

Abstract
Hepatic ischemia-reperfusion (IR) injury is a serious clinical problem. Minimizing the adverse effect of ischemia-reperfusion injury after liver surgery or trauma is an urgent need. It has been proved that besides the effect of regulating the lipid and lipoprotein metabolism, PPARα also undertakes the task of organ protection. In this paper, related literature has been summarized and we come to the conclusion that administration of PPARα agonists can strengthen the antioxidant and anti-inflammation defense system by the upregulation of the expression of antioxidant enzymes and inhibition of NF-κB activity. This may provide a potential clinical treatment for hepatic ischemia-reperfusion injury.
1. Introduction
Hepatic ischemia reperfusion (IR) injury is an important clinical problem complicating liver surgery and transplantation [1]. Depending on whether the ischemia occurs in a warm setting, such as surgical resection or trauma surgery, or cold ischemia, as occurs during liver transplantation, it can be categorized into warm IR and cold storage reperfusion injury [2]. In warm IR, the pathophysiology underlying the injury of hepatic ischemia-reperfusion is complex encompassing a number of mechanisms including oxidant stress, inflammation, and apoptosis. Recently, some researchers have focused on the use of peroxisome proliferator-activated receptor alpha (PPARα) agonist to ameliorate this injury [3, 4]. In this paper, the mechanisms of hepatic ischemia-reperfusion injury, the characteristics of PPARα, and the role of PPARα in warm hepatic ischemia-reperfusion injury have been discussed in the following sections.
2. Mechanisms of Warm Hepatic Ischemia-Reperfusion Injury
With the growth in the field of hepatobiliary surgery, the technique of partial or total vascular occlusion in room temperature has been adapted, and it has enabled surgeons to perform complex procedures such as large liver resections and repairs that otherwise would have resulted in massive hemorrhage and certain death. Apart from the apparent superiority of the technique, there are still some limitations that can cause substantial morbidity and mortality named warm hepatic ischemia-reperfusion injury. Warm hepatic ischemia-reperfusion injury is a complex cascade of events involving a multitude of pathophysiological processes, more than 50% of hepatocytes and sinusoidal endothelial cells (SEC) that formerly considered to undergo apoptosis during the first 24 hours of reperfusion [5, 6]; however, work done by team of Jaspreets Gujral suggested that apoptotic cell death, if it occurs at all, is a very minor aspect of the entire cell death [7, 8]. Based on it we can conclude that the oxidant stress and inflammation are the most critical mechanisms which contribute to the organ pathophysiology after warm hepatic ischemia reperfusion. Work done by Jaeschke et al. [9–12] indicated that there are two distinct phases of liver injury after warm ischemia and reperfusion. The initial phase of injury (<2 hours after reperfusion) is characterized by Kupffer cells activated, and the activated Kupffer cells are a primary source of reactive oxygen-derived free radicals [10, 13]. These free radicals and reactive oxygen species (ROS) are generated to create a severe enough disturbance of the cellular homeostasis. Mitochondria must be a primary target, and its dysfunction may impair the electron flow and enhance superoxide formation [14, 15]. All these will eventually trigger mitochondrial dysfunction and oxidant stress and eventually kill the cell [16, 17]. Studies have shown that it attenuates early hepatocellular injury after hepatic IR that Kupffer cells activity is suppressed by gadolinium chloride or methyl palmitate in mice [18]. Conversely, chemically upregulating Kupffer cell activation aggravates cellular injury and production of reactive oxygen species [19]. In addition, complement is a key factor that contributes to the early activation of Kupffer cells after IR [20]. Kupffer cell generation of superoxide has been shown to be a decisive factor in the injury observed in the early reperfusion period [20, 21]. In addition to Kupffer cell-induced oxidant stress, with increasing length of the ischemic episode, intracellular generation of reactive oxygen by xanthine oxidase and, in particular, mitochondria [22] may also lead to impaired adenosine triphosphate (ATP) production and acidosis result in liver dysfunction and cell injury during reperfusion [23]. Nevertheless, liver architecture assessed histologically shows only minor changes during the period of ischemia and early reperfusion. In the late phase of injury (>6 hours after reperfusion), events occurring during the initial phased serve to initiate and propagate a complex inflammatory response that culminates with the hepatic accumulation of neutrophils [24]. Kupffer cells which can not only directly activate and recruit neutrophils but also serve as the principal source of the oxidant stress during the first period phase of reperfusion injury, the production, and the release of reactive oxygen species can lead to an oxidative shift in the hepatic redox state [10, 11, 25], that is thought to activate redox-sensitive transcription factor NF-κB, which provides the signal for activation of proinflammatory genes, such as IL-12 and TNF-α [26–29]. Productions of these mediators lead to inducing the expression of secondary mediators, including neutrophil-attracting CXC chemokines and endothelial cell adhesion molecules which mediate the adhesion and transmigration of neutrophils from the vascular space into the hepatic parenchyma [30–32]. Neutralizing antibodies to CXC chemokines proven to be effective against neutrophil-induced liver injury during ischemia reperfusion [33] and partial hepatectomy [34]. The priming of neutrophils during this time may be an important factor for the later neutrophil-induced injury phase [11]. Activated neutrophils generate two major cytotoxic mediators, that is, reactive oxygen species and proteases [21]. In addition to the NADPH oxidase-derived superoxide and its dismutation product hydrogen peroxide, data from Tadashi Hasegawa and his co-workers provide a direct evidence for a specific neutrophil-mediated oxidant stress [hypo-chlorite (HOCl)-modified epitopes] during reperfusion when a relevant number of neutrophils have extravasated into the parenchyma from sinusoids [21]. HOCl, generated only from H2O2 and Cl− by myeloperoxidase (MPO), can diffuse into hepatocytes and cause formation of chloramines, which are potent oxidants and cytotoxic agents involved in hepatocytes killing and responsible for maintaining the inflammatory response [35]. In addition, neutrophils store various proteases in granules and can release these proteolytic enzymes during activation. Protease inhibitors are shown to attenuate neutrophil-induced liver injury [36]. Moreover, reactive oxygen species are indispensable for a protease-mediated injury mechanism under in vivo conditions. Therefore, accumulated neutrophils release oxidants and proteases that directly injure hepatocytes and vascular endothelial cells and may also obstruct hepatic sinusoids resulting in hepatic hypoperfusion [37]. During the second phase of reperfusion injury, neutrophils work as the most acute cytotoxic inflammatory cells activated and recruited, and the damage caused by neutrophils is recognized as a major mechanism of during reperfusion [24, 38, 39]. A recent study by Beraza et al. has suggested that the hepatic inflammatory response to IR is driven largely by NF-κB activation in hepatocytes [40]. Nuclear factor (NF)-κB is a broad term used to describe a number of dimeric combinations of proteins of the Rel family [41, 42]. In unstimulated cells, NF-κB is sequestered in the cytoplasm by inhibitors of κB (IκB) proteins which prevent nuclear localization of NF-κB by masking its nuclear localization signal peptide and block NF-κB from binding to DNA by allosteric inhibition [43]. Once is freed from IκB, NF-κB translocates to the nucleus where it initiates the transcription of target genes such as tumor necrosis factor-a (TNF-a), interleukin (IL)-1b, and IL-6 [24, 44, 45]. The NF-κB inhibitory protein A20 demonstrates hepatoprotective abilities through curtailing inflammation by inhibiting NF-κB activation [46]. On the contrary, A20 knockout mice are born cachectic and die within 3 weeks of birth as a result of unfettered liver inflammation [47]. Therefore, above the literature provides compelling evidence that inhibition of oxidant-stress and inflammation in hepatocytes during IR injury is an essential mechanism of protection.
3. Characteristics of PPARα
PPAR is originally identified by Issemann and Green [48] after screening the liver cDNA library with a cDNA sequence located in the highly conserved C domain of nuclear hormone receptors. A notability subtype of PPAR will be discussed here is PPARα. In human body, PPARα gene which spans ~93.2 κb is located on chromosome 22q12-q13.1 and encodes a protein of 468 amino acids [49]. While in mice, PPARα gene is located on chromosome 15E2, and it also encodes a protein of 468 amino acids [50]. PPARα contains four major functional domains, which are the N-terminal ligand-independent transactivation domain (A/B domain), the DNA binding domain (DBD or C domain), the cofactor docking domain (D domain), and the C-terminal E/F domain (including the ligand binding domain (LBD) and the ligand-dependent transactivation domain (AF-2 domain) [51, 52]. The divergent amino acid sequence in the LBD of PPARα is thought to provide the molecular basis for ligand selectivity. A large ligand-binding pocket (1300 Å) exists in PPARα, allowing diverse and structurally distinct compounds access to the LBD [51] and enabling PPARα to sense a broad range of endogenous substances, including fatty acids and their derivatives, or exogenous ligands, such as fibrates, Wy14643 [53, 54], and so on. Analogous with several other nuclear hormone receptors, PPARα also is a ligand-activated transcription factor which upon heterodimerization with the retinoic X receptor (RXR), recognizes PPAR response elements (PPRE), located in the promoter of target genes [55]. PPARα is highly expressed in the liver, kidney, and heart muscle, which are all organs that possess high mitochondrial and β-oxidation activity. PPARα basically function, as sensor for fatty acid derivatives and controls essential metabolic pathways involved in lipid and energy metabolism, and it also plays a significant role in various pathophysiologic conditions, such as inflammation and apoptosis caused by injury [56]. Our group and others have demonstrated that PPARα is an important regulator of postischemic liver injury [3, 4].
4. Roles of PPARα on Warm Hepatic Ischemia-Reperfusion Injury
Reactive oxygen species and inflammation factors are critical mediators that exert a toxic effect during warm hepatic ischemia-reperfusion injury. Therefore, the most significant mechanisms of PPARα hepatoprotective abilities have been demonstrated through antioxidant stress and anti-inflammation functions (Figure 1).
Figure 1.
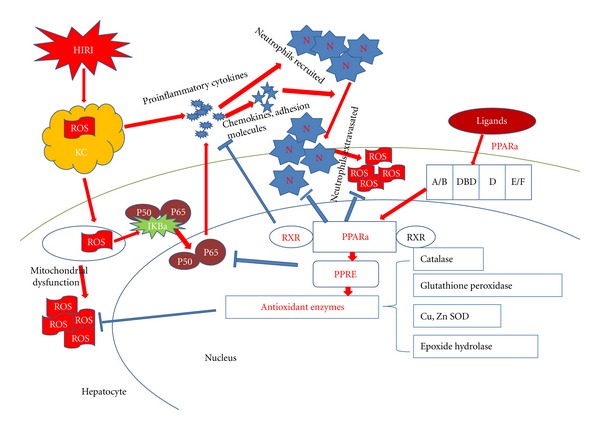
Protection mechanism of PPARα in the liver during IR injury. Ischemic stress results in the generation of reactive oxygen species (ROS) in Kupffer cells. ROS activates NF-κB and induces mitochondrial dysfunction in neighboring hepatocytes. Activation of NF-κB consequences in the production of proinflammatory cytokines, chemokines and adhesion molecules which can recruit neutrophils and propagate the inflammatory response. This vicious circle is breaked by PPARα which is a ligand-activated transcription factor that upon heterodimerization with the retinoic X receptor (RXR), recognizes PPAR response elements (PPRE), located in the promoter of target genes. Abbreviations: Neutrophil (N), Kupffer cell (KC).
4.1. Antioxidant Stress
Interruption of blood flow to liver and subsequent reperfusion lead to an acute oxidant stress response that may cause significant cellular damage and organ dysfunction. Hepatocellular injury during both the initial and later phases of reperfusion is caused in large part by reactive oxygen species including hypochlorite (HOCl) [10, 11, 25]. Accordingly, antioxidant therapy can limit the ischemia-reperfusion injury [57, 58]. The discovery of the colocalization of catalase with H2O2-generating oxidases in peroxisomes is the chief indication of their involvement in the metabolism of oxygen metabolites [59]. Peroxisomes, which are subcellular organelles within the hepatocyte, contain a battery of antioxidant enzymes and may help protect hepatocytes from oxidative damage. Catalase is the classical marker enzyme of peroxisomes metabolizing both H2O2 and a variety of substrates such as ethanol, methanol, phenol, and nitrites by peroxidatic activity [60], so it plays an important protective function against the toxic effects of peroxides and removes them with high efficiency [61]. PPARα stimulation by Wy14643 induces expression and activation of antioxidant enzymes such as superoxide dismutase (SOD), catalase, and glutathione (GSH), which protects hepatocytes against hepatic IR injury mice model in vivo [3]. From the vitro experimental results of our group, the protective effect of Wy14643 is demonstrated herein by reducing ALT, AST, ROS levels and ameliorating ultrastructure alterations of hepatocytes; this protection is associated with an inhibition of oxidative stress and upregulation of hepatocytes PPARα-mRNA expression [62]. PPARα has also been implicated in the expression or activation of antioxidant enzymes such as catalase and Cu2 +, Zn2 + superoxide dismutase (SOD1) [63]. Work done by Tetsuya Toyama has indicated that PPARa ligands (WY14643) play an antifibrotic action through disrupting the vicious cycle between hepatic damage and oxidative stress by activating antioxidant enzymes such as catalase, and resolving the oxidative stress in the rat TAA model of liver cirrhosis [63]. The PPARα agonist is also highly effective in the treatment of dietary steatohepatitis in mice which results from the action of ROS on accumulated lipids and excessive formation of lipoperoxides in the liver [64]. Simultaneously, the improved antioxidant defense system after PPARα activation can thus additionally protect against neutrophil cytotoxicity, and the related mechanism can be concluded that formation of ROS plays a key role in leading to hepatocytes necrosis mediated by neutrophil. After mice with 90 min ischemia and 8 h reperfusion, Tomohisa Okaya and Alex B. Lentsch have suggested that PPARα −/− mice have augmented hepatocellular injury compared with wild-type mice, which is proved to be associated with a marked increase in the amount of neutrophils recruited to the liver, because at this time point much of the injury to hepatocytes is thought to be due to reactive oxygen species and proteases released from recruited neutrophils. On the contrary, treatment of C57BL/6 mice with 10 mg/kg iv WY-14643 1 h before ischemia resulted in a modest but significant reduction in hepatocellular injury [3]. Furthermore, antioxidants and other interventions directed toward detoxification of reactive oxygen species also attenuated inflammatory liver injury [12, 65–67]. The mechanism of PPARα response to ROS is much more complex that it requirs further research. At the same time, the regulation of antioxidant enzymes is closely related to apoptosis signaling [68]. Therefore, elevation of anti-oxidative enzymes can suppress apoptosis and this can also promote carcinogenesis [69].
4.2. Anti-Inflammation
Neutrophils are recruited from the vascular space through a complex series of events that involve upregulated expression of cellular adhesion molecules on hepatic vascular endothelial cells and increased production of CXC chemokines [31, 70]. Accumulated neutrophils release oxidants and proteases that directly and drastically injure hepatocytes and vascular endothelial cells [24]. The first evidence indicating a potential role for PPARα in the inflammatory response is the demonstration that leukotriene B4 (LTB4), a proinflammatory eicosanoid, binds to PPARα and induces the transcription of genes involved in ω- and β-oxidation which leads to the induction of its own catabolism [71]. In this respect, the activation of PPARα by leukotriene B4 serves to limit the inflammatory process, providing a physiological mechanism to stop the damaging effects associated with inflammation [72]. Numerous recent studies have been aimed at delineating the cellular and molecular mechanisms explaining the control of the inflammatory response by PPARα in hepatic ischemia-reperfusion injury. Primarily according to previous studies by our group, the protective effects of PPARα agonists (WY14643) in postischemic liver injury are possibly associated with reductions in neutrophil accumulation, oxidative stress, and tumor necrosis factor (TNF) and interleukin-1 (IL-1) expression in livers during IR [4]. The results also have been supported by additional experimental results [38, 63]. Furthermore, works done by Tomohisa Okaya and Alex B. Lentsch suggest that livers from PPARα −/− mice have significantly more postischemic injury compared with those from wild-type mice. A possible reason may be the augmented liver neutrophil accumulation and the modest increases in activation of the transcription factor NF-κB. Treatment of cultured murine hepatocytes with WY-14643, a specific agonist of PPARα, protected cells against oxidant-induced injury. However, there are no differences in proinflammatory mediator production between PPARα −/− and wild-type mice. These data suggest that PPARα is an important regulator of the hepatic inflammatory response to ischemia reperfusion in a manner that is independent of proinflammatory cytokines [3]. What's more, evidence from in vitro experiments for an anti-inflammatory action of PPARα in endothelial cells and monocytes also demonstrate, that PPARα ligands inhibit cytokine-induced genes, such as expression of vascular cell adhesion molecule-1 and tissue factor by downregulating the transcription of these genes [73–75]. Researches address the molecular mechanisms of this anti-inflammatory action demonstrate that PPARα negatively regulate the transcription of inflammatory response genes by antagonizing the nuclear factor-κB (NF-κB) signaling pathway [76]. In addition to the antagonistic action on NF-κB signalling, PPARα activators are known to induce inhibitor of NF-κB, IκB-α, in primary smooth muscle cells and hepatocytes, which is associated with reduced NF-κB DNA binding triggered by PPARa [58]. N-3 polyunsaturated fatty acids (n-3 PUFA), eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) are considered as PPARα agonists and can also decrease the expression of proinflammatory genes by preventing IκB phosphorylation and NF-κB translocation into the nucleus [77]. Recent studies have suggested that liver preconditioning against IR injury by n-3PUFA supplementation is mediated by PPARα diminishing NF-κB DNA binding through direct protein-protein interaction with NF-κB subunit p65, leading to the recovery of NF-κB signalling activity and reestablishment of inflammatory cytokine homeostasis [78]. The zinc finger protein A20 [79], which is an intracellular ubiquitin-editing enzyme, plays a significant role in the negative feedback regulation of NF-κB activation in response to a diverse range of stimuli [80]. In lethal liver ischemia-reperfusion injury model, the survival rate of mice treated with A20 reached 67% compared with 10%–25% of control mice injected with saline. This major survival advantage in A20-treated mice is associated with protecting against liver IR injury by increasing hepatic expression of PPARα. A20-mediated protection of hepatocytes from hypoxia/reoxygenation and H2O2-mediated necrosis is reverted by pretreatment with the PPARα inhibitor MK886 [81]. However, the reverse evidence published by Yu et al. [82] reported that inhibition of NF-κB activation by A20 aggravated partial hepatic ischemia-reperfusion injury. According to this report, Xu et al. suggested that NF-κB inactivation in hepatocytes switches the TNF-α response from proliferation to apoptosis, so decreased NF-κB activity sensitizes hepatocytes to TNF-α-induced cytotoxicity and may contribute to increased liver dysfunction [83]. In nonalcoholic steatohepatitis (NASH) and simple steatosis, besides reducing steatosis by regulation lipid and lipoprotein metabolism, treatment of mice with the PPARα activator Wy14643 protects steatotic livers against IR injury, with the benefits of this treatment potentially occuring through dampening vascular cellular adhesion molecule-1 and cytokine responses and activation of NF-κB and IL-6 production [84]. The former contradictory results have suggested that the role of NF-κB activation during hepatic ischemia-reperfusion injury is controversial because it induces both protective and proinflammatory genes [85] and NF-κB inactivation either protects against hepatic IR injury [86–88] or aggravates such injury [45, 89, 90]. Studies done by Nozomu Sakai and Heather have indicated that activation of NF-κB in Kupffer cells promotes inflammation through cytokine expression, whereas activation in hepatocytes may be cell protective, based on the fact that they further proved that exogenous administration of receptor activator of NF-κB ligand (RANKL) reduces liver injury in a manner associated with increased hepatocyte NF-κB activation [91]. Supplementary works needed to be completed to explore how the PPARα plays a role in directing a clinical outcome which may lead to better prospects of more rational approaches to reduce postischemic liver injury. In addition to all of the mentioned above, PPARα is also a target of the hypothalamic hormone signaling as it plays an important role in the anti-inflammatory action of glucocorticoids [92].
5. Perspectives
As outlined in this paper, there is ample evidence for a critical involvement of oxidant-stress and inflammatory response in various animal models of hepatic ischemia-reperfusion injury. ROS are generated during initial reperfusion, where the initial cell damage triggers an inflammatory response with activation of tissue macrophages and recruitment of neutrophils both of them cause cell death and liver injury. Growing evidences for a role of PPARα in a variety of physiological and pathological processes, particularly the participation in the pathophysiology of inflammation and the protective role of hepatic ischemia-reperfusion injury via limiting oxidative injury as well as inhibiting inflammation response, has emerged in prevenient researches. However, species-specific differences in response to PPARα activators still exist between human and animals. Rats and mice are highly susceptible to peroxisome proliferation and are susceptible to hepatocarcinogenesis due to the antiapoptosis of PPARα activators. Whereas, PPARα agonists are clinically and functionally relevant as fibrate therapeutics against hyperlipidemia and agents for reducing the complications of peripheral vascular disease in diabetic patients [93]. Yet, there are no sufficient evidence to show that hepatic cancer, hypertrophy, or peroxisome proliferation is relevant to it. On the contrary, PPARα receptor activation can interrupt the development process of chronic hepatitis C and NASH to liver cancer by regulating the lipid and lipoprotein metabolism and enhancing the antioxidant stress. This is because obesity-related metabolic abnormalities [94], especially insulin resistance, may be a decisive factor in the pathogenesis of chronic hepatitis C as recently suggested in nonalcoholic steatohepatitis (NASH), along with impairment in lipid metabolism [95]. And the second reason is that oxidative stress not only damages hepatocytes and increases the rate of hepatocyte death, but also inhibits the replication of mature hepatocytes [96]. In order to balance the decreased replication capacity of mature hepatocytes, hepatic progenitors accumulate in the liver; this abnormal regenerative process may contribute to Hepatocellular Carcinoma.
One crucial difference is that the level of expression of PPARα in the human liver is lower than that found in rats and mice [97]. It is also worth pointing out that long term exposure to these drugs can result in oxidative DNA damage, among other effects [92, 98]. So despite their potentially beneficial roles, PPARα agonists should be used judiciously.
References
- 1.Hong JC, Koroleff D, Xia V, et al. Regulated hepatic reperfusion mitigates ischemia-reperfusion injury and improves survival after prolonged liver warm ischemia: a pilot study on a novel concept of organ resuscitation in a large animal model. Journal of the American College of Surgeons. 2012;214(4):505–515. doi: 10.1016/j.jamcollsurg.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 2.Clarke CN, Tevar AD, Lentsch AB. Molecular Pathology of Liver Diseases. 2011. Hepatic ischemia/reperfusion injury; pp. 397–410. [Google Scholar]
- 3.Okaya T, Lentsch AB. Peroxisome proliferator-activated receptor-α regulates postischemic liver injury. American Journal of Physiology. 2004;286(4):G606–G612. doi: 10.1152/ajpgi.00191.2003. [DOI] [PubMed] [Google Scholar]
- 4.Xu SQ, Li YH, Hu SH, Chen K, Dong LY. Effects of Wy14643 on hepatic ischemia reperfusion injury in rats. World Journal of Gastroenterology. 2008;14(45):6936–6942. doi: 10.3748/wjg.14.6936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cursio R, Gugenheim J, Ricci JE, et al. A caspase inhibitor fully protects rats against lethal normothermic liver ischemia by inhibition of liver apoptosis. The FASEB Journal. 1999;13(2):253–261. doi: 10.1096/fasebj.13.2.253. [DOI] [PubMed] [Google Scholar]
- 6.Kohli V, Madden JF, Bentley RC, Clavien PA, Selzner M. Endothelial cell and hepatocyte deaths occur by apoptosis after ischemia-reperfusion injury in the rat liver. Transplantation. 1999;67(8):1099–1105. doi: 10.1097/00007890-199904270-00003. [DOI] [PubMed] [Google Scholar]
- 7.Gujral JS, Bucci TJ, Farhood A, Jaeschke H. Mechanism of cell death during warm hepatic ischemia-reperfusion in rats: apoptosis or necrosis? Hepatology. 2001;33(2):397–405. doi: 10.1053/jhep.2001.22002. [DOI] [PubMed] [Google Scholar]
- 8.Jaeschke H, Lemasters JJ. Apoptosis versus oncotic necrosis in hepatic ischemia/reperfusion injury. Gastroenterology. 2003;125(4):1246–1257. doi: 10.1016/s0016-5085(03)01209-5. [DOI] [PubMed] [Google Scholar]
- 9.Jaeschke H, Smith CV, Mitchell JR. Reactive oxygen species during ischemia-reflow injury in isolated perfused rat liver. The Journal of Clinical Investigation. 1988;81(4):1240–1246. doi: 10.1172/JCI113441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jaeschke H, Farhood A. Neutrophil and Kupffer cell-induced oxidant stress and ischemia-reperfusion injury in rat liver. American Journal of Physiology. 1991;260(3):G355–G362. doi: 10.1152/ajpgi.1991.260.3.G355. [DOI] [PubMed] [Google Scholar]
- 11.Jaeschke H, Bautista AP, Spolarics Z, Spitzer JJ. Superoxide generation by Kupffer cells and priming of neutrophils during reperfusion after hepatic ischemia. Free Radical Research Communications. 1991;15(5):277–284. doi: 10.3109/10715769109105223. [DOI] [PubMed] [Google Scholar]
- 12.Jaeschke H. Reactive oxygen and ischemia/reperfusion injury of the liver. Chemico-Biological Interactions. 1991;79(2):115–136. doi: 10.1016/0009-2797(91)90077-k. [DOI] [PubMed] [Google Scholar]
- 13.Jaeschke H, Bautista AP, Spolarics Z, Spitzer JJ. Superoxide generation by neutrophils and Kupffer cells during in vivo reperfusion after hepatic ischemia in rats. Journal of Leukocyte Biology. 1992;52(4):377–382. doi: 10.1002/jlb.52.4.377. [DOI] [PubMed] [Google Scholar]
- 14.Jaeschke H, Mitchell JR. Mitochondria and xanthine oxidase both generate reactive oxygen species in isolated perfused rat liver after hypoxic injury. Biochemical and Biophysical Research Communications. 1989;160(1):140–147. doi: 10.1016/0006-291x(89)91632-x. [DOI] [PubMed] [Google Scholar]
- 15.Jaeschke H. Antioxidant defense mechanisms. Comprehensive Toxicology. 2010;9:319–337. [Google Scholar]
- 16.Nieminen AL, Byrne AM, Herman B, Lemasters JJ. Mitochondrial permeability transition in hepatocytes induced by t- BuOOh: NAD(P)H and reactive oxygen species. American Journal of Physiology. 1997;272(4):C1286–C1294. doi: 10.1152/ajpcell.1997.272.4.C1286. [DOI] [PubMed] [Google Scholar]
- 17.Uchiyama A, Kim JS, Kon K, et al. Translocation of iron from lysosomes into mitochondria is a key event during oxidative stress-induced hepatocellular injury. Hepatology. 2008;48(5):1644–1654. doi: 10.1002/hep.22498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu P, McGuire GM, Fisher MA, Farhood A, Smith CW, Jaeschke H. Activation of Kupffer cells and neutrophils for reactive oxygen formation is responsible for endotoxin-enhanced liver injury after hepatic ischemia. Shock. 1995;3(1):56–62. [PubMed] [Google Scholar]
- 19.Kurose I, Wolf R, Grisham MB, Granger DN. Modulation of ischemia/reperfusion-induced microvascular dysfunction by nitric oxide. Circulation Research. 1994;74(3):376–382. doi: 10.1161/01.res.74.3.376. [DOI] [PubMed] [Google Scholar]
- 20.Jaeschke H, Farhood A, Bautista AP, Spolarics Z, Spitzer JJ. Complement activates Kupffer cells and neutrophils during reperfusion after hepatic ischemia. American Journal of Physiology. 1993;264(4):G801–G809. doi: 10.1152/ajpgi.1993.264.4.G801. [DOI] [PubMed] [Google Scholar]
- 21.Jaeschke H, Smith CW. Mechanisms of neutrophil-induced parenchymal cell injury. Journal of Leukocyte Biology. 1997;61(6):647–653. doi: 10.1002/jlb.61.6.647. [DOI] [PubMed] [Google Scholar]
- 22.Grattagliano I, Vendemiale G, Lauterburg BH. Reperfusion injury of the liver: role of mitochondria and protection by glutathione ester. Journal of Surgical Research. 1999;86(1):2–8. doi: 10.1006/jsre.1999.5620. [DOI] [PubMed] [Google Scholar]
- 23.Kumamoto Y, Suematsu M, Shimazu M, et al. Kupffer cell-independent acute hepatocellular oxidative stress and decreased bile formation in post-cold-ischemic rat liver. Hepatology. 1999;30(6):1454–1463. doi: 10.1002/hep.510300601. [DOI] [PubMed] [Google Scholar]
- 24.Lentsch AB, Kato A, Yoshidome H, McMasters KM, Edwards MJ. Inflammatory mechanisms and therapeutic strategies for warm hepatic ischemia/reperfusion injury. Hepatology. 2000;32(2):169–173. doi: 10.1053/jhep.2000.9323. [DOI] [PubMed] [Google Scholar]
- 25.Jaeschke H. Molecular mechanisms of hepatic ischemia-reperfusion injury and preconditioning. American Journal of Physiology. 2003;284(1):G15–G26. doi: 10.1152/ajpgi.00342.2002. [DOI] [PubMed] [Google Scholar]
- 26.Colletti LM, Remick DG, Burtch GD, Kunkel SL, Strieter RM, Campbell DA. Role of tumor necrosis factor-α in the pathophysiologic alterations after hepatic ischemia/reperfusion injury in the rat. The Journal of Clinical Investigation. 1990;85(6):1936–1943. doi: 10.1172/JCI114656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lentsch AB, Yoshidome H, Kato A, et al. Requirement for interleukin-12 in the pathogenesis of warm hepatic ischemia/reperfusion injury in mice. Hepatology. 1999;30(6):1448–1453. doi: 10.1002/hep.510300615. [DOI] [PubMed] [Google Scholar]
- 28.Loop T, Pahl H. Nuclear Factor κB: Regulation and Role in Disease. 2003. Activators and target genes of Rel/NF-κB transcription factors; pp. 1–48. [Google Scholar]
- 29.Zwacka RM, Zhang Y, Zhou W, Halldorson J, Engelhardt JE. Ischemia/reperfusion injury in the liver of BALB/c mice activates AP-1 and nuclear factor κB independently of IκB degradation. Hepatology. 1998;28(4):1022–1030. doi: 10.1002/hep.510280417. [DOI] [PubMed] [Google Scholar]
- 30.Colletti LM, Cortis A, Lukacs N, Kunkel SL, Green M, Strieter RM. Tumor necrosis factor up-regulates intercellular adhesion molecule 1, which is important in the neutrophil-dependent lung and liver injury associated with hepatic ischemia and reperfusion in the rat. Shock. 1998;10(3):182–191. doi: 10.1097/00024382-199809000-00006. [DOI] [PubMed] [Google Scholar]
- 31.Colletti LM, Kunkel SL, Walz A, et al. Chemokine expression during hepatic ischemia/reperfusion-induced lung injury in the rat. The role of epithelial neutrophil activating protein. The Journal of Clinical Investigation. 1995;95(1):134–141. doi: 10.1172/JCI117630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lentsch AB, Yoshidome H, Cheadle WG, Miller FN, Edwards MJ. Chemokine involvement in hepatic ischemia/reperfusion injury in mice: roles for macrophage inflammatory protein-2 and KC. Hepatology. 1998;27(4):1172–1177. doi: 10.1002/hep.510270440. [DOI] [PubMed] [Google Scholar]
- 33.Colletti LM, Kunkel SL, Walz A, et al. The role of cytokine networks in the local liver injury following hepatic ischemia/reperfusion in the rat. Hepatology. 1996;23(3):506–514. doi: 10.1002/hep.510230315. [DOI] [PubMed] [Google Scholar]
- 34.Colletti LM, Kunkel SL, Green M, Burdick M, Strieter RM. Hepatic inflammation following 70% hepatectomy may be related to up-regulation of epithelial neutrophil activating protein-78. Shock. 1996;6(6):397–402. doi: 10.1097/00024382-199612000-00002. [DOI] [PubMed] [Google Scholar]
- 35.Bilzer M, Lauterburg BH. Effects of hypochlorous acid and chloramines on vascular resistance, cell integrity, and biliary glutathione disulfide in the perfused rat liver: modulation by glutathione. Journal of Hepatology. 1991;13(1):84–89. doi: 10.1016/0168-8278(91)90868-c. [DOI] [PubMed] [Google Scholar]
- 36.Li XK, Mohammad Matin AF, Suzuki H, Uno T, Yamaguchi T, Harada Y. Effect of protease inhibitor on ischemia/reperfusion injury of the rat liver. Transplantation. 1993;56(6):1331–1336. doi: 10.1097/00007890-199312000-00008. [DOI] [PubMed] [Google Scholar]
- 37.Zhang JX, Jones DV, Clemens MG. Effect of activation on neutrophil-induced hepatic microvascular injury in isolated rat liver. Shock. 1994;1(4):273–278. doi: 10.1097/00024382-199404000-00005. [DOI] [PubMed] [Google Scholar]
- 38.Di Paola R, Cuzzocrea S. Peroxisome proliferator-activated receptors ligands and ischemia-reperfusion injury. Naunyn-Schmiedeberg’s Archives of Pharmacology. 2007;375(3):157–175. doi: 10.1007/s00210-007-0141-2. [DOI] [PubMed] [Google Scholar]
- 39.Serracino-Inglott F, Habib NA, Mathie RT. Hepatic ischemia-reperfusion injury. The American Journal of Surgery. 2001;181(2):160–166. doi: 10.1016/s0002-9610(00)00573-0. [DOI] [PubMed] [Google Scholar]
- 40.Beraza N, Lüdde T, Assmus U, Roskams T, Borght SV, Trautwein C. Hepatocyte-specific IKKγ/NEMO expression determines the degree of liver injury. Gastroenterology. 2007;132(7):2504–2517. doi: 10.1053/j.gastro.2007.03.045. [DOI] [PubMed] [Google Scholar]
- 41.Ghosh S, May MJ, Kopp EB. NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses. Annual Review of Immunology. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 42.May MJ, Ghosh S. Signal transduction through NF-κB. Immunology Today. 1998;19(2):80–88. doi: 10.1016/s0167-5699(97)01197-3. [DOI] [PubMed] [Google Scholar]
- 43.Scheidereit C. IκB kinase complexes: gateways to NF-κB activation and transcription. Oncogene. 2006;25(51):6685–6705. doi: 10.1038/sj.onc.1209934. [DOI] [PubMed] [Google Scholar]
- 44.Takahashi Y, Ganster RW, Gambotto A, et al. Role of NF-κB on liver cold ischemia-reperfusion injury. American Journal of Physiology. 2002;283(5):G1175–G1184. doi: 10.1152/ajpgi.00515.2001. [DOI] [PubMed] [Google Scholar]
- 45.Okaya T, Lentsch AB. Hepatic expression of S32A/S36A IκBα does not reduce postischemic liver injury. Journal of Surgical Research. 2005;124(2):244–249. doi: 10.1016/j.jss.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 46.Wartz IE, O’Rourke KM, Zhou H, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature. 2004;430(7000):694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 47.Lee EG, Boone DL, Chai S, et al. Failure to regulate TNF-induced NF-κB and cell death responses in A20-deficient mice. Science. 2000;289(5488):2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347(6294):645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- 49.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocrine Reviews. 1999;20(5):649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 50.Pyper SR, Viswakarma N, Yu S, Reddy JK. PPARα: energy combustion, hypolipidemia, inflammation and cancer. Nuclear Receptor Signaling. 2010;8, article e002 doi: 10.1621/nrs.08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moras D, Gronemeyer H. The nuclear receptor ligand-binding domain: structure and function. Current Opinion in Cell Biology. 1998;10(3):384–391. doi: 10.1016/s0955-0674(98)80015-x. [DOI] [PubMed] [Google Scholar]
- 52.Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA. Peroxisome proliferator-activated receptors α and γ are activated by indomethacin and other non-steroidal anti-inflammatory drugs. The Journal of Biological Chemistry. 1997;272(6):3406–3410. doi: 10.1074/jbc.272.6.3406. [DOI] [PubMed] [Google Scholar]
- 53.Guan YF, Breyer MD. Peroxisome proliferator-activated receptors (PPARs): novel therapeutic targets in renal disease. Kidney International. 2001;60(1):14–30. doi: 10.1046/j.1523-1755.2001.00766.x. [DOI] [PubMed] [Google Scholar]
- 54.Cowart LA, Wei S, Hsu MH, et al. The CYP4A isoforms hydroxylate epoxyeicosatrienoic acids to form high affinity peroxisome proliferator-activated receptor ligands. The Journal of Biological Chemistry. 2002;277(38):35105–35112. doi: 10.1074/jbc.M201575200. [DOI] [PubMed] [Google Scholar]
- 55.Fruchart JC, Staels B, Duriez P. The role of fibric acids in atherosclerosis. Current Atherosclerosis Reports. 2001;3(1):83–92. doi: 10.1007/s11883-001-0015-x. [DOI] [PubMed] [Google Scholar]
- 56.Maeda T, Kishioka S. PPAR and Pain. International Review of Neurobiology. 2009;85:165–177. doi: 10.1016/S0074-7742(09)85013-7. [DOI] [PubMed] [Google Scholar]
- 57.Wheeler MD, Katuna M, Smutney OM, et al. Comparison of the effect of adenoviral delivery of three superoxide dismutase genes against hepatic ischemia-reperfusion injury. Human Gene Therapy. 2001;12(18):2167–2177. doi: 10.1089/10430340152710513. [DOI] [PubMed] [Google Scholar]
- 58.Glantzounis GK, Salacinski HJ, Yang W, Davidson BR, Seifalian AM. The contemporary role of antioxidant therapy in attenuating liver ischemia-reperfusion injury: a review. Liver Transplantation. 2005;11(9):1031–1047. doi: 10.1002/lt.20504. [DOI] [PubMed] [Google Scholar]
- 59.Schrader M, Fahimi HD. Peroxisomes and oxidative stress. Biochimica et Biophysica Acta. 2006;1763(12):1755–1766. doi: 10.1016/j.bbamcr.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 60.Oshino N, Chance B, Sies H, Bücher T. The role of H2O2 generation in perfused rat liver and the reaction of catalase compound I and hydrogen donors. Archives of Biochemistry and Biophysics. 1973;154(1):117–131. doi: 10.1016/0003-9861(73)90040-4. [DOI] [PubMed] [Google Scholar]
- 61.Siraki AG, Pourahmad J, Chan TS, Khan S, O’Brien PJ. Endogenous and endobiotic induced reactive oxygen species formation by isolated hepatocytes. Free Radical Biology and Medicine. 2002;32(1):2–10. doi: 10.1016/s0891-5849(01)00764-x. [DOI] [PubMed] [Google Scholar]
- 62.Ke C, Yuan-Hai L, Si-Qi X, et al. Protective effects of peroxisome proliferator-activated receptor-α agonist, Wy14643, on hypoxia/reoxygenation injury in primary rat hepatocytes. PPAR Research. 2012;2012:8 pages. doi: 10.1155/2012/547980.547980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Toyama T, Nakamura H, Harano Y, et al. PPARα ligands activate antioxidant enzymes and suppress hepatic fibrosis in rats. Biochemical and Biophysical Research Communications. 2004;324(2):697–704. doi: 10.1016/j.bbrc.2004.09.110. [DOI] [PubMed] [Google Scholar]
- 64.Ip E, Farrell GC, Robertson G, Hall P, Kirsch R, Leclercq I. Central role of PPARα-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology. 2003;38(1):123–132. doi: 10.1053/jhep.2003.50307. [DOI] [PubMed] [Google Scholar]
- 65.Hasegawa T, Malle E, Farhood A, Jaeschke H. Generation of hypochlorite-modified proteins by neutrophils during ischemia-reperfusion injury in rat liver: attenuation by ischemic preconditioning. American Journal of Physiology. 2005;289(4):G760–G767. doi: 10.1152/ajpgi.00141.2005. [DOI] [PubMed] [Google Scholar]
- 66.Wu TW, Hashimoto N, Au JX, Wu J, Mickle DAG, Carey D. Trolox protects rat hepatocytes against oxyradical damage and the ischemic rat liver from reperfusion injury. Hepatology. 1991;13(3):575–580. [PubMed] [Google Scholar]
- 67.Halliwell B. Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment. Drugs and Aging. 2001;18(9):685–716. doi: 10.2165/00002512-200118090-00004. [DOI] [PubMed] [Google Scholar]
- 68.McCormick F. Signalling networks that cause cancer. Trends in Genetics. 1999;15(12):M53–M56. [PubMed] [Google Scholar]
- 69.Corcoran GB, Fix L, Jones DP, et al. Apoptosis: molecular control point in toxicity. Toxicology and Applied Pharmacology. 1994;128(2):169–181. doi: 10.1006/taap.1994.1195. [DOI] [PubMed] [Google Scholar]
- 70.Colletti LM, Cortis A, Lukacs N, Kunkel SL, Green M, Strieter RM. Tumor necrosis factor up-regulates intercellular adhesion molecule 1, which is important in the neutrophil-dependent lung and liver injury associated with hepatic ischemia and reperfusion in the rat. Shock. 1998;10(3):182–191. doi: 10.1097/00024382-199809000-00006. [DOI] [PubMed] [Google Scholar]
- 71.Devchand PR, Keller H, Peters JM, Vazquez M, Gonzalez FJ, Wahli W. The PPARα-leukotriene B4 pathway to inflammation control. Nature. 1996;384(6604):39–43. doi: 10.1038/384039a0. [DOI] [PubMed] [Google Scholar]
- 72.Devchand PR, Ziouzenkova O, Plutzky J. Oxidative stress and peroxisome proliferator-activated receptors: reversing the curse? Circulation Research. 2004;95(12):1137–1139. doi: 10.1161/01.RES.0000151331.69399.b2. [DOI] [PubMed] [Google Scholar]
- 73.Delerive P, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors in inflammation control. Journal of Endocrinology. 2001;169(3):453–459. doi: 10.1677/joe.0.1690453. [DOI] [PubMed] [Google Scholar]
- 74.Marx N, Sukhova GK, Collins T, Libby P, Plutzky J. PPARα activators inhibit cytokine-induced vascular cell adhesion molecule-1 expression in human endothelial cells. Circulation. 1999;99(24):3125–3131. doi: 10.1161/01.cir.99.24.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Neve BP, Corseaux D, Chinetti G, et al. PPARα agonists inhibit tissue factor expression in human monocytes and macrophages. Circulation. 2001;103(2):207–212. doi: 10.1161/01.cir.103.2.207. [DOI] [PubMed] [Google Scholar]
- 76.Delerive P, Gervois P, Fruchart JC, Staels B. Induction of IκBα expression as a mechanism contributing to the anti-inflammatory activities of peroxisome proliferator-activated receptor-α activators. The Journal of Biological Chemistry. 2000;275(47):36703–36707. doi: 10.1074/jbc.M004045200. [DOI] [PubMed] [Google Scholar]
- 77.Adkins Y, Kelley DS. Mechanisms underlying the cardioprotective effects of omega-3 polyunsaturated fatty acids. The Journal of Nutritional Biochemistry. 2010;21(9):781–792. doi: 10.1016/j.jnutbio.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 78.Zúñiga J, Cancino M, Medina F, et al. N-3 PUFA supplementation triggers PPAR-α activation and PPAR-α/NF-κB interaction: anti-inflammatory implications in liver ischemia-reperfusion injury. PLoS ONE. 2011;6(12) doi: 10.1371/journal.pone.0028502.e28502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Opipari A, Jr., Boguski MS, Dixit VM. The A20 cDNA induced by tumor necrosis factor α encodes a novel type of zinc finger protein. The Journal of Biological Chemistry. 1990;265(25):14705–14708. [PubMed] [Google Scholar]
- 80.Coornaert B, Carpentier I, Beyaert R. A20: central gatekeeper in inflammation and immunity. The Journal of Biological Chemistry. 2009;284(13):8217–8221. doi: 10.1074/jbc.R800032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ramsey HE, Da Silva CG, Longo CR, et al. A20 protects mice from lethal liver ischemia/reperfusion injury by increasing peroxisome proliferator-activated receptor-α expression. Liver Transplantation. 2009;15(11):1613–1621. doi: 10.1002/lt.21879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yu J, Lee HS, Lee SM, et al. Aggravation of post-ischemic liver injury by overexpression of A20, an NF-κB suppressor. Journal of Hepatology. 2011;55(2):328–336. doi: 10.1016/j.jhep.2010.11.029. [DOI] [PubMed] [Google Scholar]
- 83.Xu Y, Bialik S, Jones BE, et al. NF-κB inactivation converts a hepatocyte cell line TNF-α response from proliferation to apoptosis. American Journal of Physiology. 1998;275(4, part 1):C1058–C1066. doi: 10.1152/ajpcell.1998.275.4.C1058. [DOI] [PubMed] [Google Scholar]
- 84.Teoh NC, Williams J, Hartley J, Yu J, McCuskey RS, Farrell GC. Short-term therapy with peroxisome proliferation-activator receptor-agonist Wy-14,643 protects murine fatty liver against ischemia-reperfusion injury. Hepatology. 2010;51(3):996–1006. doi: 10.1002/hep.23420. [DOI] [PubMed] [Google Scholar]
- 85.Luedde T, Trautwein C. Intracellular survival pathways in the liver. Liver International. 2006;26(10):1163–1174. doi: 10.1111/j.1478-3231.2006.01366.x. [DOI] [PubMed] [Google Scholar]
- 86.Luedde T, Assmus U, Wüstefeld T, et al. Deletion of IKK2 in hepatocytes does not sensitize these cells to TNF-induced apoptosis but protects from ischemia/reperfusion injury. The Journal of Clinical Investigation. 2005;115(4):849–859. doi: 10.1172/JCI23493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Matsui N, Kasajima K, Hada M, et al. Inhibiton of NF-κB activation during ischemia reduces hepatic ischemia/reperfusion injury in rats. The Journal of Toxicological Sciences. 2005;30(2):103–110. doi: 10.2131/jts.30.103. [DOI] [PubMed] [Google Scholar]
- 88.Suetsugu H, Iimuro Y, Uehara T, et al. Nuclear factor κB inactivation in the rat liver ameliorates short term total warm ischaemia/reperfusion injury. Gut. 2005;54(6):835–842. doi: 10.1136/gut.2004.043034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kuboki S, Okaya T, Schuster R, et al. Hepatocyte NF-κB activation is hepatoprotective during ischemia-reperfusion injury and is augmented by ischemic hypothermia. American Journal of Physiology. 2007;292(1):G201–G207. doi: 10.1152/ajpgi.00186.2006. [DOI] [PubMed] [Google Scholar]
- 90.Kuboki S, Sakai N, Clarke C, et al. The peptidyl-prolyl isomerase, Pin1, facilitates NF-κB binding in hepatocytes and protects against hepatic ischemia/reperfusion injury. Journal of Hepatology. 2009;51(2):296–306. doi: 10.1016/j.jhep.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sakai N, Van Sweringen HL, Schuster R, et al. Receptor activator of nuclear factor-κB ligand (RANKL) protects against hepatic ischemia/reperfusion injury in mice. Hepatology. 2012;55(3):888–897. doi: 10.1002/hep.24756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Crisafulli C, Bruscoli S, Esposito E, et al. PPAR-α contributes to the anti-inflammatory activity of 17β-estradiol. Journal of Pharmacology and Experimental Therapeutics. 2009;331(3):796–807. doi: 10.1124/jpet.109.156646. [DOI] [PubMed] [Google Scholar]
- 93.Bulhak AA, Jung C, Östenson CG, Lundberg JO, Sjoquist PO, Pernow J. PPAR-α activation protects the type 2 diabetic myocardium against ischemia-reperfusion injury: involvement of the PI3-kinase/Akt and NO pathway. American Journal of Physiology. 2009;296(3):H719–H727. doi: 10.1152/ajpheart.00394.2008. [DOI] [PubMed] [Google Scholar]
- 94.Yang S, Lin HZ, Hwang J, Chacko VP, Diehl AM. Hepatic hyperplasia in noncirrhotic fatty livers: is obesity-related hepatic steatosis a premalignant condition? Cancer Research. 2001;61(13):5016–5023. [PubMed] [Google Scholar]
- 95.Koike K. Hepatitis C as a metabolic disease: implication for the pathogenesis of NASH. Hepatology Research. 2005;33(2):145–150. doi: 10.1016/j.hepres.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 96.Yang SQ, Mandal AK, Huang J, Diehl AM. Disrupted signaling and inhibited regeneration in obese mice with fatty livers: implications for nonalcoholic fatty liver disease pathophysiology. Hepatology. 2001;34(4, part 1):694–706. doi: 10.1053/jhep.2001.28054. [DOI] [PubMed] [Google Scholar]
- 97.Holden PR, Tugwood JD. Peroxisome proliferator-activated receptor alpha: role in rodent liver cancer and species differences. Journal of Molecular Endocrinology. 1999;22(1):1–8. doi: 10.1677/jme.0.0220001. [DOI] [PubMed] [Google Scholar]
- 98.Rusyn I, Asakura S, Pachkowski B, et al. Expression of base excision DNA repair genes is a sensitive biomarker for in vivo detection of chemical-induced chronic oxidative stress. Cancer Research. 2004;64(3):1050–1057. doi: 10.1158/0008-5472.can-03-3027. [DOI] [PubMed] [Google Scholar]