

Abstract
A number of previous studies suggested the presence of deleterious amino acid altering nonsynonymous single-nucleotide polymorphisms (nSNPs) in human populations. However, the proportions of deleterious nSNPs among rare and common variants are not known. To estimate these, >77 000 SNPs from human protein-coding genes were analyzed. Based on two independent methods, this study reveals that up to 53% of rare nSNPs (minor allele frequency (MAF)<0.002) could be deleterious in nature. The fraction of deleterious nSNPs declines with the increase in their allele frequencies and only 12% of the common nSNPs (MAF>0.4) were found to be harmful. This shows that even at high frequencies significant fractions of deleterious polymorphisms are present in human populations. These results could be useful for genome-wide association studies in understanding the relative contributions of rare and common variants in causing human genetic diseases.
Keywords: human evolution, deleterious mutations, disease-associated mutations, genome-wide association, rare and common variants, SNP and population genetics theory
Introduction
Although harmful mutations affect fitness of an organism they are nevertheless present in human populations and contribute to the diversity due to random genetic drift.1 However natural selection eliminates such deleterious mutations over time and thus they are prevented from reaching high frequencies. Therefore low-frequency single-nucleotide polymorphisms (SNPs) typically comprise deleterious as well as neutral polymorphisms, whereas high frequency SNPs are largely neutral in nature. As amino-acid-changing SNPs might be detrimental to proper protein function, a significant proportion of them could be harmful. A number of previous studies have shown an enrichment of low-frequency nonsynonymous SNPs (nSNPs) compared with those with high frequencies,2, 3, 4, 5 which indirectly suggests that these nSNPs are deleterious and removed over time by natural selection. However the fraction of deleterious nSNPs with respect to their allele frequencies is unclear. In other words the proportion of deleterious nSNPs among low (or high) frequency variants has not been quantified. To estimate this, the present investigation has gathered over 77 000 SNPs from human protein-coding genes and grouped them based on their minor allele frequencies. Two independent methods were used to estimate the proportion of deleterious nSNPs and the frequency distribution of these harmful nSNPs was examined.
Materials and methods
SNP data
First, SNPs of all human protein-coding genes (dbSNP build130) were obtained from the UCSC genome resource (http://genome.ucsc.edu/). Then using the rsIDs of SNPs, their corresponding minor allele frequencies were obtained from the dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP/). For consistency, only the SNPs and their allele frequencies reported by the 1000 genome project2 (1000 Genome phase 1 – May 2011 data release) were used for further analysis. This final data set consisted of 37 123 nSNPs and 40 599 synonymous SNPs (sSNPs). These SNPs were grouped into 10 categories based on their minor allele frequencies and the proportion of deleterious nSNPs was computed for each category as described below.
Estimation of the deleterious proportion of nSNPs
McDonald and Kreitman6 showed that under neutral evolution the ratio of nonsynonymous (Pn) to synonymous (Ps) polymorphisms (Pn/Ps) within species is expected to be equal to the ratio of nonsynonymous (Dn) to synonymous (Ds) substitutions between species, that is,
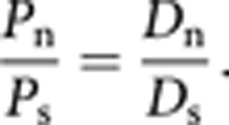 |
However, it is clear from Table 1 that the ratios of SNPs are always higher than that of the substitutions between human–chimp, that is,
Table 1. Human polymorphisms, substitutions (between human–chimp) and deleterious fractions of nSNPs.
| Minor allele frequency (%) | Nonsynonymous SNPs (Pn) | Synonymous SNPs (Ps) | Pn/Ps | δ (SE) | Damaging SNPs | Benign SNPs | ρ (SE) |
|---|---|---|---|---|---|---|---|
| <0.2 | 1290 | 919 | 1.40 | 0.534 (0.021) | 375 | 721 | 0.342 (0.014) |
| 0.2–0.5 | 2618 | 2156 | 1.21 | 0.461 (0.015) | 773 | 1466 | 0.345 (0.010) |
| 0.5–1 | 3633 | 3330 | 1.09 | 0.400 (0.014) | 938 | 2181 | 0.301 (0.008) |
| 1–2 | 4819 | 4696 | 1.03 | 0.362 (0.013) | 1148 | 2942 | 0.281 (0.007) |
| 2–5 | 6915 | 7516 | 0.92 | 0.289 (0.011) | 1461 | 4406 | 0.249 (0.006) |
| 5–10 | 5335 | 6051 | 0.88 | 0.258 (0.014) | 992 | 3422 | 0.225 (0.006) |
| 10–20 | 4965 | 5978 | 0.831 | 0.212 (0.016) | 716 | 3364 | 0.175 (0.006) |
| 20–30 | 3061 | 3914 | 0.782 | 0.163 (0.020) | 360 | 2148 | 0.144 (0.007) |
| 30–40 | 2362 | 3173 | 0.744 | 0.121 (0.024) | 222 | 1723 | 0.114 (0.007) |
| 40–50 | 2125 | 2866 | 0.741 | 0.118 (0.025) | 236 | 1530 | 0.134 (0.008) |
| Human–Chimp1 | 47 079 (Dn) | 71 956 (Ds) | 0.654 | — | — | — | — |
−Dn/Ds ratio estimated for the human–chimp pair is significantly smaller than all Pn/Ps ratios (G test, P<0.0001).
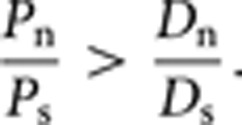 |
This is due to the presence of deleterious nSNPs in the human populations as predicted by previous theoretical studies.1, 7 Hence to subtract the fraction of deleterious nSNPs (δ) the equation could be written as
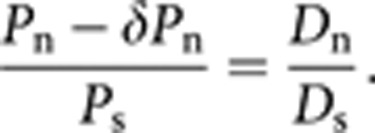 |
This equation could be simplified to estimate the fraction of deleterious nSNPs (δ) as:
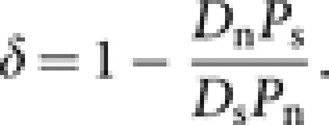 |
The measure δ is the fraction of deleterious nSNPs that are segregating in the population.8 The numbers of nonsynonymous (Dn=47 079) and synonymous (Ds=71 956) substitutions (based on 13 454 orthologous human–chimpanzee protein coding genes) were obtained from a previous study.9 To obtain the standard error, a bootstrap procedure was used by resampling the SNPs (1000 replications).
Quantification of the fraction of damaging nSNPs
To determine the deleterious nature of each nSNP, the online software tool Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/bgi.shtml) was used.10 Using protein secondary structures, functional motifs, and relative conservation of each amino acid in the protein, the above program predicts the possible impact of an amino-acid replacement polymorphism on the structure and/or function of a human protein. For each nSNP, this program predicted whether the given type of amino acid change is benign, possibly damaging or probably damaging. The fraction of damaging nSNPs (ρ) was computed by adding the counts of possibly and probably damaging nSNPs and dividing this by the total nSNP count. The binomial variance was used to estimate the SE.
Results
As some of the amino-acid polymorphisms are deleterious, selection prevent such nSNPs from spreading in a population. Therefore nSNPs are expected to be more abundant at low frequencies than at high frequencies. In contrast, all sSNPs are largely neutral and hence they are likely to be present in equal proportions at low and high frequencies. Therefore sSNPs could be used as a normalizing factor and thus the ratio of nSNPs to sSNPs (Pn/Ps) will reflect the excess fraction of nSNPs. Table 1 shows that this ratio has a negative relationship with the minor allele frequencies of SNPs. This ratio is roughly two times (1.4 vs 0.74) higher for the SNPs with a minor allele frequency (MAF) of <0.002 compared with those with a MAF>0.4. It should be noted that the discovery of very-low-frequency variants (MAF<0.002) might be error prone as the observed number of minor alleles was small (<4).2 However the method used to estimate the fraction of deleterious SNPs is based on the ratio of nSNPs and sSNPs. Hence this estimate will not be significantly affected as the error rate is expected to be fairly the same for both types of SNPs.
The ratio of nonsynonymous to synonymous substitutions (Dn/Ds) estimated for the human–chimp pair (0.65) is significantly smaller than all Pn/Ps ratios (G test, P<0.0001). This suggests an overabundance of nSNPs with respect to the nonsynonymous substitutions and this excess fraction of nSNPs is deleterious as they were prevented from becoming fixed (see Rand and Kann11). This deleterious fraction was estimated as described in the Materials and Methods section. Clearly the deleterious proportion of nSNPs (δ) shows a negative relationship with minor allele frequencies (Figure 1a). Deleterious nSNPs constitute as high as 53% of the nSNPs with a MAF <0.002, whereas for common nSNPs (MAF=0.4–0.5) the deleterious fraction is only 12%.
Figure 1.

(a) Deleterious fraction of nSNPs (δ) estimated for variants with different minor allele frequencies (MAF). SNPs were grouped into 10 categories based on their MAF and only the upper values of the ranges are shown (X-axis). Error bars denote the SE, which was estimated using a bootstrap procedure (1000 replications). (b) The proportion of damaging nSNPs (ρ) was estimated using amino acid variants belonging to 10 MAF categories. Error bars indicate the SE computed using the binomial variance.
I also used an independent method to quantify the fraction of deleterious nSNPs using the online software tool Polyphen-2.10 This program determines the deleterious nature of amino-acid-changing nSNPs based on their effect on protein structure and/or function and based on their location in the protein. Using this software the fraction of damaging nSNPs (ρ) was estimated as explained in the Materials and Methods section. Interestingly, the relationship between ρ and MAF shown in Figure 1b is very similar to that observed for δ and MAF (Figure 1a). The estimate ρ obtained for low-frequency nSNPs (MAF<0.002) was 2.6 times higher than that estimated for high-frequency nSNPs with a MAF=0.4–0.5 (0.34 vs 0.13). Here the estimate ρ includes the nSNPs that are predicted by Polyphen-2 as ‘possibly damaging' and ‘probably damaging' with probabilities of >50% and >95%, respectively, to disrupt the structure and/or function of a protein. However using only ‘probably damaging' nSNPs also produced a negative relationship with similar magnitude and the ρ of low-frequency nSNPs was three times higher than that of high-frequency SNPs (0.19 vs 0.06).
Discussion
Based on two independent methods this study estimated the proportion of deleterious amino acid variants in human populations. The first method showed a much higher fraction of deleterious nSNPs among the rare variants (MAF<0.002) compared with the second method (53% vs 34% Table 1). As the second method (using Polyphen-2) depends on the relevant information available for a protein (to predict the deleterious nature of an SNP), this method is rather subjective. More detailed information about proteins in the future might result in redefining some of the harmless nSNPs to harmful ones. In contrast the first method is based on a ratio, which is objective and not depended on the availability of protein specific information.
The high fraction of deleterious nSNPs reported for the low-frequency nSNPs suggests that rare variants are more likely to be associated with diseases than common variants.5, 12 On the other hand the results also showed that a significant fraction of high-frequency nSNPs could be deleterious in nature. This suggests a likely association of some of the common variants to human genetic diseases.13 The deleterious fraction of nSNPs reported here could be an underestimate of deleterious mutations in humans as it does not include lethal or strongly deleterious mutations. On the other hand, these estimates might include false positive SNPs due to sequencing errors.14
The present study has estimated the proportion of deleterious SNPs (δ) only for protein-coding regions. However, the same formula could be used to estimate δ for SNPs in constrained noncoding regions such as UTRs, promotors, enhancers, and silencers. For such a calculation, Pn and Dn are the number of SNPs and substitutions observed in the noncoding region (eg, promotor), and Ps and Ds are the number of SNPs and substitutions in synonymous positions or intron(s). The findings of this study might have implications in genome-wide association studies in understanding the respective contributions of rare as well as common variants to human diseases.
Acknowledgments
The author is grateful to David Lambert and thanks Leon Huynen and two anonymous reviewers for valuable comments.
The author declares no conflict of interest.
References
- Kimura M. The Neutral Theory of Molecular Evolution. Cambridge: Cambridge University press; 1983. [Google Scholar]
- 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cargill M, Altshuler D, Ireland J, et al. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet. 1999;22:231–238. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- Frazer KA, Ballinger DG, Cox DR, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Ge D, Maia JM, et al. A genome-wide comparison of the functional properties of rare and common genetic variants in humans. Am J Hum Genet. 2011;88:458–468. doi: 10.1016/j.ajhg.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- Kryazhimskiy S, Plotkin JB. The population genetics of dN/dS. Plos Genet. 2008;4:e1000304. doi: 10.1371/journal.pgen.1000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S. High proportions of deleterious polymorphisms in constrained human genes. Mol Biol Evol. 2011;28:49–52. doi: 10.1093/molbev/msq287. [DOI] [PubMed] [Google Scholar]
- Mikkelsen TS, Hillier LW, Eichler EE, et al. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand DM, Kann LM. Excess amino acid polymorphism in mitochondrial DNA: contrasts among genes from Drosophila, mice, and humans. Mol Biol Evol. 1996;13:735–748. doi: 10.1093/oxfordjournals.molbev.a025634. [DOI] [PubMed] [Google Scholar]
- Pritchard JK. Are rare variants responsible for susceptibility to complex diseases. Am J Hum Genet. 2001;69:124–137. doi: 10.1086/321272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich DE, Lander ES. On the allelic spectrum of human disease. Trends Genet. 2001;17:502–510. doi: 10.1016/s0168-9525(01)02410-6. [DOI] [PubMed] [Google Scholar]
- MacArthur DG, Tyler-Smith C. Loss-of-function variants in the genomes of healthy humans. Hum Mol Genet. 2010;19:R125–R130. doi: 10.1093/hmg/ddq365. [DOI] [PMC free article] [PubMed] [Google Scholar]