

Abstract
Silvery hair is a common presentation of rare group of autosomal recessive disorders called Silvery hair syndromes including Griscelli syndrome (GS), Chediak-Higashi syndrome, and Elejalde syndrome. GS is characterized by a silvery grey sheen to hair, large clumped melanosomes in hair shaft, partial albinism, and variable cellular immunodeficiency. We report two cases of GS with classical clinical features and confirmatory findings by microscopic skin and hair examination.
Keywords: Griscelli syndrome, immunodeficiency, silvery hair
INTRODUCTION
Griscelli syndrome (GS) is a rare autosomal recessive disorder characterized by pigmentary dilution of skin and hair, variable cellular and humoral immunodeficiency.[1] The condition is fatal. Recurrent episodes of fever and lymphohistiocytic infiltration of organs lead to hepatosplenomegaly, lymphadenopathy, pancytopenia, and neurological impairment. It was first described by Griscelli and Prunieras in 1978.[2] Approximately 60 cases have been reported, mostly from Turkish and Mediterranean population.[3] Only few cases have been reported from India. We report two cases of GS.
CASE REPORTS
Case 1
An 8-year-old male child born of non-consanguineous marriage was referred for evaluation of silvery grey hair all over the body from birth. His history was remarkable for recurrent respiratory infections. His developmental milestones were normal and immunized till date. On examination, vitals were normal. Child had silvery grey hair over the entire body including scalp, eyebrows, and eyelashes [Figure 1]. Systemic examination was unremarkable. On investigations, complete hemogram showed hemoglobin of 6.5 g/dl, total leukocyte of 4200/cu.mm, and platelet count of 8000 cu.mm. No prominent granules were noted in leukocytes. Liver function tests, renal function tests, and chest radiograph was normal. His hair microscopic examination revealed irregular pigment clumps in hair shaft [Figure 2]. Skin biopsy showed clumped pigment deposition in the basal epithelial layer. Bone marrow examination was normal.
Figure 1.
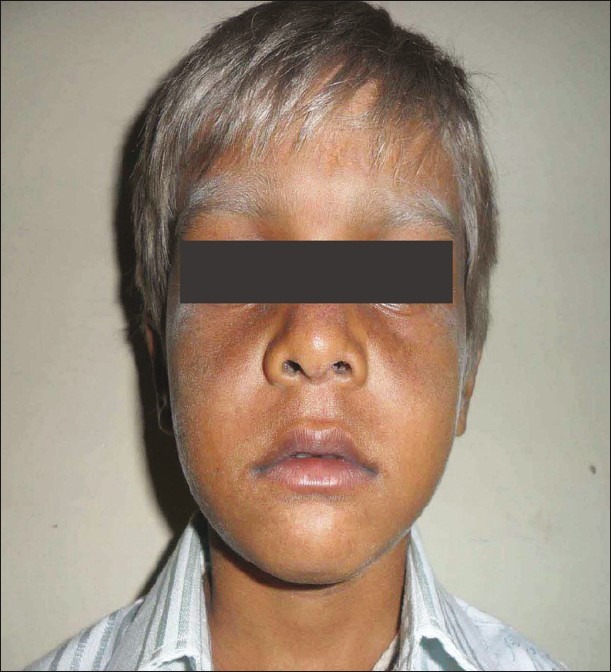
Eight-year-old boy with silvery grey hair
Figure 2.
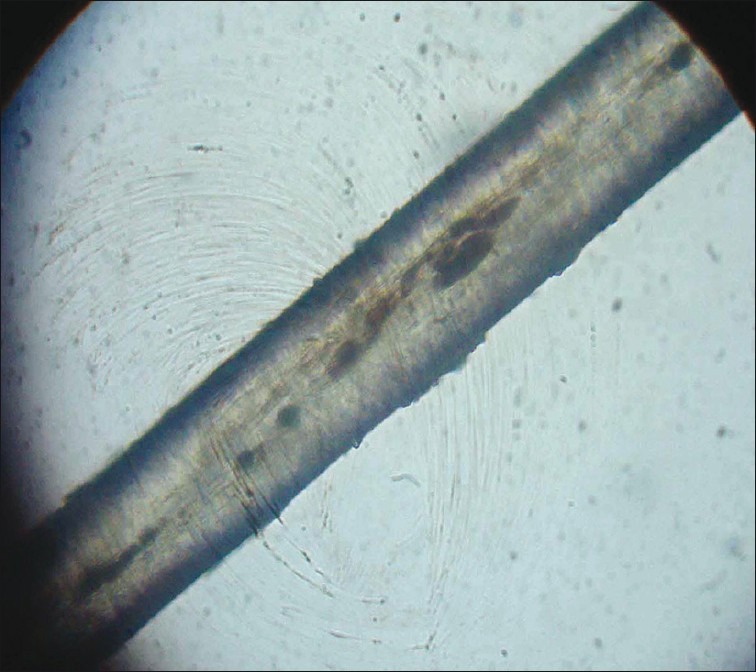
Hair shaft showing irregular pigment clumps
Case 2
A 1-year-old female child born of non-consanguineous marriage presented with recurrent episodes of fever, abdominal distension, and vomiting since 6 months. Child had delayed milestones with growth retardation. On examination, child had pallor, vitals were normal. Cutaneous examination showed hypopigmented macule on face with silvery brown hair all over the body [Figure 3]. A complete hemogram showed hemoglobin of 8.5 g/ dl, total leukocyte of 3200/cu.mm, and platelet count of 4000 cu.mm. However, peripheral smear was normal. Liver enzymes were two-fold increased. Ultrasonography of abdomen revealed hepatosplenomegaly. Other investigations were normal. Microscopic examination of hair shaft showed unevenly distributed melanin aggregates. Child died a few weeks later due to septicemia.
Figure 3.
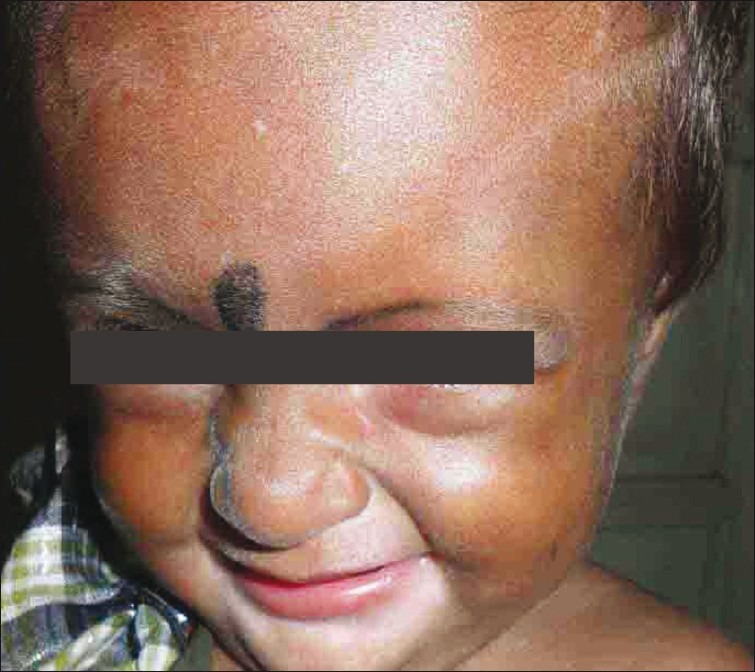
Silvery grey hair with hypopigmented macule on the face
Based on the clinical, laboratory findings and presence of clumped melanosomes on microscopy of hair shaft, a diagnosis of GS was made.
DISCUSSION
GS is a rare autosomal disorder that was first described in 1978. It is characterized by partial albinism of hair and skin, variable cellular and humoral immunodeficiency, and the occurrence of accelerated phases consisting of hemophagocytosis, pancytopenia, elevation of serum triglyceride levels, hypofibrinogenemia, and hypoproteinemia.[4] Three different subgroups are now clearly delineated that share cutaneous characteristics but differ in their extracutaneous manifestations.
Type 1 GS is associated with severe neurological impairment with no immunodeficiency. It results from mutations in MYO5A, a motor molecule involved in melanosome movement as well as neurosecretory vesicles. Type 2 GS is associated with hemophagocytosis and immunodeficiency and results from mutations in Rab27a, which has been implicated in the regulation of membrane trafficking including melanosome transport and various regulated secretion events.[5] Type 3 GS manifestations are restricted to skin and hair and results from mutations in the gene that encodes melanophilin (Mlph).[6]
Dermatologic findings may be limited to hair and skin and occasionally retinal pigmentation may be seen. Other clinical manifestations include severe hypotonia that may progress to spastic quadriparesis or flaccid quadriplegia, seizures, ataxic movement, and mental retardation.[7] Microscopic examination of hair shaft reveals uneven clusters of aggregated melanin pigments, accumulated mainly in the medullary area of hair shaft. Histopathological examination of skin biopsy shows hyperpigmented oval melanocytes with poorly pigmented keratinocytes. Electron microscopic evaluation of skin specimens shows epidermal melanocytes filled with numerous stage IV melanosomes.[8]
GS must be considered as one of the conditions in the differential diagnosis of silvery grey hair syndrome. The other two conditions include Chediak Higashi and Elejalde syndrome. The difference between the three conditions has been summarized in Table 1.[9]
Table 1.
Investigations to differentiate between Griscelli syndrome, Chediak-Higashi syndrome, and Elejalde disease

The prognosis of GS is grave. Prenatal diagnosis has been accomplished by examination of hair from fetal scalp and skin biopsies performed at 20 weeks gestation.[8] Bone marrow transplantation or peripheral blood stem cell transplantation is the only curative therapy for GS and is advised as early as possible in the course of the disease. This suggests that the cells of hematopoietic origin are responsible for the fatal outcome in GS.[10]
The diagnosis of GS in our cases was based on clinical findings and hematological abnormalities with characteristic findings of hair on light microscopy. Silvery grey hair provides clue to underlying immunodeficiency and hence finding of silvery grey hair in childhood when associated with fever, pancytopenia, and systemic involvement should alert the clinician to consider GS since early diagnosis and treatment can improve prognosis.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Mancini AJ, Chan LS, Paller AS. Partial albinism with immunodeficiency: Griscelli syndrome: Report of a case and review of literature. J Am Acad Dermatol. 1998;38:295–300. doi: 10.1016/s0190-9622(98)70568-7. [DOI] [PubMed] [Google Scholar]
- 2.Griscelli C, Prunieras M. Pigment dilution and immunodeficiency: A new syndrome. Int J Dermatol. 1978;17:788–91. doi: 10.1111/j.1365-4362.1978.tb05980.x. [DOI] [PubMed] [Google Scholar]
- 3.Sheela SR, Latha M, Injody SJ. Griscelli syndrome: Rab27a mutation. Indian Pediatr. 2004;41:944–7. [PubMed] [Google Scholar]
- 4.Manglani M, Adhvaryu K, Seth B. Griscelli syndrome - a case report. Indian Pediatr. 2004;41:734–7. [PubMed] [Google Scholar]
- 5.Fukuda M. Versatile role of Rab27 in membrane trafficking: focus on the Rab27 effector families. J Biochem. 2005;137:9–16. doi: 10.1093/jb/mvi002. [DOI] [PubMed] [Google Scholar]
- 6.Menasche G, Ho CH, Sanal O, Feldmann J, Tezcan I, Ersoy F, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5AF exon deletion (GS1) J Clin Invest. 2003;112:450–6. doi: 10.1172/JCI18264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hurvitz H, Gillis R, Klaus S, Klar A, Gross-Kieselstein F, Okon E. A kindred with Griscelli disease: Spectrum of neurologic involvement. Eur J Pediatr. 1993;152:402–5. doi: 10.1007/BF01955897. [DOI] [PubMed] [Google Scholar]
- 8.Kumar TS, Ebenazar S, Moses PD. Griscelli syndrome. Indian J Dermatol. 2006;51:269–71. [Google Scholar]
- 9.Inamdar AC, Palit A. Silvery hair with bronze tan in child: A case of Elejalde disease. Indian J Dermatol Venereol Leprol. 2007;73:417–9. doi: 10.4103/0378-6323.37063. [DOI] [PubMed] [Google Scholar]
- 10.Schneider LC, Berman RS, Shea CR, Perez-Atayde AR, Weinstein H, Geha RS. Bone marrow transplantation for the syndrome of pigmentary dilution and lymphohistiocytosis (Griscelli syndrome) J Clin Immunol. 1990;10:146–53. doi: 10.1007/BF00917914. [DOI] [PubMed] [Google Scholar]