

Abstract
A cDNA encoding the Arabidopsis thaliana uridine 5′-monophosphate (UMP)/cytidine 5′-monophosphate (CMP) kinase was isolated by complementation of a Saccharomyces cerevisiae ura6 mutant. The deduced amino acid sequence of the plant UMP/CMP kinase has 50% identity with other eukaryotic UMP/CMP kinase proteins. The cDNA was subcloned into pGEX-4T-3 and expressed as a glutathione S-transferase fusion protein in Escherichia coli. Following proteolytic digestion, the plant UMP/CMP kinase was purified and analyzed for its structural and kinetic properties. The mass, N-terminal sequence, and total amino acid composition agreed with the sequence and composition predicted from the cDNA sequence. Kinetic analysis revealed that the UMP/CMP kinase preferentially uses ATP (Michaelis constant [Km] = 29 μm when UMP is the other substrate and Km = 292 μm when CMP is the other substrate) as a phosphate donor. However, both UMP (Km = 153 μm) and CMP (Km = 266 μm) were equally acceptable as the phosphate acceptor. The optimal pH for the enzyme is 6.5. P1, P5-di(adenosine-5′) pentaphosphate was found to be a competitive inhibitor of both ATP and UMP.
Pyrimidines are intimately involved in the physiology of cells. They participate at multiple levels in intermediary and secondary metabolism from nucleotide and macromolecule biosynthesis to the biosynthesis of complex carbohydrates and the metabolic regulation of intermediary metabolism. All pyrimidines within the cell are derived from UMP, which arises either from the de novo pyrimidine biosynthetic pathway or from salvage of preformed pyrimidines. UMP/CMP kinase converts uridine and cytidine monophosphates into the corresponding uridine and cytidine diphosphates. Because all pyrimidines are derived from UMP, UMP kinase is the first committed step and one of the central enzymes in the further anabolism of pyrimidine nucleotides.
The importance of pyrimidine monophosphokinases to cell physiology has been firmly established. In both bacteria and yeast, the roles of pyrimidine monophosphokinases with respect to cell proliferation and physiology have been widely studied. In Escherichia coli, the product of the UMP kinase gene (pyrH/smbA) has been shown to influence cell proliferation. Yamanaka et al. (1992), working with the mukB gene, isolated a suppressor of mukB that they termed smbA. The smbA phenotype is pleiotropic. First, the smbA mutant ceased macromolecular synthesis, was hypersensitive to SDS, and showed a novel morphological phenotype under nonpermissive conditions. Later, it was found that the wild-type smbA gene is identical to the pyrH gene and that the smbA2 mutant protein encodes an unstable UMP kinase with impaired catalytic and regulatory functions.
In yeast, mutations in the UMP kinase gene have been shown to cause a conditional lethal phenotype (Liljelund and Lacroute, 1986). When grown at the nonpermissive temperature, the UTP and CTP pools decline to 10% of their wild-type levels, which affects both RNA and protein synthesis and ultimately results in cell death. Complementation of this mutant permitted the first isolation of a eukaryotic UMP kinase gene (Liljelund and Lacroute, 1986). This is the same mutant that we have complemented in this study to isolate the Arabidopsis thaliana UMP/CMP kinase cDNA.
Other pyrimidine monophosphokinases are also important in normal cellular physiology. The cdc8 mutant of Saccharomyces cerevisiae was isolated as a cell-cycle-deficient mutant that was defective in nuclear division (Newlon and Fangman, 1975). When incubated at the restrictive temperature, the cdc8 mutant cells arrest in S phase with a typical dumbbell morphology. This mutation also produces other pleiotropic effects, including inhibition of normal cellular DNA replication (Birkenmeyer et al., 1984), involvement in error-prone repair (Prakash et al., 1979; Baranowska and Zuk, 1991), and involvement in repair of single-stranded breaks (Baranowska et al., 1990). The CDC8 gene was isolated (Birkenmeyer et al., 1984; Kuo and Campbell, 1983) and found to encode dTMP kinase (Jong et al., 1984). Known suppressors of the CDC8 gene have also been isolated and characterized. One of these, SOC8, encodes the UMP kinase gene ura6 (Jong et al., 1993). Thus, nucleotide monophosphokinases can have profound effects on cellular morphology and physiology.
Substrate utilization by eukaryotic UMP kinases has also received much study (Weismüller et al., 1990; Jong et al., 1993; Müller-Dieckmann and Schultz, 1994). Whereas all eukaryotic enzymes have specificity for both UMP and CMP, the yeast enzyme also has specificity for AMP. Indeed, the yeast UMP kinase has such a high affinity for AMP that the ura6 gene has been isolated as a multicopy suppressor of yeasts deficient in adenylate kinase (Schricker et al., 1992). The yeast UMP kinase gene also complements the E. coli adenylate kinase enzyme. In contrast, the cellular slime mold (Dictyostelium discoideum) enzyme does not complement either the yeast or the E. coli adenylate kinase mutants. UMP kinase is also required for the metabolic activation of several important anti-tumor drugs, including 5-fluorouracil and Ara-C (Seagrave and Reyes, 1987). Consequently, UMP kinase plays a central and very important role in pyrimidine anabolism.
In addition to studies of the eukaryotic enzyme, the prokaryotic enzyme has received much attention (Valentin-Hansen, 1978; Yamanaka et al., 1992; Serina et al., 1995, 1996). Because of this, significant differences between the enzymatic activities of the prokaryotic and eukaryotic enzymes have been identified. The prokaryotic enzyme is allosterically regulated by both GTP and UTP (Serina et al., 1995). GTP functions to stimulate the enzyme when there is an overabundance of purine triphosphates, and UTP down-regulates the enzyme when pyrimidine triphosphates have accumulated to a high level. In addition, UMP kinase is further regulated by divalent metal ions in a novel mechanism related to metal-free-UTP binding (Serina et al., 1996). The binding of metal-free UTP causes a gel-sol transition that affects the state of UMP kinase aggregation and, subsequently, the enzyme activity.
In humans, UMP kinase is associated with an autoimmune deficiency that results in susceptibility to respiratory infections such as invasive Hemophilus influenzae type B disease in Alaskan Eskimos (Petersen et al., 1985) and South American Indians (Gallango and Suinaga, 1978; Gallango et al., 1978). This underexpression of UMP kinase results in a syndrome similar to the immune defect resulting from adenosine deaminase deficiency (Giblett et al., 1974), which is thought to be due to the toxic build-up of substrates.
In plants, UMP kinase has received only marginal study. UMP kinase is elevated during seedling development (Deng and Ives, 1972; Mazus and Buchowicz, 1972) and fruit ontogeny (Rudd and Fites, 1972; Deng and Ives, 1975). Because UMP kinase is likely to be as important in plants as it is in microorganisms, we have isolated the cDNA for the A. thaliana UMP/CMP kinase, expressed the coding region in E. coli, and characterized the resulting plant enzyme.
MATERIALS AND METHODS
The pGEX-4T-3 and glutathione-Sepharose 4B were purchased from Pharmacia. The vector pT7-Blue was from Novagen (Madison, WI). Enterokinase was from Biozyme Laboratories (San Diego, CA). Restriction enzymes, T4 ligase, and Taq polymerase were from Promega. All other enzymes and reagents were obtained from Sigma unless otherwise noted. The Arabidopsis thaliana cDNA library in a yeast transformation vector was previously described (Minet et al., 1992). Oligonucleotides were synthesized at the Iowa State University (Ames) Nucleic Acid Facility.
Strains
Saccharomyces cerevisiae FL100a was the wild-type strain used for all yeast-related manipulations. The S. cerevisiae ura6 strain was derived from FL100a and displayed a conditional thermosensitive and 5-fluorouracil-resistant phenotype (Liljelund and Lacroute, 1986). The strain used in these studies was also auxotrophic for His and Trp. Genotypes of the yeast strains were determined by plating at either the permissive or restrictive temperature on media lacking various ingredients. The Escherichia coli strain XL1-Blue was used for all bacterial manipulations.
Yeast Methods
Yeast transformation was conducted as described previously (Gietz et al., 1992). S. cerevisiae auxotrophic mutants were complemented by A. thaliana cDNAs as previously described (Minet et al., 1992). To rescue the plasmids from yeast, a loopful of yeast cells was suspended in 0.4 mL of 10 mm Tris, pH 8.0, 1 mm EDTA, and 0.4 m NaCl and vortexed (2 min) with 200 μL of glass beads. Following vortexing, an equal volume of phenol/chloroform (1:1) was added and the vortexing was repeated. After the sample was centrifuged, 250 μL of supernatant was removed and DNA was precipitated by the addition of 500 μL of ethanol. The sample was washed with 70% ethanol, and the DNA was resuspended in water and used for electroporation of E. coli (Ausubel et al., 1993).
Recombinant DNA Methods
DNA-sequencing reactions were performed using the Prism Dye-Deoxy cycle sequencing kit (Applied Biosystems). The reactions were run on a DNA sequencer (Prism 377, Perkin-Elmer). Sequencing was initiated from known vector sequences. On the basis of these runs, oligonucleotide primers specific to the A. thaliana UMP/CMP kinase gene sequence were constructed. Both strands were completely sequenced.
The 606-bp UMP kinase-coding region was PCR amplified from the pAt-URA6 clone using a pair of oligonucleotides, LZ2080 (5′-GCGGATCCGATGACGATGACAAGATGGGATCTGTTGATGCTGCT-3′) and LZ2081 (5′-CGGCTCGAGCTACTAGGCTTCAACCTTCTCAGC-3′). The PCR product was cloned into the pT7-Blue(R) T-vector to form the vector pRT379. These oligonucleotide primers introduced into the PCR produced unique BamHI and XhoI sites at the ends of the PCR fragment, an enterokinase site for cleavage of the GST fusion protein, and an additional stop codon. Following cloning of the PCR product in pRT379, the sequence of the coding region was confirmed by completely sequencing the insert. The expression vector pRT380 was subsequently prepared by inserting the 637-bp BamHI/XhoI fragment from pRT379 into the BamHI/XhoI sites of pGEX-4T-3. Again, the sequence of the coding region was confirmed by completely sequencing the insert.
Expression in E. coli
The vector pRT380 was transformed into E. coli XL1-Blue for the production of the fusion protein. To induce the fusion protein, 5 mL of overnight culture was diluted into 500 mL of 2YT medium (1.6% bactotryptone, 1.0% yeast extract, and 0.5% NaCl, pH 6.0) and grown at 37°C until mid-log phase. IPTG was then added to a final concentration of 1 mm. Growth continued for 3 h at 37°C. Cells were harvested by centrifugation at 4000g for 10 min. The cell pellet was stable when stored at −20°C.
Purification of UMP/CMP Kinase
Frozen cells were thawed in 10 mL of PBS containing 25 mg of lysozyme. After 30 min at room temperature, the cells were sonicated on ice. DNase I (2 mg) in 0.8 m MgCl2 was added and the cell sonicate was incubated at room temperature for 10 min. Cellular debris were removed by centrifugation at 12,000g for 40 min, and the supernatant was used to resuspend 0.3 mL of packed PBS-washed glutathione-Sepharose beads. The beads and supernatant were incubated overnight at room temperature on a rotating wheel. The beads were removed by centrifugation and washed with PBS until the supernatant was clear (usually five times). The fusion protein could be eluted by the addition of 10 mm glutathione. However, for most studies, the UMP/CMP kinase domain was removed from the bound fusion protein by resuspending the beads in 0.35 mL of enterokinase buffer (25 mm Tris, pH 7.5, and 10 mm CaCl2) and digesting with 1000 units of enterokinase until completion at room temperature on a rotating wheel.
After digestion, the supernatant was recovered by centrifugation and applied to a reverse-phase C18 HPLC column (250 × 10 mm, i.d.; Vydac, Hesperia, CA). The column was eluted at a rate of 4 mL min−1 with a programmed elution profile. Solvent A was 0.1% trifluoroacetic acid in water and solvent B was 0.08% trifluoroacetic acid in acetonitrile. The enzyme was eluted by a series of linear gradients: 0 to 5 min, gradient from 20 to 40% solvent B; 5 to 23 min, gradient from 40 to 43% solvent B; the column was cleaned and equilibrated for further separations by two gradients: 23 to 26 min, gradient from 43 to 100% solvent B; and 26 to 28 min, gradient back to 20% solvent B. The UMP/CMP kinase was eluted at 41% solvent B. Following elution, the enzyme fraction was dialyzed against 20 mm Mes, pH 6.5, at 4°C for 24 h and then concentrated to about 0.5 mL using a centrifugal concentrator (Centricon-10, Amicon, Beverly, MA).
Protein Methods
Protein concentration was determined by the method of Bradford (1976) with BSA as a standard. SDS-PAGE was performed according to the method of Laemmli (1970). N-terminal amino acid analysis was performed in the Iowa State University Protein Facility (Ames) by sequential Edman degradation on a protein sequencer (model 477A, Applied Biosystems) and a protein analyzer (model 120A, Applied Biosystems). Amino acid composition was performed with phenyl isothiocyanate-derivatized amino acids following hydrolysis of purified UMP/CMP kinase in 6 n HCl.
Matrix-assisted laser-desorption ionization MS was used for determining molecular mass. Protein samples of 0.5 to 1.0 μL containing about 0.5 to 1 μg of protein were loaded with 0.5 μL of freshly prepared 3,5-dimethoxy-4-hydroxy cinnamic acid matrix onto a time-of-flight mass analyzer (Lasermat 2000 MALDI, Finnigan, Madison, WI). The collected data were analyzed using data processing software (Lasermat 2000, Finnigan). Lysozyme was used as an internal calibration standard.
Enzymatic activity was determined spectrophotometrically by measuring the formation of ADP and UDP at 23°C with a coupled-enzyme assay (Agarwal et al., 1978). Quantitation was performed by following the decrease of NADH A340. The initial-rate data were analyzed for kinetic mechanisms (Fromm, 1975) by using a computer program written in the MINITAB language with an α-value of 2.0 (Siano et al., 1975).
RESULTS
Cloning by Complementation
Yeast genetic crosses were made between ura2− and ura6-15(Ts) to provide progeny blocked in de novo pyrimidine biosynthesis as well as in the conversion of UMP into UDP. The inclusion of the ura2− mutation provided a better screen for ura6–15(Ts). These progeny were unable to make pyrimidines via the de novo pyrimidine biosynthetic pathway, but could be rescued at the permissive temperature by the addition of uracil through the salvage pathway. Strain ura6-15A, which has an FL100a, ura6-15Ts,ura2−,trp−,his− genotype, was used for all transformation experiments.
The yeast strain was transformed with an Arabidopsis cDNA library in the vector pFL61 (Minet et al., 1992). Approximately 54,000 individual transformants were screened for colonies at the restrictive temperature. Two colonies were found from the 54,000 transformants. DNA was isolated from each of these yeast strains and E. coli was transformed by electroporation. Both colonies yielded the identical cDNA clone when analyzed by DNA sequence analysis. After growth in E. coli, both clones were reisolated and used to transform the original ura6–15A strain to verify that the clones produced the URA+ phenotype. The plasmid containing this clone was termed pAt-ura6.
Analysis of the UMP/CMP Kinase mRNA
The DNA sequence of the pAt-ura6 insert is presented in Figure 1. This presumptive Arabidopsis UMP/CMP kinase cDNA sequence has been deposited in GenBank as accession no. AF000147 and is 895 nucleotides long. It shows a 60-nucleotide 5′-untranslated region, a coding region of 606 nucleotides, and a 175-nucleotide 3′-untranslated region extending to the poly(A+) site at position 874. This cDNA did not show a typical polyadenylation signal. The closest to the consensus is AATTTT, which is duplicated at positions 836 to 841 and 853 to 858. DNA matrix analysis revealed no significant regions of internal duplication or inverted repeats within the cDNA sequence.
Figure 1.

cDNA sequence of the A. thaliana UMP/CMP kinase. The nucleotide number is presented at the end of each row. The deduced amino acid sequence is presented below the DNA sequence and the amino acids are numbered above the DNA sequence. Asterisks indicate the amber (UAG) codon.
The translation start codon correlates well with the rules of Kozak (1986). The sequence at the translation start differs from the eukaryotic consensus at only a single nucleotide (ACAATGG). Consequently, this cDNA is expected to be translated very efficiently.
Analysis of the UMP/CMP Kinase Protein
The deduced amino acid sequence of the Arabidopsis UMP/CMP kinase is also presented in Figure 1. The protein is 202 amino acids long. There is a pair of conserved Cys residues that are shared among all of the eukaryotic UMP kinases. However, it has been proposed that the conserved Cys residue found at position 31 in the Arabidopsis protein has a free SH group (Weismüller et al., 1990).
The Arabidopsis UMP/CMP kinase amino acid sequence is very similar to UMP kinases from other eukaryotic sources (Fig. 2). UMP kinases have been isolated from yeast (S. cerevisiae), a cellular slime mold (D. discoideum), and a mammal (Sus scrofa). Each of these enzymes shows 47 to 53% amino acid identity with each other. It was interesting that 32.2% of the amino acid residues are identical among all of the four proteins. If conservative substitutions are permitted, then the homology among the four proteins increases to 47.5%.
Figure 2.

Comparison of pyrimidine monophosphokinases. A, Comparison of the amino acid identity between the various pyrimidine monophosphate kinases was performed using the PileUp program from the Genetics Computer Group (Madison, WI). For this analysis, both UMP kinases and TMP kinases were included. UMP kinases were included from archebacterial, eubacterial, and eukaryotic (plant, mammal, yeast, and mold) sources. The GenBank accession numbers for the sequences used in this study are as follows: Saccharomyces pombe TMP kinase, L04126; S. cerevisiae TMP kinase, K02116; Homo sapiens TMP kinase, L16991; Thermus aquaticus UMP kinase, X83598; E. coli UMP kinase, X78809; S. cerevisiae UMP kinase, M69295; S. scrofa UMP kinase, D29655; D. discoideum UMP kinase, M34568; and A. thaliana UMP/CMP kinase, AF000147. B, Alignment of amino acid sequences of eukaryotic UMP kinases from a plant (A. thaliana), a mammal (S. scrofa), a yeast (S. cerevisiae), and a cellular slime mold (D. discoideum). Identical residues are marked by dots (•) and conservative substitutions are indicated with plus signs (+).
There is a conserved region near the N terminus that is maintained in nucleotide-binding proteins (Möller and Amons, 1985). This sequence, GGPGS/AGK, is also preserved in the eukaryotic nucleotide monophosphokinases. In crystallographic studies of adenylate kinase, Pai et al. (1977) found that this loop anchors the γ-phosphate moiety of ATP.
Expression in E. coli
After characterization of the Arabidopsis UMP/CMP kinase cDNA, we constructed a vector to express the Arabidopsis protein in E. coli. The structure of the GST fusion protein used in this work is shown in Figure 3A. The nucleotide sequence of the GST-kinase and the kinase-vector junction in pRT380 are presented in Figure 3B. Because the plant UMP/CMP kinase does not contain an N-terminal signal sequence, the vector was designed so that enterokinase would cleave at the exact N terminus of the enzyme to yield the full-length UMP/CMP kinase protein.
Figure 3.

Sequence of portions of the construct pRT380. A, Structural model of the GST-kinase fusion protein showing the amino acid sequence at the site of fusion. B, Nucleotide and translated amino acid sequences of the pRT380 construct at the site of GST-UMP/CMP kinase fusion and the 3′ end of the fusion protein. The sequences up through the BamHI site are from the pGEX-4T-3 vector and encode the C terminus of the GST. The aspartate-rich sequence extending from the BamHI site to the Met is the site of enterokinase recognition. Cleavage occurs after the Lys and is indicated by the arrow. The sequence Met-Gly-Ser-Val-Asp represents the first five amino acids of the Arabidopsis UMP/CMP kinase. The sequence of the remainder of the protein is identical to that presented in Figure 1. After the stop codon, we introduced a second TAG stop codon and an XhoI site. Downstream of the XhoI site is the pGEX-4T-3 vector.
After transfer of the vector to XL1-Blue cells, the induction of these cells by IPTG resulted in the accumulation of a 51-kD protein (Fig. 4, lane 2) that was not present in host cells without plasmid (data not shown) or uninduced cells containing pRT380 (Fig. 4, lane 1). After sonication of the IPTG-induced cells, enzymatic assays showed the presence of a new UMP kinase activity that was not present in control cells (data not shown). This new UMP kinase activity arises from the GST-kinase fusion protein.
Figure 4.

SDS-PAGE of UMP/CMP kinase. All samples were analyzed on a 12% polyacrylamide gel and stained with Coomassie brilliant blue R-250. Lane 1, Cell sonicates of E. coli XL1-Blue cells containing the plasmid pRT380 with no added IPTG; lane 2, cell sonicates of E. coli XL1-Blue cells containing the plasmid pRT380 after the addition of IPTG; lane 3, GST-UMP/CMP kinase fusion protein eluted from glutathione-Sepharose beads; lane 4, GST-UMP/CMP kinase fusion protein eluted from glutathione-Sepharose beads and then digested with enterokinase; lane 5, digestion of GST-UMP/CMP kinase fusion protein with enterokinase while fusion protein was still attached to the beads; lane 6, HPLC purified UMP/CMP kinase. Molecular mass markers used were: phosphorylase b, 97.4 kD; BSA, 66 kD; ovalbumin, 45 kD; carbonic anhydrase, 31 kD; trypsin inhibitor, 21.5 kD; and lysozyme, 14.4 kD.
Purification of UMP/CMP Kinase
The various steps in the purification of the enzyme were monitored by SDS-PAGE, as shown in Figure 4. A 51-kD protein was observed upon induction of the cells containing pRT380 with IPTG (Fig. 4, compare lane 2 [induced] with lane 1 [uninduced]). This fusion protein could be selectively bound and eluted from the glutathione-Sepharose beads (Fig. 4, lane 3). Enterokinase digestion of the fusion protein resulted in the conversion of the 51-kD protein into two subunits (Fig. 4, lane 4). For most studies, however, the GST-kinase fusion protein was proteolyzed while still bound to glutathione-Sepharose beads, which releases the 22.5-kD protein while the 29-kD protein remains bound to the glutathione-Sepharose (Fig. 4, lane 5). After enzymatic cleavage of the fusion protein the UMP/CMP kinase was purified by HPLC. A typical HPLC elution profile is shown in Figure 5. The eluted UMP/CMP kinase protein was collected, dialyzed, and concentrated. An aliquot of the purified UMP/CMP kinase after HPLC elution is shown in Figure 4, lane 6.
Figure 5.

HPLC purification of UMP/CMP kinase. The GST-UMP/CMP kinase fusion protein was digested with enterokinase while still attached to the glutathione-Sepharose beads. The eluate was applied to a 250- × 10-mm, i.d., reverse-phase C18 column. A214 was monitored. The eluted protein peak containing UMP/CMP kinase activity was collected (B = 41%). The hatched area under the peak illustrates the collected pool of UMP/CMP kinase.
About 0.5 to 1 μg of the HPLC-purified UMP/CMP kinase was subjected to MS (Fig. 6). The subunit molecular mass of the purified UMP/CMP kinase [MH]+ peak was 22,405 ± 185 D (n = 4). Secondary peaks [MH2]2+ and [2MH]+ were identified at 11,286 ± 94 and 44,894 ± 333 D. Therefore, the subunit molecular mass found from the average of these masses, 22, 448 ± 44 D, is in good agreement with the molecular mass of UMP/CMP kinase predicted from the cDNA (22,482 D).
Figure 6.

Typical mass spectrum of A. thaliana UMP/CMP kinase. The [MH2]2+ peak is at 11,286, the [MH]+ peak is at 22,405, and the [2MH]+ peak is at 44,984.
Characterization of the Purified UMP/CMP Kinase Protein
To confirm that the expressed protein was indeed the UMP/CMP kinase, the N terminus of the purified protein was sequenced. The N-terminal sequence MGSVDAANGSGKKPT was found to be identical to the amino acid sequence deduced from the cDNA. This sequence also indicates that the enterokinase cuts exactly at the predicted site, leaving no extra amino acids at the N terminus.
The amino acid composition of the expressed protein was also determined. About 1 μg of HPLC-purified UMP/CMP kinase was hydrolyzed in 6 n HCl for 65 min at 150°C (PICO-TAG Workstation, Waters). The free amino acids were derivatized under basic conditions with phenyl isothiocyanate in a derivatizer (model 420A, Applied Biosystems) and separated on a narrow-bore C18 column. The phenylthiocarbamyl chromophore was detected at A254. Quantitation was performed from the A254 and comparison with a norleucine internal standard. The total amino acid composition was determined independently two times (data not shown). Gln and Asn could not be detected because both amino acids were converted to the corresponding carboxylic acids. Therefore, the resulting Asp and Glu were the sum of Asp and Asn (Asx) and of Glu and Gln (Glx), respectively. The number of Trp and Cys residues could not be determined by this method because they were destroyed during acid hydrolysis. There was good agreement between the independent analyses. A χ2 analysis indicated with greater than 99% confidence that the composition found in the purified protein was the same as that of the predicted protein, confirming that the purified protein was the Arabidopsis UMP/CMP kinase.
Initial Rate Studies
Once we were convinced that the purified protein was indeed the Arabidopsis UMP/CMP kinase, the kinetic parameters of this purified protein were determined (Table I). The enzyme can utilize CMP as a phosphate donor almost as well as it does UMP. The Km for UMP was 5- to 6-fold higher than that for ATP. Therefore, the enzyme binds ATP much more tightly than UMP. However, the Km for CMP was about equal to the Km for ATP, indicating that the enzyme binds CMP as tightly as ATP. The kcat obtained for each substrate was relatively the same.
Table I.
Kinetic parameters of A. thaliana UMP/CMP kinase
| Activity | Specific Activity |
Km
|
kcat | ||
|---|---|---|---|---|---|
| ATP | UMP | CMP | |||
| units mg−1 × 10−2 | μm | s−1 | |||
| UMP kinase | 3.67 ± 0.12 | 29.3 ± 2.8 | 152.9 ± 14.5 | – | 7.64 ± 0.24 |
| CMP kinase | 4.12 ± 0.05 | 291.7 ± 24.3 | – | 266.4 ± 21.7 | 8.57 ± 0.11 |
The standard enzyme reaction contained 50 mm Mes, pH 6.5, 50 mm KCl, 2 mm MgCl2, 1 mm PEP, 0.2 mm NADH, 3.5 units of pyruvate kinase, and 5 units of lactate dehydrogenase in a final volume of 1 mL. When ATP was used as the variable substrate, UMP and CMP were fixed at 400 and 450 μm, respectively. When UMP or CMP were used as the variable substrate, ATP-Mg2+ was fixed at 300 μm for UMP and at 800 μm for CMP. The reaction was started by the addition of UMP/CMP kinase. The change in A340 was recorded. One unit of UMP kinase is defined as the amount of enzyme that catalyzed the formation of 1 μmol of UDP or CDP per min.
pH Dependence of the Enzyme
The values of kcat were determined at a series of different pH values from 5.5 to 8.0. All pH buffers contained both 50 mm Mes and 50 mm Hepes. When ATP was the variable substrate, the concentration of UMP was fixed at 400 μm. When UMP was the variable substrate, the concentration of ATP was fixed at 300 μm. The values of kcat were obtained at pH 5.5, 6.5, 7.0, 7.5, and 8.0 when ATP or UMP was the variable substrate (data not shown). These analyses showed that optimal activity was achieved at pH 6.5 for both ATP and UMP.
Thermal Stability
The enzyme was heated for 10 min at various temperatures between 30 and 100°C. Residual activity was determined with 300 μm ATP and 400 μm UMP (data not shown). One-half of the enzymatic activity was maintained when the enzyme was heated at 58°C. Although still high, this is about 10°C lower than that of the E. coli enzyme.
Kinetic Mechanism
To better understand the kinetic mechanism, enzyme assays were conducted in which one substrate was varied at different fixed concentrations of other substrates. Double-reciprocal plots of these data showed that, when the ATP concentration was varied at different fixed levels of UMP, a family of lines intersecting in the second quadrant was obtained (Fig. 7A). A similar family of lines was obtained for various 1/UMP concentrations at different fixed levels of ATP (Fig. 7B). These families of lines are theoretical, obtained from Equation 1 when n = 1. The data obtained from the experimental evaluation matches the theoretical family of lines for both ATP and UMP. Based on the closeness of this fit, we conclude that the enzyme fits a random Bi-Bi mechanism. The data fit the random Bi-Bi mechanism shown in Equation 1 (Fromm, 1975):
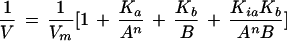 |
1 |
where V, Vm, A, B, Ka, Kb, and Kia represent the initial velocity, maximum velocity, concentration of free ATP, concentration of free UMP, Michaelis constant for ATP, Michaelis constant for UMP, and dissociation constant for ATP, respectively. The Hill coefficient for ATP is represented by n. Kib is the dissociation constant for UMP and substitutes into Equation 1 in place of Kia. Kia and Kib were determined experimentally. Kia (for ATP) = 14.9 ± 1.3 μm and Kib (for UMP) = 110.2 ± 8.7 μm.
Figure 7.

Analysis of kinetic mechanism. A, Double-reciprocal plot of initial velocity versus ATP concentrations. For these experiments the concentrations of UMP were 40 (▪), 60 (+), 90 (▴), and 120 μm (□). The lines are theoretical based on Equation 1 where n = 1. The points were experimentally determined. B, Double-reciprocal plot of initial velocity versus UMP concentrations. For these experiments, the concentrations of ATP were 15 (▪), 25 (+), 40 (▴), and 65 μm (□). The lines are theoretical based on Equation 1 where n = 1. The points were experimentally determined.
Alternative Substrates
To examine the specificity of the UMP/CMP kinase in more detail, various phosphate donors and acceptors were evaluated for their ability to function in this enzyme assay. To evaluate the phosphate acceptors, 300 μm ATP was used as the donor with different monophosphate acceptors at 400 μm. Only UMP and CMP are effective phosphate acceptors. Neither orotidine monophosphate nor TMP were effective as a phosphate acceptor. The presence of the 2′ hydroxyl on the Rib moiety is also important because dUMP and dCMP are 30-fold less active than their ribosyl analogs (data not shown). None of the purine monophosphates tested (AMP, GMP, inosine monophosphate, and xanthine monophosphate) functioned effectively as a phosphate acceptor. Similarly, other nucleotide triphosphates were examined for their ability to function as phosphate donors to UMP. For evaluation, 300 μm phosphate donors were used with UMP at 400 μm. In this analysis, only ATP and dATP were effective phosphate donors, with dATP only half as effective as ATP. Other purine triphosphates (GTP, dGTP, inosine triphosphate, and xanthine triphosphate) were 20- to 30-fold less active than ATP. Pyrimidine triphosphates (UTP, CTP, dCTP, and deoxyribothymine triphosphate) were 70-fold less active than ATP.
It is known that the E. coli UMP kinase is allosterically regulated by both GTP and UTP (Serina et al., 1995). With the E. coli enzyme, 100 μm GTP activates the enzyme 5-fold and 100 μm UTP down-regulates the enzyme 5-fold. We therefore explored whether these nucleotides affected the activity of the Arabidopsis enzyme. Neither of these nucleotides significantly affected the activity of the Arabidopsis UMP/CMP kinase, even at extremely high levels. At 3 mm GTP enzyme activity increased by 25%, but this is much lower than the 5-fold activation of the E. coli enzyme that occurs with lower levels of GTP (Serina et al., 1995). Conversely, UTP also affects the UMP/CMP kinase activity but, again, only by 25% at UTP concentrations as high as 3 mm. Therefore, whereas GTP and UTP both affect the plant enzyme, the effect of each of these nucleotides even at very high levels is significantly less than the effect on the prokaryotic enzyme.
Kinetics of Ap5A Inhibition
We also examined the inhibition of the Arabidopsis UMP/CMP kinase with the bifunctional inhibitor Ap5A (Fig. 8). The inhibitor concentration required for 50% inhibition was determined by fixing ATP at 300 μm and UMP at 400 μm and varying the Ap5A concentration between 0 and 100 μm. The inhibitor concentration for 50% displacement of Ap5A on the Arabidopsis enzyme was 14 μm, which is lower than that found on the enzyme from either D. discoideum (Weismüller et al., 1990) or E. coli (Serina et al., 1995). The inhibition mechanism of Ap5A was determined by fixing one substrate at a saturating concentration and varying the concentration of the other substrate at different fixed concentrations of Ap5A. The families of lines shown in Figure 8 are theoretically fit to Equation 2 (Fromm, 1975) and the points were experimentally derived.
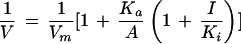 |
2 |
where I, Ka, and Ki represent the concentration of free Ap5A, the Michaelis constant for ATP (or UMP), and the inhibition constant for Ap5A, respectively. The inhibition by Ap5A when ATP was the variable substrate is shown in Figure 8A, and the inhibition of Ap5A when UMP was the variable substrateis shown in Figure 8B. These data clearly demonstrate that Ap5A is a competitive inhibitor of both ATP and UMP. The Ki values for ATP and UMP were 1.20 ± 0.02 and 6.53 ± 0.03 μm, respectively.
Figure 8.

Kinetics of Ap5A inhibition. A, Double-reciprocal plot of initial velocity versus ATP concentrations in the presence of Ap5A inhibitor. UMP concentrations were fixed at 400 μm. The concentrations of Ap5A were 0 (▪), 2 (+), 4 (▴), and 6 μm (□). The lines are theoretical based on Equation 2. The points were experimentally determined. B, Plot of reciprocal of initial velocity of UMP/CMP kinase versus reciprocal of UMP concentrations in the presence of Ap5A inhibitor. ATP concentrations were fixed at 300 μm. The concentrations of Ap5A were 0 (▪), 8 (+), 16 (▴), and 24 μm (□). The lines are theoretical based on Equation 2. The points were experimentally determined.
DISCUSSION
We have isolated the cDNA encoding the A. thaliana UMP/CMP kinase by complementation of an S. cerevisiae UMP kinase mutant. Complementation of known yeast mutants is a very effective way to obtain eukaryotic genes. Following isolation, the plant cDNA was characterized by sequencing. The full-length cDNA encodes a 202-amino acid protein that is closely related to UMP kinases from other eukaryotic sources. The plant enzyme, however, did not share high identity with the bacterial or archebacterial UMP kinases. In previous studies, the E. coli UMP kinase was found to belong to the aspartokinase family (Serina et al., 1995).
When the intracellular location of the Arabidopsis UMP kinase was examined using the online analysis tool PSORT (Nakai and Kanehisa, 1992), a cytosolic localization for the enzyme was predicted. A cytosolic localization has also been found for the rice adenylate kinase enzyme (Kawai and Uchimiya, 1995). The cytosolic localization is also consistent with the finding that uridine nucleotides are predominantly located in the cytosol (Dancer et al., 1990).
After characterization of the cDNA, the coding region was expressed by fusing it to GST. Following expression, the fusion protein was cleaved and the Arabidopsis UMP/CMP kinase was purified by HPLC. The molecular mass, amino acid composition, and N-terminal sequence of the expressed protein all demonstrated that it was indeed UMP/CMP kinase. When examined for enzyme activity, the purified enzyme was found to have both UMP kinase and CMP kinase activity. Kinetic parameters were determined for the plant enzyme and several differences from the E. coli enzyme were noted.
First, the E. coli enzyme shows remarkable thermal stability in the absence of protective agents, being stable up to 65°C. Although relatively thermostable, the Arabidopsis enzyme did not show the same degree of stability. Second, the plant enzyme is not allosterically regulated in the same way that the prokaryotic enzyme is regulated. At concentrations that dramatically affect the enzyme activity of the E. coli UMP kinase, neither GTP nor UTP have a significant effect on the plant enzyme. Indeed, at 30 times the level that affects the bacterial enzyme, the plant enzyme is affected by only about 25%. Bourne et al. (1991) have identified a pair of sequences, Asp-77-His-Met-Gly-80 and Thr-165-Lys-Val-Asp-168, that are conserved in GTP-binding proteins. Both of these sequences are conserved in the E. coli UMP kinase (Serina et al., 1995). However, neither of them is present in the Arabidopsis UMP/CMP kinase. Thus, by both sequence identity and experimental observation, allosteric regulatory sites are not present in the plant enzyme, and the Arabidopsis enzyme seems to be both functionally and structurally different from the prokaryotic enzyme.
The Km values for UMP and CMP indicate that the Arabidopsis enzyme can utilize both pyrimidine monophosphates equally well as phosphate acceptors. This is similar to the enzyme from rat Novikoff ascites tumors (Orengo and Maness, 1978), but the mouse enzyme utilizes UMP nearly twice as effectively as CMP as a phosphate acceptor (Andersen, 1978a). The deoxy forms are almost totally ineffective as phosphate acceptors in the Arabidopsis enzyme. Two important conclusions can be drawn from this finding. First, a different enzyme must be responsible for the conversion of dCMP into dCDP. This is different from the mouse and the Tetrahymena pyriformis enzyme, in which dCMP can also act as a phosphate acceptor (Andersen, 1978b). Second, because the nucleotide monophosphate-binding pocket does not discriminate between UMP and CMP, the exclusion of deoxynucleotides results not only in the exclusion of dCMP but also in the exclusion of dUMP. Thus, the conversion of dUMP into dUDP does not occur, thereby forcing the conversion of dUMP into TMP by thymidylate synthase. Furthermore, TMP is ineffective as a phosphate acceptor, indicating that, as in yeast (Jong et al., 1984), a separate TMP kinase must also exist.
Structural studies of adenylate kinase from both E. coli (Müller and Schulz, 1992) and beef heart (Diederichs and Schulz, 1991) revealed that the 2′ hydroxyl of the phosphate acceptor forms a strong hydrogen bond with an α-chain carboxyl. It is likely that similar interactions are required in the plant UMP/CMP kinase, thereby biasing the specificity against the deoxynucleotides.
Eukaryotic UMP kinases in general have a higher specificity for ATP as the phosphate donor, with dATP effective at about 10% the level of ATP (Orengo and Maness, 1978). This was also found for the plant enzyme. Other nucleotide triphosphates were essentially ineffective as phosphate donors.
The plant UMP kinase has a high degree of identity with other eukaryotic UMP kinases and are expected to share significant structural identity. The UMP kinase enzyme from yeast has been purified and characterized (Ma et al., 1990). The crystal structure of this enzyme has been solved with substrates in place (Müller-Dieckmann and Schulz, 1994, 1995). The substrates are held in position by numerous favorable contacts with the protein. Most of these contacting residues are conserved between the yeast enzyme and the plant enzyme.
Structural studies have also demonstrated that the UMP-binding pocket of the yeast enzyme is of sufficient size to accommodate an AMP moiety (Müller-Dieckmann and Schulz, 1994, 1995). This explains the high activity of the yeast UMP kinase for AMP. The finding that Ap5A is a competitive inhibitor of UMP with a micromolar Ki indicates that the Arabidopsis enzyme has a UMP-binding pocket that is also sufficiently large to accommodate an AMP moiety. However, the Arabidopsis enzyme has less than 0.5% activity with AMP. Therefore, the structure of the UMP-binding pocket of the Arabidopsis enzyme will be particularly interesting to understand. The radiography-crystallographic studies of the yeast UMP kinase have failed to explain the specificity of this enzyme for UMP. Those residues that have been shown to line the uracil-binding pocket of the yeast UMP kinase (Ala-47, Leu-51, Ile-75, Val-76, Thr-81, Phe-105, Arg-107, and Gln-111) are, with only one exception (Asn substitutes for Gln), completely conserved in the Arabidopsis enzyme. Note that this same substitution (Asn for Gln) is found in the D. discoideum enzyme, which also shows a high degree of substrate discrimination for UMP over AMP (Weismüller et al., 1990).
The enzymatic mechanism is also relatively well understood for the yeast enzyme. The transition state of phosphoryl transfer is maintained by a scaffold of interactions, including the Cα-backbones of the CORE domains and a series of six positively charged residues (Lys-29, Arg-52, Arg-107, Arg-142, Arg-148, and Arg-159) positioned to coordinate the phosphates. All of these residues are conserved in the Arabidopsis enzyme. The interaction of the substrate-fixed phosphates with the yeast UMP kinase is virtually identical to E. coli adenylate kinase (Müller-Dieckmann and Schultz, 1994), indicating the widespread and general conservation of this enzymatic mechanism. Because of the high degree of conservation in these important contacting residues, it is probable that the Arabidopsis enzyme also shares this enzymatic mechanism.
ACKNOWLEDGMENTS
We thank Dr. Michelle Minet for her help with yeast molecular biology techniques and Dr. Herbert Fromm for his help with analysis of the kinetics of UMP/CMP kinase. We would also like to thank Dr. Roger S. Goody (MPI fuer Molekulare Physiologie, Reinlanddamm) for kindly supplying the Ap5A.
Abbreviations:
- Ap5A
P1, P5-di(adenosine-5′) pentaphosphate
- GST
glutathione S-transferase
- IPTG
isopropyl-β-d-thiogalactoside
Footnotes
This research was supported by grant no. 91-37301-6208 from the U.S. Department of Agriculture. This is paper no. J-17416 from the Iowa Agriculture and Home Economics Experiment Station.
LITERATURE CITED
- Agarwal KC, Miech RP, Parks RE. Guanylate kinase from human erythrocytes, hog brain and rat liver. Methods Enzymol. 1978;51:483–490. doi: 10.1016/s0076-6879(78)51066-5. [DOI] [PubMed] [Google Scholar]
- Anderson EP. Uridine-cytidine kinase from a murine neoplasm. Methods Enzymol. 1978a;51:314–321. doi: 10.1016/s0076-6879(78)51042-2. [DOI] [PubMed] [Google Scholar]
- Anderson EP. UMP-CMP kinase from Tetrahymena pyriformis. Methods Enzymol. 1978b;51:331–337. doi: 10.1016/s0076-6879(78)51044-6. [DOI] [PubMed] [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (1993) Current Protocols in Molecular Biology. John Wiley & Sons, New York, pp 1.8.4
- Baranowska H, Zaborowska D, Jachymczyk WJ, Zuk J. Role of the CDC8 gene in the repair of single strand breaks in DNA of the yeast Saccharomyces cerevisiae. Curr Genet. 1990;18:175–179. doi: 10.1007/BF00318376. [DOI] [PubMed] [Google Scholar]
- Baranowska H, Zuk J. Chemical mutagenesis and DNA synthesis in cdc8, a DNA replication mutant of Saccharomyces cerevisiae. Curr Genet. 1991;20:471–474. doi: 10.1007/BF00334774. [DOI] [PubMed] [Google Scholar]
- Birkenmeyer LG, Hill JC, Dumas LB. Saccharomyces cerevisiae CDC8 gene and its product. Mol Cell Biol. 1984;4:583–590. doi: 10.1128/mcb.4.4.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: conserved structure and molecular mechanism. Nature. 1991;349:117–127. doi: 10.1038/349117a0. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principles of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Dancer J, Neuhaus HE, Stitt M. Subcellular compartmentation of uridine nucleotides and nucleoside-5′-diphosphate kinase in leaves. Plant Physiol. 1990;92:637–641. doi: 10.1104/pp.92.3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q-I, Ives DH. Modes of nucleoside phosphorylation in plants: studies on the apparent thymidine kinase and true uridine kinase of seedlings. Biochim Biophys Acta. 1972;277:235–244. [PubMed] [Google Scholar]
- Deng Q-I, Ives DH. Non-allosteric regulation of the uridine kinase from seeds of Zea mays. Biochim Biophys Acta. 1975;377:84–94. doi: 10.1016/0005-2744(75)90289-2. [DOI] [PubMed] [Google Scholar]
- Diederichs K, Schulz GE. The refined structure of the complex between adenylate kinase from beef heart mitochondrial matrix and its substrate AMP at 1.85 Å resolution. J Mol Biol. 1991;217:541–549. doi: 10.1016/0022-2836(91)90756-v. [DOI] [PubMed] [Google Scholar]
- Fromm HJ (1975) Initial Rate Enzyme Kinetics. Springer-Verlag, Berlin, pp 36–37
- Gallango ML, Muller A, Suinaga R. Biochemical characterization of a red cell UMP kinase variant found in the Warao Indians of Venezuela. Biochem Genet. 1978;16:1085–1093. doi: 10.1007/BF00484529. [DOI] [PubMed] [Google Scholar]
- Gallango ML, Suinaga R. Uridine monophosphate kinase polymorphism in two Venezuelan populations. Am J Hum Genet. 1978;30:215–218. [PMC free article] [PubMed] [Google Scholar]
- Giblett ER, Anderson JE, Chen S-H, Teng Y-S, Cohen F. Uridine monophosphate kinase: a new genetic polymorphism with possible clinical implications. Am J Hum Genet. 1974;26:627–635. [PMC free article] [PubMed] [Google Scholar]
- Gietz D, St. Jean A, Woods RA, Schiestl RH. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 1992;20:1425–1428. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jong A, Yeh Y, Ma JJ. Characteristics, substrate analysis and intracellular location of Saccharomyces cerevisiae UMP kinase. Arch Biochem Biophys. 1993;304:197–204. doi: 10.1006/abbi.1993.1339. [DOI] [PubMed] [Google Scholar]
- Jong AY, Kuo C-L, Campbell JL. The CDC8 gene of yeast encodes thymidylate kinase. J Biol Chem. 1984;259:11052–11059. [PubMed] [Google Scholar]
- Kawai M, Uchimiya H. Biochemical properties of rice adenylate kinase and subcellular location in plant cells. Plant Mol Biol. 1995;27:943–951. doi: 10.1007/BF00037022. [DOI] [PubMed] [Google Scholar]
- Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986;44:283–292. doi: 10.1016/0092-8674(86)90762-2. [DOI] [PubMed] [Google Scholar]
- Kuo CL, Campbell JL. Cloning of Saccharomyces cerevisiae DNA replication genes: isolation of the CDC8 gene and two genes that compensate for the cdc8–1 mutation. Mol Cell Biol. 1983;3:1730–1737. doi: 10.1128/mcb.3.10.1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Liljelund P, Lacroute F. Genetic characterization and isolation of the Saccharomyces cerevisiae gene coding for uridine monophosphokinase. Mol Gen Genet. 1986;205:74–81. doi: 10.1007/BF02428034. [DOI] [PubMed] [Google Scholar]
- Ma JJ, Huang SH, Jong AY. Purification and characterization of Saccharomyces cerevisiae uridine monophosphate kinase. J Biol Chem. 1990;265:19122–19127. [PubMed] [Google Scholar]
- Mazus B, Buchowicz J. Activity of enzymes involved in pyrimidine metabolism in the germinating wheat grains. Phytochemistry. 1972;11:77–82. [Google Scholar]
- Minet M, Dufour M-E, Lacroute F. Complementation of Saccharomyces cerevisiae auxotrophic mutants by Arabidopsis thaliana cDNAs. Plant J. 1992;2:417–422. doi: 10.1111/j.1365-313x.1992.00417.x. [DOI] [PubMed] [Google Scholar]
- Möller W, Amons R. Phosphate-binding sequences in nucleotide binding proteins. FEBS Lett. 1985;186:1–7. doi: 10.1016/0014-5793(85)81326-0. [DOI] [PubMed] [Google Scholar]
- Müller CW, Schulz GE. Structure of the complex between adenylate kinase from Escherichia coli and the inhibitor Ap5A refined at 1.9 Å resolution. J Mol Biol. 1992;224:159–177. doi: 10.1016/0022-2836(92)90582-5. [DOI] [PubMed] [Google Scholar]
- Muller-Dieckmann HJ, Schultz GE. The structure of uridylate kinase with its substrates, showing the transition state geometry. J Mol Biol. 1994;236:361–367. doi: 10.1006/jmbi.1994.1140. [DOI] [PubMed] [Google Scholar]
- Muller-Dieckmann HJ, Schultz GE. Substrate specificity and assembly of the catalytic center derived from two structures of ligated uridylate kinase. J Mol Biol. 1995;246:522–530. doi: 10.1006/jmbi.1994.0104. [DOI] [PubMed] [Google Scholar]
- Nakai K, Kanehisa M. A knowledge base for predicting protein localization sites in eukaryotic cells. Genomics. 1992;14:897–911. doi: 10.1016/S0888-7543(05)80111-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newlon CS, Fangman WL. Mitochondrial DNA synthesis in cell cycle mutants of Saccharomyces cerevisiae. Cell. 1975;5:423–428. doi: 10.1016/0092-8674(75)90061-6. [DOI] [PubMed] [Google Scholar]
- Orengo A, Maness P. Pyrimidine nucleoside monophosphate kinase from rat liver and rat Novikoff ascites hepatoma. Methods Enzymol. 1978;51:321–331. doi: 10.1016/s0076-6879(78)51043-4. [DOI] [PubMed] [Google Scholar]
- Pai EF, Sachsenheimer W, Schirmer RH, Schulz GE. Substrate positions and induced-fit in crystalline adenylate kinase. J Mol Biol. 1977;114:37–45. doi: 10.1016/0022-2836(77)90281-9. [DOI] [PubMed] [Google Scholar]
- Petersen GM, Silimperi DR, Scott EM, Hall DB, Rotter JI, Ward JI. Uridine monophosphate kinase 3: a genetic marker for susceptibility to Haemophilus influenzae type B disease. Lancet. 1985;2:417–418. doi: 10.1016/s0140-6736(85)92738-2. [DOI] [PubMed] [Google Scholar]
- Prakash L, Hinkle D, Prakash S. Decreased UV mutagenesis in cdc8, a DNA replication mutant of Saccharomyces cerevisiae. Mol Gen Genet. 1979;172:249–258. doi: 10.1007/BF00271724. [DOI] [PubMed] [Google Scholar]
- Rudd TP, Fites RC. Association of thymidine and uridine kinase activities with changes in nucleic acid levels during peanut fruit ontogeny. Phytochemistry. 1972;11:1631–1636. [Google Scholar]
- Schricker R, Magdolen V, Kaniak A, Wolf K, Bandlow W. The adenylate kinase family in yeast: identification of URA6 as a multicopy suppressor of deficiency in major AMP kinase. Gene. 1992;122:111–118. doi: 10.1016/0378-1119(92)90038-q. [DOI] [PubMed] [Google Scholar]
- Seagrave J, Reyes P. Pyrimidine nucleoside monophosphate kinase from rat bone marrow cells: a kinetic analysis of the reaction mechanism. Arch Biochem Biophys. 1987;254:518–525. doi: 10.1016/0003-9861(87)90132-9. [DOI] [PubMed] [Google Scholar]
- Serina L, Blondin C, Krin E, Sismeiro O, Danchin A, Sakamoto H, Giles A-M, Bârzu O. Escherichia coli UMP-kinase, a member of the aspartokinase family is a hexomer regulated by guanine nucleotides and UTP. Biochemistry. 1995;34:5066–5074. doi: 10.1021/bi00015a018. [DOI] [PubMed] [Google Scholar]
- Serina L, Bucurenci N, Giles A-M, Surewicz WK, Favian H, Mantsch HH, Takahashi M, Petrescu I, Batelier G, Bârzu O. Structural properties of UMP-kinase from Escherichia coli: modulation of protein solubility by pH and UTP. Biochemistry. 1996;35:7003–7011. doi: 10.1021/bi960062v. [DOI] [PubMed] [Google Scholar]
- Siano DB, Zyskind JW, Fromm HJ. A computer program for fitting and statistically analyzing initial rate data applied to bovine hexokinase type III isozyme. Arch Biochem Biophys. 1975;170:587–600. doi: 10.1016/0003-9861(75)90154-x. [DOI] [PubMed] [Google Scholar]
- Valentin-Hansen P. Uridine-cytidine kinase from Escherichia coli. Methods Enzymol. 1978;51:308–314. doi: 10.1016/s0076-6879(78)51041-0. [DOI] [PubMed] [Google Scholar]
- Weismüller L, Noegel AA, Bârzu O, Gerisch G, Schleicher M. cDNA derived sequence of UMP-CMP kinase from Dictyostelium discoideum and expression of the enzyme in Escherichia coli. J Biol Chem. 1990;265:6339–6345. [PubMed] [Google Scholar]
- Yamanaka K, Ogura T, Niki H, Hiraga S. Identification and characterization of the smbA gene, a suppressor of the mukB null mutant of Escherichia coli. J Bacteriol. 1992;174:7517–7526. doi: 10.1128/jb.174.23.7517-7526.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]