and without AQP0 (lower curve) subjected to a 25 mOsm sucrose gradient at pH 6.5. This osmolarity was chosen specifically for this figure to demonstrate the clear difference in kinetics. For actual data collection a 50 mOsm sucrose gradient was used for AQP0-BCP vesicles and a 300 mOsm gradient for pure BCP vesicles as described in Supporting Information Materials and Methods. At a gradient of 25 mOsm, the kinetics for PB12 vesicles with AQP0 took ∼70 ms to saturate while it took ∼500 ms for pure PB12 vesicles. (B) The approximately 7-fold higher water permeability of vesicles containing AQP0 compared to that of pure PB12 vesicles indicates that the incorporated water channels are functional. The error bars represent standard deviation of three measurements. (C) Determination of the activation energy (Ea) yielded 12.8 kcal/mol for pure PB12 vesicles (lower curve) and 5.7 kcal/mol for PB12 vesicle with AQP0 (upper curve). The values reported in the text are averages of three independent measurements.
Reconstitution of AQP0 at a PoPR of 15 increased the water permeability of the PB12 vesicles to 1409 ± 409.5 μm/s (Figure 3B). To rule out the possibility that the increase in water permeability of the AQP0-containing PB12 vesicles is due to residual detergent, we performed permeability measurements of pure PB12 vesicles in the presence of different OG concentrations (Supporting Information Figure S7). Since the measured water permeability did not change up to an OG concentration of 0.01% and since the residual OG concentration remaining in AQP0–PB12 vesicle samples after dialysis was measured to be only ∼0.0001% (Supporting Information Figure S3), the increase in water permeability must be due to the incorporation of functional AQP0 channels.
Measurement of the water permeability at different temperatures allowed determination of the activation energy. The activation energy of water conduction by pure PB12 vesicles, 13.2 ± 0.9 kcal/mol, is similar to that of vesicles formed by other BCPs and lipids5,30 and indicative of passive diffusion of water across the PB12 membrane. Incorporation of AQP0 into the PB12 vesicles lowered the activation energy to 7.6 ± 1.7 kcal/mol (Figure 3C), comparable to previously determined values for the activation energy of AQP0-mediated water conduction (5 and 6.9 kcal/mol; refs (32) and (37)). These results show that AQP0 function is preserved in BCP membranes, even at the high protein density of the vesicles used in this study.
Discussion
The results obtained in this study show that membrane proteins can be incorporated into BCP membranes at high density, and that incorporation of membrane proteins affects the morphology of the resulting BCP–membrane protein aggregates. Furthermore, in some cases, AQP0 formed 2D crystals in BCP membranes, which, after further optimization, may allow structure determination of membrane proteins in BCP membranes and investigation of BCP–protein interactions analogous to ongoing studies of lipid–protein interactions.25 Such studies will be important for proposed applications of membrane protein-containing BCP membranes as it will help determine factors that contribute to compatibility between BCPs and membrane proteins. The present 2D crystals already indicate that the structure and organization of AQP0 tetramers in BCP membranes are the same as in lipid-based 2D crystals. The 2D crystals also show that a high density of membrane proteins can be incorporated into BCP membranes with planar architecture, ideal for engineering applications. Finally, functional studies with AQP0 incorporated into BCP vesicles show that, by using the dialysis approach presented here, it is possible to reconstitute functional membrane proteins into BCP vesicles at packing densities not reported before. These findings and their implications are further discussed in the following paragraphs.
Dialysis Greatly Increases the Efficiency of Membrane Protein Incorporation into BCP Membranes
The film rehydration method relies on separation of a BCP film from a glass surface to form membranes with simultaneous insertion of membrane proteins.38 Even though this method has been successful, one study showed that protein insertion is limited to an equivalent PoPR of ∼100 (ref (5)). In an alternative protein incorporation method, preformed BCP vesicles are destabilized by addition of detergent to allow insertion of detergent-solubilized membrane proteins.39 This procedure may also limit the number of membrane proteins that can be incorporated, because inserting membrane proteins into preformed membranes is energetically expensive, especially if the hydrophobic region of the membrane protein does not match that of the BCP membrane.19
Dialysis is a method often used in the reconstitution of membrane proteins into lipid membranes to form either proteoliposomes for functional studies (reviewed in ref (40)) or 2D crystals for structural studies (reviewed in refs (41−43)). However, BCPs are substantially less soluble in detergents than lipids,19,20 requiring additional considerations. If the ternary BCP/protein/detergent solution is not well mixed prior to the start of dialysis or if the rate of detergent removal is too high, the polymer can precipitate out of solution and self-assemble by itself without significant membrane protein incorporation. Complete dissolution of the BCP in detergent before mixing it with the detergent-solubilized membrane protein and slowing the dialysis rate to allow the ternary BCP/protein/detergent mixture to slowly transition through the cmc of the detergent is thus critical for efficient membrane protein insertion. This strategy is also frequently used for 2D crystallization of membrane proteins in lipid membranes.41,44 We thus propose that controlled, slow detergent removal by dialysis may currently be the most efficient method for membrane protein incorporation into BCP membranes. The drawback of this method is the large amount of detergent needed to dissolve BCPs and to control the dialysis rate, but it may be possible to further optimize the procedure and to reduce the amount of detergent needed.
The Amount of Incorporated Membrane Protein Affects the Morphology of BCP–protein Aggregates
The morphology of self-assembled aggregates formed by pure BCPs has been correlated with the volume ratio of their hydrophilic and hydrophobic blocks.45−47 This dependence has been attributed to BCP molecules with different hydrophilic volume ratios having different shapes and symmetries in solution based on the volumes occupied by each block. For example, wedge-shaped BCPs with hydrophobic blocks that occupy smaller volumes than their hydrophilic blocks form aggregates with spherical morphologies in aqueous solutions. On the other hand, rod-shaped BCPs with hydrophilic and hydrophobic blocks that occupy similar volumes, reflected in equal volume fractions for the two blocks, form planar membranes.48 Inclusion of a molecule that interacts with only the hydrophilic or the hydrophobic block will change the volume fraction of that block, thus providing a means to control the morphology of the aggregates that form.48 Nanoparticles that interact with the hydrophilic block have been shown to affect the morphology of resulting BCP structures by increasing the strength of segregation.49 Simulations and calculations using self-consistent field theory suggested that a large number of membrane proteins could be inserted into BCP membranes, even if the hydrophobic lengths of the membrane protein and the BCP membrane are mismatched,50,51 but this prediction had not been experimentally realized.
Increasing the amount of membrane protein incorporated into lipid bilayers can change the morphology of the membranes; for example, from vesicles to planar membranes and then to 2D crystals.44 Here, we report similar changes in the morphology of self-assembled BCP structures with an increase in incorporated AQP0. However, the PoPRs at which the transitions occur seem to span a wide range and are very different from those seen when membrane proteins are reconstituted into lipid membranes. For the case of aquaporins, lipid membranes transition from densely packed to 2D crystalline at a molar lipid-to-protein ratio (LPR) in the range of about 8 to 50 (Supporting Information Table S1), while this transition occurs at a PoPR in the range of about 1.3 and 0.6 for PB12 and ABA42, respectively, the two BCPs that undergo this transition. We compared the morphological transitions in BCPs with those of a model lipid, dioleoyl phosphatidyletahnolamine (DOPE) (Figure 4A). The native structure formed by DOPE in the absence of protein is a vesicle. With increasing incorporation of AQP0, vesicles transition into mostly planar membranes at an LPR of ∼13, and then to crystals at an LPR of ∼12 (Supporting Information Table S2 and Figure S8), a much tighter transition range than seen for the BCPs tested in this study.
Figure 4.

The morphology of BCP–AQP0 aggregates depends on the AQP0 volume fraction. The transitions between different morphologies of BCP–protein aggregates are compared with those of lipid (DOPE)–protein aggregates for one complete data set (Supporting Information Table S2). (A) Plot of molar PoPRs against morphology transitions. N, native structures; V, vesicles; M, planar membranes; C, 2D crystals. (B) Plot for the same data set of transitions between aggregate morphologies against the calculated AQP0 volume fraction. The transition for diblock copolymers, triblock copolymers and the lipid investigated in this study occur at similar hydrophilic volume ratios. All studied amphiphiles transitioned to vesicular structures at an AQP0 volume fraction of 16% or higher (solid gray line) and to planar membranes at an AQP0 volume fraction of 65% or higher (dashed line). (C) Plot for the same data set of the transitions between aggregate morphologies, in which the MW of the “aggregate unit” is plotted against the calculated AQP0 volume fraction. The values for the MW of the aggregate unit, the MW of one polymer or lipid molecule with the associated fraction of the MW of AQP0, were calculated by adding the MW of a lipid or polymer molecule to the fraction of the MW of AQP0 associated with the lipid or polymer based on the LPR or PoPR, respectively. The lines indicate the approximate transition boundaries.
The volume fraction of the incorporated AQP0 in the BCP–protein aggregates provides a more consistent basis for understanding morphological transitions for the various BCPs and lipid than PoPRs/LPR (Figure 4B). Assuming that the hydrophobic part of AQP0 exerts the predominant effect on the self-assembly of the BCP–protein aggregates, we considered only the hydrophobic volume of the AQP0 molecule, which we estimated from its atomic structure to be 18.91 nm3 (Supporting Information). This value allowed us to calculate the AQP0 volume fraction in BCP–protein and lipid–protein aggregates (the hydrophilic and hydrophobic volume ratios of the lipid were calculated from the structure of hydrated DOPE molecules;52−54Supporting Information). Transitions between different aggregate morphologies seem to occur for all systems at similar AQP0 volume fractions (Figure 4B). While further studies are required, this result may indicate that the morphology of BCP–protein and lipid–protein aggregates is driven by segregation of the two blocks of the amphiphilic molecules, which is enhanced by the presence of hydrophobic membrane proteins interacting with the hydrophobic block. Figure 4B shows clear transitions from one to another dominant morphology in particular AQP0 volume fraction ranges. Figure 4C shows the same data when the AQP0 volume ratio is plotted against the MW of the aggregate unit (the MW of the polymer or lipid molecule forming the membrane and the associated fraction of the MW of AQP0). Again clear transitions are seen between dominant morphologies in particular AQP0 volume fraction ranges.
The change in morphology of BCP aggregates resulting from different amounts of incorporated protein has relevance for the design of hybrid BCP–protein materials. Furthermore, the unit cell constants of crystalline AQP0 arrays in BCP membranes, which are identical to those in lipid-based AQP0 2D crystals, indicate that the overall structure and organization of AQP0 is maintained in these BCP membranes. The formation of planar BCP membranes rich in structurally and functionally intact membrane protein, which could then be supported on suitable substrates, has applications in many areas.
BCPs may eventually be used to grow 2D crystals of membrane proteins that allow structure determination by electron crystallography.43 The 2D crystals of AQP0 in both diblock and triblock copolymer membranes presented here provide only a proof of concept, but after optimization of polymer chemistry and crystallization conditions, it may be possible to grow AQP0 2D crystals that are sufficiently well ordered to reveal the interaction of BCPs with membrane proteins, as lipid-based AQP0 2D crystals are currently providing insights into lipid–protein interactions.24,25 Furthermore, high-resolution structures of application-relevant membrane proteins in BCP membranes would help to explain similarities and/or differences in the activity seen for membrane proteins incorporated into BCP membranes of different hydrophobic block thicknesses, such as those seen for NADH-ubiquinone oxidase incorporated into BCP membranes.39 Eventually, such structural information may allow it to deduce principles for the design of materials that optimize membrane protein activity.
Functional Membrane Proteins Can Be Packed into BCP Membranes at High Densities
In previous studies, full function of aquaporins in BCP membranes has only been demonstrated at low packing densities. The highest packing density showing the expected function was demonstrated for AqpZ reconstituted into a BCP membrane at a molar PoPR (adjusted for triblock architecture) of 100. We show here that full AQP0 function still persists at a PoPR of 15. We also show that the reconstitution method is critical, but polymer block lengths and chemistries may also be important factors that determine how much protein can be functionally reconstituted into BCP membranes. The possibility to obtain a high density of functional membrane proteins in BCP membranes has significant implications for applications of such systems. Overall, this work provides a framework for developing highly efficient membrane protein devices.
Conclusions
The results obtained in this study show that membrane proteins can be functionally incorporated into BCP membranes at high density, and that incorporation of membrane proteins affects the morphology of the resulting BCP–membrane protein aggregates. The effect of AQP on the morphology of self-assembled structures for all four BCPs and one lipid tested followed similar trends depending on the volume fraction occupied by AQP0. Furthermore, in some cases, AQP0 formed 2D crystals in BCP membranes, representing the limit of membrane protein packing in bilayer-like membranes. Concentration-dependent morphology evolution allows for the design of membrane protein devices and membranes of defined form factor, and the high densities shown possible to be achieved provides for orders of magnitude improvement in sensitivity or transport rates of such devices, allowing for miniaturization or other unique designs.
Acknowledgments
We thank Dr. Zongli Li for support with electron microscopy, Dr. Timothy Springer for providing access to light scattering equipment, Dr. Scott Milner for advice concerning the analysis of morphology transitions, and Dr. Andreas Schenk for help with AQP0 volume calculations. Financial support to W.P.M. by the Swiss National Science Foundation, the NCCR Nanosciences, and the Swiss Nanoscience Institute is gratefully acknowledged. W.P.M. also thanks in particular P. Vajkoczy. Work on AQP0 in the Walz laboratory is supported by NIH grant R01 EY015107. The molecular electron microscopy facility at Harvard Medical School was established with a generous donation from the Giovanni Armenise Harvard Center for Structural Biology and is maintained with funds from NIH grant P01 GM62580 (to Stephen C. Harrison). T.W. is an investigator in the Howard Hughes Medical Institute.
Supporting Information Available
Materials and Methods, Calculations (hydrophilic ratios of block copolymers, hydrophobic volume of AQP0 and hydrophobic fraction of AQP0 in block copolymer aggregates), Table summarizing data on the 2D crystallization of aquaporins with lipids (Table S1), Supplementary Figures 1 through 8. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Author Present Address
† Department of Chemical Engineering, Pennsylvania State University, University Park, PA 16802, USA
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Gu L.; Braha O.; Conlan S.; Cheley S.; Bayley H. Nature 1999, 398, 686–690. [DOI] [PubMed] [Google Scholar]
- Bayley H.; Cremer P. S. Nature 2001, 413, 226–230. [DOI] [PubMed] [Google Scholar]
- Astier Y.; Bayley H.; Howorka S. Curr. Opin. Chem. Biol. 2005, 9, 576–584. [DOI] [PubMed] [Google Scholar]
- Suzuki H.; Tabata K.; Kato-Yamada Y.; Noji H.; Takeuchi S. Lab. Chip 2004, 4, 502–505. [DOI] [PubMed] [Google Scholar]
- Kumar M.; Grzelakowski M.; Zilles J.; Clark M.; Meier W. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 20719–20724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubert A.; Napoli A.; Meier W. Curr. Opin. Chem. Biol. 2004, 8, 598–603. [DOI] [PubMed] [Google Scholar]
- Discher B.; Won Y.; Ege D.; Lee J.; Bates F.; Discher D.; Hammer D. Science 1999, 284, 1143–1146. [DOI] [PubMed] [Google Scholar]
- Discher D. E.; Eisenberg A. Science 2002, 297, 967–973. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Eisenberg A. Science 1995, 268, 1728–1731. [DOI] [PubMed] [Google Scholar]
- Grzelakowski M.; Onaca O.; Rigler P.; Kumar M.; Meier W. Small 2009, 5, 2545–2548. [DOI] [PubMed] [Google Scholar]
- Egli S.; Nussbaumer M. G.; Balasubramanian V.; Chami M.; Bruns N.; Palivan C.; Meier W. J. Am. Chem. Soc. 2011, 133, 4476–4483. [DOI] [PubMed] [Google Scholar]
- Nardin C.; Hirt T.; Leukel J.; Meier W. Langmuir 2000, 16, 1035–1041. [Google Scholar]
- Peer D.; Karp J. M.; Hong S.; Farokhzad O. C.; Margalit R.; Langer R. Nat. Nanotechnol. 2007, 2, 751–760. [DOI] [PubMed] [Google Scholar]
- Kita-Tokarczyk K.; Grumelard J.; Haefele T.; Meier W. Polymer 2005, 46, 3540–3563. [Google Scholar]
- Ho D.; Chang S.; Montemagno C. D. Nanomedicine 2006, 2, 103–112. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Perez A.; Stibius K. B.; Vissing T.; Nielsen C. H.; Mouritsen O. G. Langmuir 2009, 25, 10447–10450. [DOI] [PubMed] [Google Scholar]
- Langer R. Science 1990, 249, 1527–1533. [DOI] [PubMed] [Google Scholar]
- Mueller P.; Rudin D. O.; Tien H. T.; Wescott W. C. J. Phys. Chem. 2011, 67, 534–535. [Google Scholar]
- Pata V.; Ahmed F.; Discher D.; Dan N. Langmuir 2004, 20, 3888–3893. [DOI] [PubMed] [Google Scholar]
- Marsden H. R.; Quer C. B.; Sanchez E. Y.; Gabrielli L.; Jiskoot W.; Kros A. Biomacromolecules 2010, 11, 833–838. [DOI] [PubMed] [Google Scholar]
- Kumar M.; Payne M.; Poust S.; Zilles J.. Polymer-based biomimetic membranes for desalination. In Biomimetic Membranes for Sensor and Separation Applications; Neilsen C. H., Eds.; 2012; pp 43–62. [Google Scholar]
- Zampighi G. A.; Eskandari S.; Hall J. E.; Zampighi L.; Kreman M. Exp. Eye Res. 2002, 75, 505–519. [DOI] [PubMed] [Google Scholar]
- Zampighi G.; Simon S.; Robertson J.; Mcintosh T.; Costello M. J. Cell. Biol. 1982, 93, 175–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonen T.; Cheng Y.; Sliz P.; Hiroaki Y.; Fujiyoshi Y.; Harrison S. C.; Walz T. Nature 2005, 438, 633–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hite R. K.; Li Z.; Walz T. EMBO J. 2010, 29, 1652–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann T. C.; Engel A.; Rémigy H.-W. Biophys. J. 2006, 90, 310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S.; Bates F. Science 2003, 300, 460–464. [DOI] [PubMed] [Google Scholar]
- Gipson B.; Zeng X.; Zhang Z. Y.; Stahlberg H. J. Struct. Biol. 2007, 157, 64–72. [DOI] [PubMed] [Google Scholar]
- Kozono D.; Ding X.; Iwasaki I.; Meng X.; Kamagata Y.; Agre P.; Kitagawa Y. J. Biol. Chem. 2003, 278, 10649–10656. [DOI] [PubMed] [Google Scholar]
- Borgnia M.; Kozono D.; Calamita G.; Maloney P.; Agre P. J. Mol. Biol. 1999, 291, 1169–1179. [DOI] [PubMed] [Google Scholar]
- Zeidel M. L.; Ambudkar S. V.; Smith B. L.; Agre P. Biochemistry 1992, 31, 7436–7440. [DOI] [PubMed] [Google Scholar]
- Mulders S. M.; Preston G. M.; Deen P. M.; Guggino W. B.; van Os C. H.; Agre P. J. Biol. Chem. 1995, 270, 9010–9016. [DOI] [PubMed] [Google Scholar]
- Mathai J. C.; Tristram-Nagle S.; Nagle J. F.; Zeidel M. L. J. Gen. Physiol. 2007, 131, 69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigaud J.-L.; Lévy D. Methods Enzymol. 2003, 372, 65–86. [DOI] [PubMed] [Google Scholar]
- Varadaraj K.; Kumari S.; Shiels A.; Mathias R. T. Invest. Ophthalmol. Visual Sci. 2005, 46, 1393–1402. [DOI] [PubMed] [Google Scholar]
- Nemeth-Cahalan K. L.; Hall J. E. J. Biol. Chem. 2000, 275, 6777–6782. [DOI] [PubMed] [Google Scholar]
- Yang B.; Verkman A. S. J. Biol. Chem. 1997, 272, 16140–16146. [DOI] [PubMed] [Google Scholar]
- Kita-Tokarczyk K.; Grumelard J.; Haefele T.; Meier W. Polymer 2005, 46, 3540–3563. [Google Scholar]
- Graff A.; Fraysse-Ailhas C.; Palivan C. G.; Grzelakowski M.; Friedrich T.; Vebert C.; Gescheidt G.; Meier W. Macromol. Chem. Phys. 2010, 211, 229–238. [Google Scholar]
- Seddon A. M.; Curnow P.; Booth P. J. Biochim. Biophys. Acta 2004, 1666, 105–117. [DOI] [PubMed] [Google Scholar]
- Hasler L.; Heymann J. B.; Engel A.; Kistler J.; Walz T. J. Struct. Biol. 1998, 121, 162–171. [DOI] [PubMed] [Google Scholar]
- Hite R. K.; Raunser S.; Walz T. Curr. Opin. Struct. Biol. 2007, 17, 389–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raunser S.; Walz T. Annu. Rev. Biophys. 2009, 38, 89–105. [DOI] [PubMed] [Google Scholar]
- Dolder M.; Engel A.; Zulauf M. FEBS Lett. 1996, 382, 203–208. [DOI] [PubMed] [Google Scholar]
- Winey K.; Thomas E.; Fetters L. Macromolecules 1992, 25, 2645–2650. [Google Scholar]
- Winey K.; Thomas E.; Fetters L. Macromolecules 1991, 24, 6182–6188. [Google Scholar]
- Milner S. Macromolecules 1994, 27, 2333–2335. [Google Scholar]
- Kinning D.; Thomas E. L.; Fetters L. J. J. Chem. Phys. 1989, 90, 5806–5825. [Google Scholar]
- Lin Y.; Daga V. K.; Anderson E. R.; Gido S. P.; Watkins J. J. J. Am. Chem. Soc. 2011, 133, 6513–6516. [DOI] [PubMed] [Google Scholar]
- Srinivas G.; Discher D. E.; Klein M. L. Nano Lett. 2005, 5, 2343–2349. [DOI] [PubMed] [Google Scholar]
- Pata V.; Dan N. Biophys. J. 2003, 85, 2111–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashl R. J.; Scott H. L.; Subramaniam S.; Jakobsson E. Biophys. J. 2001, 81, 3005–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tristram-Nagle S.; Petrache H. I.; Nagle J. F. Biophys. J. 1998, 75, 917–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer J. R. Biophys. J. 1989, 55, 957–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.