

Abstract
B-Myb is a highly conserved member of the Myb transcription factor family, which plays an essential role in cell cycle progression by regulating the transcription of genes at the G2/M-phase boundary. The role of B-Myb in other parts of the cell cycle is less well-understood. By employing siRNA-mediated silencing of B-Myb expression, we found that B-Myb is required for efficient entry into S-phase. Surprisingly, a B-Myb mutant that lacks sequence-specific DNA-binding activity and is unable to activate transcription of B-Myb target genes is able to rescue the S-phase defect observed after B-Myb knockdown. Moreover, we have identified polymerase delta-interacting protein 1 (Pdip1), a BTB domain protein known to bind to the DNA replication and repair factor PCNA as a novel B-Myb interaction partner. We have shown that Pdip1 is able to interact with B-Myb and PCNA simultaneously. In addition, we found that a fraction of endogenous B-Myb can be co-precipitated via PCNA, suggesting that B-Myb might be involved in processes related to DNA replication or repair. Taken together, our work suggests a novel role for B-Myb in S-phase that appears to be independent of its sequence-specific DNA-binding activity and its ability to stimulate the expression of bona fide B-Myb target genes.
Keywords: B-Myb, cell cycle, PCNA, Pdip1/S-phase, DNA-binding
Introduction
B-Myb, a highly conserved member of the vertebrate Myb transcription factor family, is ubiquitously expressed in all proliferating cells and plays an important role in cell cycle progression.1,2 Transcription of the B-myb gene is induced at the G1/S-phase boundary by an E2F-dependent mechanism.3 The activity of B-Myb is further controlled by post-translational modification and interaction with other proteins. Notably, phosphorylation of B-Myb by cyclin A2/Cdk2 at the beginning of S-phase stimulates its transactivation potential by relieving repressive effects exerted by the C-terminal domain of the protein.4-9 Several interacting proteins affect the transactivation potential of B-Myb, including cyclin D,10,11 poly-(ADP-ribose) polymerase (PARP),12 p107,13 nucleolin,14 p300,11,15,16 TAFII25017 and N-CoR/SMRT.18,19 The interactions of some of these proteins, such as PARP,20 N-CoR/SMRT18 and p300,11 are modulated by the phosphorylation state of B-Myb, suggesting that they are involved in the phosphorylation-dependent stimulation of B-Myb activity. Phosphorylation not only activates B-Myb, but also triggers its degradation by the ubiquitin-dependent Cdc34-SCFp45Skp2 pathway.21 Although B-Myb expression levels are rather uniform in different types of proliferating cells, embryonic stem cells express the protein at much higher levels,22,23 suggesting that B-Myb is particularly important early during embryogenesis. This is also highlighted by the phenotype of B-myb-knockout mice, which show early embryonic death caused by proliferation defects of the cells of the inner cell mass of the blastocyst.24
Recent work performed in Drosophila as well as mammalian cells has demonstrated that B-Myb and its Drosophila homolog (dmMyb) are part of dynamic protein complexes that regulate genes acting at the G2/M transition of the cell cycle. The Drosophila dREAM/Myb-MuvB complex25,26 consists of dmMyb and a number of other proteins, including the Drosophila homologs of E2F2 and Rb and several Myb-interacting proteins (Mip40, Mip120 and Mip130), and has been shown to stimulate the expression of G2/M genes.27,28 In mammalian cells, a similar complex, termed LINC, consisting of E2F4 and either p130 or p107 and several other proteins, such as Lin-9, Lin-37, Lin-54 and Lin-5 (the homologs of the Drosophila Mip proteins), has been identified. In resting cells, LINC represses E2F target genes29 whereas in the S-phase, LINC switches to B-Myb to activate genes required for the G2/M transition and mitosis.30,31 Reporter assays and chromatin immunoprecipitation experiments have demonstrated that B-Myb targets LINC to the promoters of several G2/M genes, including cyclin B1, cdc2 and others, thereby activating their expression.32,33 It is thought that the crucial role of B-Myb in regulating the transcription of G2/M-phase genes is responsible for the decreased genomic stability observed in Drosophila,34-37 zebrafish38 and mammalian cell systems23,39,40 carrying mutant versions of B-Myb or showing reduced B-Myb expression. Apart from acting as a crucial transcription factor at the G2/M transition, B-Myb appears to also perform non-transcriptional roles during mitosis. In the so-called Myb-Clafi complex, B-Myb, together with clathrin and filamin is required for mitotic spindle formation,41 further emphasizing the role of B-Myb during mitosis.
In addition to its function at the G2/M-transition, there is accumulating evidence that B-Myb might also be involved in DNA replication. Work on Drosophila Myb has shown that it performs a non-transcriptional role in the control of the activity of the replication origin that mediates chorion gene amplification.42 Very recently, it was shown that decreased expression of B-Myb in embryonic stem cells disturbs the DNA replication program.43 Here, we show that B-Myb expression is necessary for the S-phase independently of its role as a sequence-specific DNA-binding protein. Furthermore, we describe the characterization of a novel B-Myb interacting protein, Pdip1, which can act as an adaptor bridging B-Myb and PCNA, a protein that plays key roles in DNA replication and repair.
Results
Knockdown of B-Myb reduces the number of cells engaged in DNA synthesis
A number of studies have provided a clear picture of B-Myb as a transcriptional regulator of G2/M-phase genes; however, relatively little is known about the role of B-Myb at the G1/S-boundary and in S-phase, when B-Myb is highly expressed. Previous work on the Myb protein of Drosophila melanogaster (dmMyb) has demonstrated a direct role of dmMyb in DNA replication.42 Among the vertebrate Myb family members, B-Myb is thought to be the homolog of dmMyb. We were therefore interested to investigate a potential influence of B-Myb on DNA synthesis in mammalian cells. As an initial experiment, we examined the effect of siRNA-mediated silencing of B-Myb expression on DNA-synthesis in HepG2 cells. Figure 1A shows that B-Myb expression was significantly reduced by two different B-myb-specific siRNAs but not by control siRNA. Cells in S-phase were detected by staining them with the nucleoside 5-ethynyl-2′-deoxyuridine (EdU). EdU is incorporated into newly replicated DNA and can subsequently be visualized by a copper-catalyzed click reaction with an azide derivative of Alexa Fluor 594. We found that the relative number of B-myb-knockdown cells that had incorporated EdU was significantly smaller than the corresponding number of the control siRNA-treated cells (Fig. 1B and C). Since we did not observe a G2/M-block in HepG2 cells after B-Myb knockdown (Fig. S1), this suggested that DNA synthesis and/or entry into S-phase was compromised in cells after B-Myb knockdown.
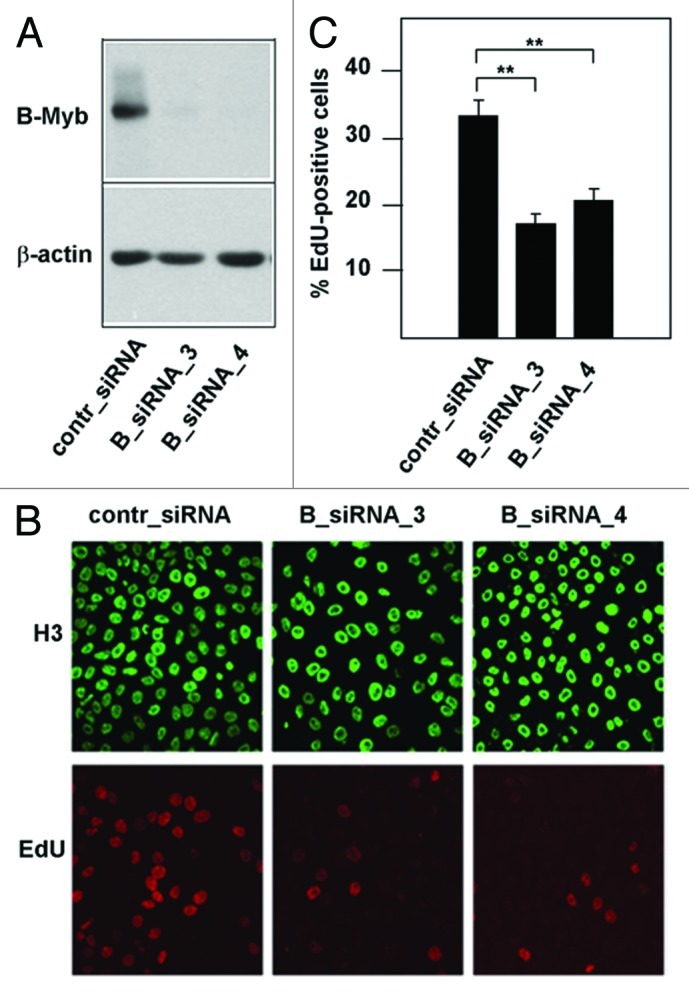
Figure 1. Knockdown of B-Myb reduces the fraction of S-phase cells. (A) HepG2 cells transfected with control siRNA or with two different B-Myb-specific siRNAs. Cells harvested 48 h after transfection were analyzed by western blotting with antibodies against B-Myb and β-actin. (B). HepG2 cells transfected with control or B-myb siRNAs were labeled for 5 min with EdU to visualize S-phase cells. Additionally, cells were stained with antibodies against histone H3 to visualize the total number of nuclei. (C). Quantification of EdU-positive cells. The bars show the percentage of EdU-positive cells in the total cell population, determined by counting 1,000 cells from different photographic fields and several independent knockdown experiments. Thin lines show standard deviations. The asterisks indicate statistical significance (** p < 0.01, Student’s-t-test).
Knockdown of B-Myb slows down S-phase entry and progression of mimosine-arrested cells
To investigate whether the decrease of the fraction of S-phase cells after B-Myb knockdown was due to a block at the entry into S-phase, HepG2 cells were transfected with control or B-myb-specific siRNA and synchronized with mimosine at the G1/S-phase boundary. Previous work has shown that mimosine-arrested cells contain assembled pre-replication complexes that are ready to establish active replication forks within minutes, both in vivo and in vitro.44-46 It is therefore thought that mimosine arrests cells at the G1/S-transition just before DNA replication starts. The successful knockdown of B-Myb in the synchronized cells is shown in Figure 2B. After release from the mimosine-arrest, most of the control siRNA transfected cells entered the S-phase to finally reach the G2-phase (Fig. 2A, upper panels). In contrast, a substantial fraction of the B-Myb siRNA transfected cells remained with a G1 DNA content, indicating that they did not proceed into S-phase. B-Myb knockdown also appeared to slow down progression of the remaining cells. This was evident 8 h after release from the mimosine block, and suggested that B-Myb knockdown also affects the progression through S-phase.

Figure 2. Effect of B-Myb knockdown on S-phase progression. (A). HepG2 cells transfected with B-Myb-specific and control siRNAs were synchronized by mimosine treatment. The cells were then released into S-phase for the indicated times, followed by flow cytometry to examine the progression through S-phase. The white and black arrows mark the G1 and G2 DNA content, respectively. (B). The successful knockdown of B-Myb was confirmed by western blotting.
A DNA binding-deficient point mutant of B-Myb rescues the defect in S-phase after B-Myb knockdown
The defect in S-phase observed after B-Myb knockdown might be explained by the lack of activation of target genes that are crucial to S-phase. For example, B-Myb has been proposed to activate c-Myc expression,43 which itself is known to play a direct role in DNA replication.47 Western blot analysis showed that c-Myc expression was not affected by the B-Myb knockdown, excluding reduced levels of c-Myc as an explanation for the S-phase defect observed after B-Myb knockdown (Fig. S2). To more systematically explore if the role of B-Myb at the onset of the S-phase is related to its function as a sequence-specific DNA-binding transcription factor, we generated a B-Myb mutant that lacks sequence-specific DNA-binding activity and stably expressed it in a doxycyclin-inducible manner in HepG2 cells. Based on the known structures of the c-Myb and B-Myb DNA-binding domains48,49 and the results of mutagenesis experiments of the c-Myb DNA-binding domain,50,51 we expected that the substitution of asparagine 174 by alanine would abolish the specific DNA-binding activity of B-Myb. EMSA assays using bacterially expressed wild-type B-Myb and the B-Myb (N174A) mutant confirmed that the mutated protein had lost sequence-specific DNA-binding activity (Fig. 3B). In addition to the N174A mutation, we introduced several nucleotide substitutions into the target site for B-Myb siRNA_4. These substitutions were designed to only affect the binding of the siRNA but not the amino acid sequence of the B-Myb protein (Fig. 3A). The resulting construct was resistant to B-myb siRNA_4 and could still be silenced by B-myb siRNA_3 (Fig. 3C). We then generated a clone of stably transfected HepG2 cells in which the expression of the mutant B-Myb was inducible by doxycyclin, as demonstrated by immunofluorescence and western blot analysis (Fig. 4A and B). The mutant B-Myb was not affected by siRNA_4 in these cells, whereas the endogenous B-Myb was silenced by the siRNA (Fig. 4B). We then used these cells to ask whether the mutated B-Myb was able to functionally replace the endogenous B-Myb in a knockdown experiment. As shown in Figure 4C, this was indeed the case. In the absence of doxycyclin, knockdown of B-Myb led to a reduction of the amount of EdU-positive cells, consistent with the results shown in Figure 1. In the presence of doxycyclin (i.e., when the mutant B-Myb was expressed), knockdown of the endogenous B-Myb had no effect on the number of cells in S-phase. This demonstrated that the specific DNA-binding activity of B-Myb is not required for B-Myb to correct the S-phase defect, suggesting that the function of B-Myb at the G1/S-boundary is independent of B-Myb’s role as a sequence-specific DNA-binding transcription factor. To demonstrate that the mutant B-Myb was unable to stimulate the expression of bona fide B-Myb target genes, we analyzed the expression of the cyclin B1 and cdc2 genes. The expression of both genes was reduced after B-Myb knockdown to similar extent both in the absence and in the presence of doxycyclin (Fig. 4C). Thus, the mutant B-Myb was able to rescue the S-phase defect but not the reduced transcription of the target genes after B-Myb knockdown. Taken together, our data argue for a role of B-Myb at the G1/S-transition that is independent of its sequence-specific DNA-binding activity and its ability to stimulate the expression of bona fide B-Myb target genes.

Figure 3. Construction of a DNA-binding defective and siRNA-resistant B-Myb mutant. (A). Schematic illustration of the DNA-binding deficient and RNAi resistant B-Myb mutant. The nucleotide and amino acid sequences of the wt and mutant B-Myb at the B-myb_siRNA4 target region are shown. (B). Equal amounts of GST-B-Myb-wt and GST B-Myb-N174A proteins were subjected to an EMSA experiment using a Myb-binding site oligonucleotide (top). Aliquots of the protein extracts were analyzed by SDS-PAGE and stained with Coomassie brilliant blue to confirm equal protein concentration (bottom). (C). HepG2 cells were first transfected with expression vector for B-Myb (N174A) mutsiRNA_4. Equal aliquots of these cells were subsequently transfected with control siRNA, B-myb_siRNA3 or B-myb_siRNA4 and analyzed by western blotting 24 h later.

Figure 4. Decreased S-phase entry after B-Myb knockdown is rescued by a DNA-binding deficient and RNAi-resistant B-Myb mutant. (A). HepG2 cells expressing doxycyclin-inducible B-Myb N174A mutsiRNA_4 were grown in the absence or presence of doxycyclin, followed by immunofluorescence analysis. Only the cell nuclei are visible. (B). The cells were additionally transfected with control or B-Myb-specific siRNA_4, followed by western blotting with antibodies against B-Myb and β-actin. (C). HepG2 cells expressing doxycyclin-inducible B-Myb N174A mutsiRNA_4 were grown in the presence or absence of doxycyclin and transfected with the indicated siRNAs. The cells were then analyzed for the percentage of EdU-positive cells (top) and by real-time PCR for the expression of the cdc2 and cyclin B1 mRNAs. The asterisks indicate statistical significance (* p < 0.05, ** p < 0.01, Student’s-t-test).
Pdip1/Ctb52/KCTD13 is a novel interaction partner of B-Myb
To further understand the function of B-Myb, we were interested to know if B-Myb interacts with proteins that are involved in DNA replication. In a previous attempt to identify novel B-Myb-interacting proteins we had incubated bacterially expressed GST/B-Myb fusion proteins with extracts of human cells to enrich for candidate B-Myb-binding proteins. This work led to the eventual identification of a protein known as Pdip1 (polymerase delta interacting protein 1), also known as KCTD13 (potassium channel tetramerization domain 13) or Ctb52,52 as a novel B-Myb binding protein (T. Bartusel and K.-H. Klempnauer, unpublished). Since Pdip1 has been shown to interact with two proteins that play important roles in DNA replication, PCNA and the p50 subunit of DNA-polymerase delta, we studied the interaction of B-Myb and Pdip1 in more detail. Figure 5A demonstrates the interaction of B-Myb and Pdip1 by co-immunoprecipitation experiments using cells that were transfected with expression vectors for both proteins. We found that Pdip1 was co-precipitated via B-Myb (lane 3) and, conversely, that B-Myb was co-precipitated via Pdip1 (lane 6). Control immunoprecipitations performed with cells transfected only with B-Myb (lanes 1 and 4) or Pdip1 (lanes 2 and 5) expression vectors demonstrated the specificity of the co-immunoprecipitation. A similar experiment showed that c-Myb was unable to interact with Pdip1 (Fig. 5B). This demonstrated the specificity of the co-immunoprecipitation and suggested that Pdip1 is a specific interaction partner of B-Myb.

Figure 5. Interaction of B-Myb and Pdip1. (A and B). QT6 cells were transfected with expression vectors for B-Myb, c-Myb and Myc-Pdip1, as indicated at the bottom. After 16 h, the cells were radiolabeled with 35S-methionine and cell extracts were subsequently immunoprecipitated with antibodies against B-Myb, c-Myb or the Myc-tag. Immunoprecipitates were then analyzed by SDS-PAGE and autoradiography. Protein bands corresponding to B-Myb, c-Myb and Pdip1 are marked. (C). Co-immunoprecipitation of endogenous B-Myb and Pdip1. Cell extract from HEK293 cells was precipitated with rabbit antiserum against Pdip1 or an unrelated protein (control IgG). The immunoprecipitates and an aliquot (5%) of the total-cell extract (TCE) were analyzed by western blotting with antibodies against B-Myb. The asterisk marks a non-specific protein band. The bottom part of (C) shows a sample of the total-cell extract stained with the Pdip1-specific antiserum.
To confirm the interaction of B-Myb and Pdip1 in cells expressing both proteins at their endogenous levels, we performed additional immunoprecipitations using untransfected cells. As shown in Figure 5C, B-Myb could be co-precipitated by Pdip1 antiserum but not by a control serum. Taken together, the data shown in Figure 5 demonstrated that Pdip1 is a novel B-Myb-interacting protein.
Mapping of domains mediating the interaction of Pdip1 and B-Myb
To characterize the B-Myb/Pdip1 interaction in more detail, we mapped the domains of both proteins involved in their interaction. To identify the part of Pdip1 that interacts with B-Myb we expressed the N- and C-terminal parts of Pdip1 individually and analyzed their interaction with B-Myb. Figure 6 shows that the C-terminal part of Pdip1 interacted very efficiently with B-Myb (see lanes 7), whereas the N-terminal part, which contains the BTB-domain, showed much weaker interaction (see lanes 5). Thus, the interaction of Pdip1 and B-Myb appears to be mediated mainly by the C-terminal part of Pdip1.

Figure 6. Mapping the binding region for B-Myb within Pdip1. Top: Schematic illustration of Pdip1 deletion constructs. The BTB-domain is highlighted. Bottom: QT6 cells were transfected with expression vectors for B-Myb and the indicated Myc-tagged Pdip1 constructs. Cells were then analyzed by immunoprecipitation as in Figure 5. Protein bands corresponding to B-Myb and Pdip1 constructs are marked.
To identify the interaction site for Pdip1 in B-Myb, we used the B-Myb constructs depicted schematically in Figure 7A. Figure 7B shows that a C-terminal deletion construct consisting mainly of the DNA-binding domain (B-MybΔPvu) interacted with Pdip1 less well than full-length B-Myb. Figure 7B also shows that deletion of the second and third repeat of the DNA-binding domain of B-Myb did not abolish the interaction with Pdip1. Likewise, swapping the N-terminal parts of B-Myb and c-Myb did not affect the interaction with Pdip1 (Fig. 7C). Taken together, these observations suggested that the B-Myb DNA-binding domain is involved in binding Pdip1; however, the main interaction between B-Myb and Pdip1 appeared to be due to the C-terminal part of B-Myb. This was confirmed by in vitro GST pull-down and in vivo “GFP trap” experiments which demonstrated binding of Pdip1 to the B-Myb transactivation domain and the adjoining C-terminal sequences (Fig. 8). These experiments showed that Pdip1 binds between amino acid residues 345 and 440 of B-Myb.

Figure 7. Mapping the binding region for Pdip1 within B-Myb. (A). Schematic illustration of B-Myb constructs. R1, R2, R3 and TAD refer to the repeats of the Myb DNA-binding domain and the transactivation domain, respectively. Small numbers refer to amino acids. c-Myb sequences are represented by black bars. (B and C). QT6 cells were transfected with expression vectors for Myc-Pdip1 and the indicated B-Myb constructs. Cells were radiolabeled with 35S-methionine and analyzed by immunoprecipitation with antibodies against B-Myb (top panels) or the Myc-tag (bottom panels), as described for Figure 5. In lanes 6 to 8 of (C), antiserum against c-Myb instead of B-Myb was used. Protein bands corresponding to Pdip1 and the B-Myb constructs are marked. Pdip1 co-precipitated via B-Myb is marked with an asterisk.

Figure 8. Pdip1 binds to sequences overlapping with the B-Myb transactivation domain. (A). Schematic illustration of GST- and YFP-B-Myb constructs. (B). GST-pull down experiments were performed with the indicated GST and GST-B-Myb fusion proteins and lysates of QT6 cells expressing Myc-Pdip1. Bound proteins (lanes 2 to 4) and an aliquot (10%) of the input sample (lane 1) were analyzed by western blotting using a Myc-specific antibody. An anti-GST western blot of the bacterial proteins used in the pull-down experiment is shown in the lower panel and demonstrates comparable protein amounts. (C). QT6 cells were transfected with the indicated combinations of plasmids encoding Myc-Pdip1, YFP-B-Myb, YFP-B-Myb D3 (345–440) and YFP. Cells were lysed 24 h post-transfection and the soluble fraction was incubated with GFP-trap beads, resulting in the binding of YFP-fusion proteins and their binding partners to the beads. Bound proteins and aliquots (6%) of the input extracts were analyzed by western blotting with antibodies against Myc and GFP.
Because Pdip1 interacts with the transactivation domain of B-Myb, we were interested to know if Pdip1 affects the transactivation potential of B-Myb. The results of reporter gene experiments using a Myb-responsive promoter construct are illustrated in Figure S3. The reporter gene was stimulated by full-length B-Myb to the same extent in the presence or absence of Pdip1. Since full-length B-Myb is a rather weak transactivator due to the suppressive effect of the C terminus,5,9 we also used a C-terminally truncated B-Myb construct (B-MybΔSac), which is more active.11 However, the activity of the truncated B-Myb was also not affected by Pdip1. Thus, it appears that Pdip1 does not influence the transactivation potential of B-Myb.
Pdip1 is able to tether B-Myb to PCNA
Previous work had shown that Pdip1 interacts with PCNA.53 We were therefore interested to investigate whether Pdip1 facilitates an interaction of B-Myb with PCNA. To address this question we co-transfected different combinations of expression vectors for B-Myb, Myc-Pdip1 and GFP-PCNA, followed by immunoprecipitation of extracts of the transfected cells. Figure 9A shows that antibodies against B-Myb were unable to co-precipitate GFP-PCNA when Pdip1 was not co-expressed (lane 3), whereas GFP-PCNA was co-precipitated with B-Myb in the presence of Pdip1 (lanes 4 and 5). This suggested that Pdip1 is able to tether PCNA to B-Myb. We then wished to confirm that B-Myb and PCNA are also co-precipitated as endogenous proteins from untransfected cells. Figure 9B shows that a fraction of B-Myb was indeed co-precipitated with antibodies against PCNA but not with unrelated control antibodies. Taken together, these experiments suggested that fractions of B-Myb and PCNA are present in a complex, with Pdip1 possibly facilitating the interaction of both proteins.

Figure 9. Co-immunoprecipitation of B-Myb and PCNA. (A). QT6 cells were transfected with expression vectors for B-Myb, GFP-PCNA and Myc-Pdip1, as indicated at the bottom. Cell extracts prepared 16 h after transfection were then immunoprecipitated with antibodies against B-Myb. The immunoprecipitates and aliquots (5%) of the total-cell extract (TCE) were analyzed by western blotting with antibodies against B-Myb (top panels) or GFP (bottom panels). Protein bands corresponding to B-Myb and GFP-PCNA are marked. (B). Co-immunoprecipitation of endogenous B-Myb and PCNA. Cell extracts from untransfected Hek293 cells were precipitated with antibodies against PCNA or with control IgG, as indicated. The immunoprecipitates and aliquots (2.5%) of the total-cell extracts (TCE) were analyzed by western blotting with antibodies against B-Myb. The asterisk marks a non-specific protein band.
Discussion
B-Myb is a highly conserved member of the Myb transcription factor family, which is involved in cell cycle regulation and performs essential functions in proliferating cells. While recent work on B-Myb has provided insight into its role in the transcriptional regulation of gene expression at the G2/M-transition, relatively little is known about the function of B-Myb at the G1/S-transition and in S-phase, when its expression is highest.54 Our results reveal two novel aspects of the function of B-Myb and, thereby, shed new light on the role of B-Myb in this part of the cell cycle. First, by employing siRNA-induced silencing of B-Myb expression we have shown that B-Myb is required for S-phase entry and progression. Surprisingly, we found that expression of a mutant of B-Myb that lacks sequence-specific DNA-binding activity and is unable to activate bona fide Myb target genes is still able to rescue the defect in S-phase entry after knockdown of endogenous B-Myb. This suggests that the effects of B-Myb knockdown on S-phase are not due to a lack of activation of specific B-Myb target genes, but rather argues for a role of B-Myb in S-phase that is independent of its function as a sequence-specific DNA-binding protein. In agreement with this, Zhu et al. showed that B-Myb binds to G2/M-regulated but not to G1/S-regulated promoters.55 The finding that DNA binding-deficient B-Myb is able to promote S-phase is reminiscent of older studies, which showed that B-Myb is able to partially overcome a G1-block induced by overexpression of the pocket protein p107 in an apparently transcription-independent manner.13 Together with these studies, our work strongly supports the notion that B-Myb carries out functions at the beginning of S-phase that are independent of its role as a classical transcription factor.
Second, we have identified the BTB-domain protein Pdip1/KCTD13/Ctb52 as a novel binding partner of B-Myb. Pdip1 is an as yet poorly characterized but highly conserved protein that was originally identified in a yeast two hybrid screen using the p50 subunit of DNA polymerase δ as bait and subsequently shown to bind to PCNA.53 PCNA plays a key role as sliding clamp at the replication fork and also interacts with and orchestrates the activities of various proteins involved in DNA repair.56,57 Pdip1 has been reported to co-localize with foci of newly replicated DNA and to stimulate the activity of DNA-polymerase δ in vitro in a PCNA-dependent manner.53 Together, these observations strongly suggest that Pdip1 is involved in the replication and/or repair of DNA, although its precise function is not known. Pdip1 is closely related to two other BTB domain-containing proteins, TNFAIP1 (tumor necrosis factor α-induced protein 1) and KCTD10 (potassium channel tetramerisation domain 10).58 Preliminary evidence based on in vivo co-immunoprecipitation studies indicates that B-Myb also interacts with TNFAIP1 (T.S., unpublished observations). The identification of Pdip1 as a novel B-Myb-interacting protein suggests the interesting possibility that B-Myb, in addition to its role as a transcriptional activator of G2/M-phase genes, might also be involved in certain aspects of DNA replication and/or repair. This idea is consistent with our co-immunoprecipitation experiments, which have provided initial evidence that Pdip1 might possibly act as an adaptor that tethers B-Myb to PCNA. That a fraction of endogenous B-Myb co-precipitates with PCNA is also consistent with our hypothesis that B-Myb might have a role in DNA replication or repair.
The idea that B-Myb might not solely act as a classical DNA-binding transcription factor during S-phase but might be involved in processes related to DNA replication or repair is in line with previous work on the Drosophila melanogaster myb homolog. Drosophila myb is thought to be the homolog of mammalian B-myb, rather than c-myb or A-myb, because B-myb is the only mammalian myb family member that is able to complement the cell cycle defects seen in Myb-null Drosophila hemocytes.59 Besides its role in the transcription of G2/M-phase genes, dmMyb interacts with Orc2, a component of the DNA replication initiation machinery, and is directly involved in the initiation of DNA replication at the chorion gene origin of DNA replication.42 Additional evidence for a link between B-Myb and DNA replication has been provided by recent work in mammalian cells, which showed that ablation of B-Myb expression in mouse embryonic stem cells results in the disorganization of the DNA replication program, including a slow-down of the speed and collapse of the replication forks and an increase of the number of replication foci.43 Lorvellec et al.43 have suggested that the effect on DNA replication observed upon B-Myb ablation might be mediated by a decrease of c-Myc expression. However, we have clearly shown that Myc expression is not affected by the B-Myb knockdown in our cell system (Fig. S1). This indicates that the effects of B-Myb knockdown on S-phase observed here are not due to changes in the level of c-Myc expression.
At present, the exact nature of the proposed novel function of B-Myb in S-phase is not known. B-Myb might play a role at certain replication origins, similar to the function of dmMyb at the chorion gene replication origin.42 Pdip1 has also been identified as interaction partner of the cullin3 ubiquitin ligase,52 which raises the possibility that B-Myb might also influence the ubiquitinylation of proteins during DNA replication or might itself be ubiquitinylated in a cullin3-dependent manner. These possibilities have to be addressed in further work. Nevertheless, the data presented here provide strong evidence for a novel role of B-Myb in S-phase that is independent of its sequence-specific DNA-binding activity and its ability to stimulate the expression of bona fide B-Myb target genes.
Materials and Methods
Cells
QT6, DF-1 and HEK293 cells are quail and chicken fibroblast lines and transformed human embryonic kidney cells, respectively. HepG2 is a human hepatocellular liver carcinoma line. HepG2 cells were synchronized by treatment with 0.5 mM mimosine for 20 h. Mimosine-arrested cells were released into S-phase by two washes with growth medium lacking mimosine. Cell cycle analysis was performed by fixing the cells with 70% ethanol in PBS for 1 h at -20°C and staining them for 1 h at room temperature with PBS containing 50 μg/ml propidium iodide and 25 μg/ml RNaseA. FACS analysis was performed using a Beckman-Coulter Cytomics FC500 flow cytometer.
Expression vectors
Expression vectors for full-length mouse B-Myb (pCDNA3-MuBMyb), derivatives lacking the second and third repeat of the B-Myb DNA-binding domain (pCDNA3-B-Myb-ΔDBD) or truncated from the C terminus (pCDNA3-B-Myb-ΔPvu) and c-Myb (pCDNA3-Mu-c-Myb) have been described.9,22 Expression vectors for the recombinant proteins B_c-Myb and c_B-Myb were generated by using AgeI restriction sites located at equivalent positions in the B-myb and c-myb coding regions to exchange the N-terminal parts of B-Myb and c-Myb. Plasmid pCDNA4-B-Myb N174A mutsiRNA_4 encodes full-length B-Myb mutated in the DNA-binding domain by replacing asparagine 174 with alanine. In addition, silent nucleotide substitutions were introduced into the target region of B-myb siRNA_4, thereby changing the B-myb sequence from GAAACATGCTGCGTTTGTA to GAAGCACGCAGCTTTCGTT. Expression vectors for GST-B-Myb-D2 and D3 were generated by fusing B-Myb aminoacids 240–371 (D2) and 345–530 (D3) to GST. Expression vectors for the GST-fused B-Myb DNA-binding domain were generated by cloning a 821 bp BamHI/EcoRI fragment from pCDNA4-B-Myb wt or N174A between the BamHI and EcoRI sites of pGEX5-X-3. The expression vector pCS2 + Ctb52 encodes Myc-tagged Pdip1.52 An expression vector for the N-terminal part of Pdip1 (pCDNA3-Pdip1-ΔSma) was generated by cloning a 750 bp BamHI/SmaI fragment from pCS2 + Ctb52 between the BamHI and EcoRV sites of pCDNA3 (Invitrogen). The expression vector for the C-terminal part of Pdip1 (pCDNA3-Pdip1-C-term) was constructed by deleting a 450 bp NcoI fragment from plasmid pCS2 + Ctb52, resulting an N-terminally truncated protein initiated at methionine 152 of Pdip1. An expression vector encoding GFP-tagged PCNA was obtained from C. Cardoso.
Immunoprecipitation and antibodies
Cells were lysed in ELB buffer (50 mM Tris/HCl pH 7,5; 120 mM NaCl; 20 mM NaF; 1 mM EDTA; 6 mM EGTA; 15 mM sodium pyrophosphate; 1 mM phenylmethylsulfonyl fluoride; 0,2% NP-40; 10% glycerol and a protease inhibitor mix containing Aprotinin, Leupeptin and Pepstatin). After incubation on ice for 30 min, lysates were centrifuged at 14000 × g for 30 min, and the supernatant was used as total protein extract. Aliquots of the total protein extract were immunoprecipitated with the appropriate antibodies for 1 and up to 12 h at 4°C. Protein-A/G Sepharose beads were then added and incubated further for 3 h at 4°C under constant agitation. Immune complexes bound to the beads were collected by centrifugation, washed five times with lysis buffer and finally subjected to SDS-PAGE.35S-methionine labeled proteins were visualized by autoradiography of the dried gels using a phosphor-imager. Unlabeled proteins were transferred to nitrocellulose membranes followed by immunostaining with the appropriate antibodies. Image acquisition was performed by exposure of stained blots to X-ray films.
Polyclonal rabbit antisera against mouse B-Myb and mouse c-Myb have been described.9,22 A monoclonal anti-mouse B-Myb antibody was kindly provided by R. Watson. The following commercial antibodies were used: polyclonal rabbit anti-human PCNA (AB18197, Abcam), monoclonal mouse anti-human PCNA (PC10, Sigma-Aldrich), polyclonal rabbit anti-human Pdip1 (AB32974, Abcam), polyclonal rabbit anti-human histone H3 (AB1791, Abcam), monoclonal mouse anti β-actin (AC-15, Sigma-Aldrich), monoclonal mouse anti GFP (Sigma-Aldrich), monoclonal mouse anti Myc-epitope (9E10), monoclonal mouse anti-Flag-epitope (M2, Sigma-Aldrich) and polyclonal rabbit anti c-Myc (sc-764, St. Cruz). Polyclonal anti GST antibodies were raised against bacterially expressed GST.
GST pull-down and GFP-trap experiments
Expression and purification of GST-proteins and in vitro pull-down experiments were performed as described.10 For GFP-trap experiments, QT6 cells were transfected with expression vectors for Pdip1 and YFP fusion proteins. Cells were lysed in ELB buffer 24 h after transfection. The cell extract was then incubated with GFP-trap beads (Chromotec, München) for 3 h at 4°C. Beads were washed three times with ELB buffer and bound proteins as well as samples of the input cell extracts were analyzed by western blotting using anti Myc or anti GFP antibodies.
Immunofluorescence analysis and staining of newly replicated DNA
Cells were fixed with 2% paraformaldehyde in PBS for 10 min, followed by incubation with 50 mM NH4Cl for 10 min at room temperature. The cells were then permeabilized with PBS containing 0.2% NP40 for 10 min at room temperature and immunostained with the desired primary antibodies. Alexa Fluor® 488-coupled anti-mouse (A11059, Invitrogen) or anti-rabbit (A11034, Invitrogen) immunoglobulins were used as secondary antibodies. For EdU staining, cells were pre-labeled for 5 min with 10 μM EdU prior to fixation. Copper-catalyzed click reactions were performed using click-iT® EdU Alexa Fluor® 594 Imaging kit (Invitrogen), following the protocol of the manufacturer.
RNA interference
B-Myb expression was silenced with siRNA duplexes targeting the sequences CUGGAACUCUACCAUCAAA (B-myb siRNA_3), GAAACAUGCUGCGUUUGUA (B-myb siRNA_4). siRNA targeting Renilla luciferase (AAACAUGCAGAAAAUGCUG) was used as negative control. Cells were grown in 6-well plates to approximately 50–80% confluence and received 1.5 ml fresh growth medium prior to transfection. Then 100 nM siRNA was transfected using MetafecteneTM Pro (Biontex), according to manufacturers' protocols. Cells were harvested 48 h later, and the level of knockdown was evaluated by western blotting, flow cytometry or immunostaining, as appropriate. If cell synchronization was required, mimosine (0.5 mM) was added 24 h after transfection and the cells were released into S-phase and analyzed by FACS 24 h later.
Electrophoretic mobility shift assay (EMSA)
Two complementary oligonucleotides (GCTCTAAAAAACCGTTATAATGTACAGATATCTT and AAGATATCTGTACATTATAACGGTTTTTTAGAG) containing a Myb binding site were annealed and radiolabeled by filling-in the ends with α32P-dCTP. Bacterially expressed GST/B-Myb (1–271) wt or GST/B-Myb (1–271) N174A was purified by binding to glutathion-sepharose followed by elution of the bound protein with glutathione. Gel retardation assays were performed as described.60
Real-time PCR
Total cellular RNA was isolated with the Nucleo Spin® RNA II Kit (Macherey-Nagel), following the instructions of the manufacturer. First-strand cDNA synthesis was performed with a cDNA kit (Fermentas). The cDNAs were analyzed by quantitative real-time PCR with the following primers. Cdc2: 5′- CATGGCTACCACTTGACCTGT-3′ and 5′- AAGCCGGGATCTACCATACC-3′, CycB1: 5′- CAGATGTTTCCATTGGGCTT-3′ and 5′- TACCTATGCTGGTGCCAGTG-3′, β-actin: 5′-CGTCCACCGCAAATGCTT-3′ and 5′-GTTTTCTGCGCAAGTTAGGT-3′.
Supplementary Material
Acknowledgments
We thank Cristina Cardoso for plasmids and Roger Watson for the monoclonal anti B-Myb antibodies. This work was supported by the DFG (grant KL461/14–1) and the Graduate School of Chemistry (GSC-MS) at the University of Muenster (K.H.K. and E.W.). This work was also supported by NIH grant 5RO1GM082940 (J.D.S.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/22386
References
- 1.Joaquin M, Watson RJ. Cell cycle regulation by the B-Myb transcription factor. Cell Mol Life Sci. 2003;60:2389–401. doi: 10.1007/s00018-003-3037-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sala A. B-MYB, a transcription factor implicated in regulating cell cycle, apoptosis and cancer. Eur J Cancer. 2005;41:2479–84. doi: 10.1016/j.ejca.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 3.Lam EW, Watson RJ. An E2F-binding site mediates cell-cycle regulated repression of mouse B-myb transcription. EMBO J. 1993;12:2705–13. doi: 10.1002/j.1460-2075.1993.tb05932.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartsch O, Horstmann S, Toprak K, Klempnauer KH, Ferrari S. Identification of cyclin A/Cdk2 phosphorylation sites in B-Myb. Eur J Biochem. 1999;260:384–91. doi: 10.1046/j.1432-1327.1999.00191.x. [DOI] [PubMed] [Google Scholar]
- 5.Lane S, Farlie P, Watson R. B-Myb function can be markedly enhanced by cyclin A-dependent kinase and protein truncation. Oncogene. 1997;14:2445–53. doi: 10.1038/sj.onc.1201086. [DOI] [PubMed] [Google Scholar]
- 6.Robinson C, Light Y, Groves R, Mann D, Marias R, Watson R. Cell-cycle regulation of B-Myb protein expression: specific phosphorylation during the S phase of the cell cycle. Oncogene. 1996;12:1855–64. [PubMed] [Google Scholar]
- 7.Sala A, Kundu M, Casella I, Engelhard A, Calabretta B, Grasso L, et al. Activation of human B-MYB by cyclins. Proc Natl Acad Sci USA. 1997;94:532–6. doi: 10.1073/pnas.94.2.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saville MK, Watson RJ. The cell-cycle regulated transcription factor B-Myb is phosphorylated by cyclin A/Cdk2 at sites that enhance its transactivation properties. Oncogene. 1998;17:2679–89. doi: 10.1038/sj.onc.1202503. [DOI] [PubMed] [Google Scholar]
- 9.Ziebold U, Bartsch O, Marais R, Ferrari S, Klempnauer KH. Phosphorylation and activation of B-Myb by cyclin A-Cdk2. Curr Biol. 1997;7:253–60. doi: 10.1016/S0960-9822(06)00121-7. [DOI] [PubMed] [Google Scholar]
- 10.Horstmann S, Ferrari S, Klempnauer KH. Regulation of B-Myb activity by cyclin D1. Oncogene. 2000;19:298–306. doi: 10.1038/sj.onc.1203302. [DOI] [PubMed] [Google Scholar]
- 11.Schubert S, Horstmann S, Bartusel T, Klempnauer KH. The cooperation of B-Myb with the coactivator p300 is orchestrated by cyclins A and D1. Oncogene. 2004;23:1392–404. doi: 10.1038/sj.onc.1207255. [DOI] [PubMed] [Google Scholar]
- 12.Cervellera MN, Sala A. Poly(ADP-ribose) polymerase is a B-MYB coactivator. J Biol Chem. 2000;275:10692–6. doi: 10.1074/jbc.275.14.10692. [DOI] [PubMed] [Google Scholar]
- 13.Joaquin M, Bessa M, Saville MK, Watson RJ. B-Myb overcomes a p107-mediated cell proliferation block by interacting with an N-terminal domain of p107. Oncogene. 2002;21:7923–32. doi: 10.1038/sj.onc.1206001. [DOI] [PubMed] [Google Scholar]
- 14.Ying GG, Proost P, van Damme J, Bruschi M, Introna M, Golay J. Nucleolin, a novel partner for the Myb transcription factor family that regulates their activity. J Biol Chem. 2000;275:4152–8. doi: 10.1074/jbc.275.6.4152. [DOI] [PubMed] [Google Scholar]
- 15.Bessa M, Saville MK, Watson RJ. Inhibition of cyclin A/Cdk2 phosphorylation impairs B-Myb transactivation function without affecting interactions with DNA or the CBP coactivator. Oncogene. 2001;20:3376–86. doi: 10.1038/sj.onc.1204439. [DOI] [PubMed] [Google Scholar]
- 16.Johnson LR, Johnson TK, Desler M, Luster TA, Nowling T, Lewis RE, et al. Effects of B-Myb on gene transcription: phosphorylation-dependent activity ans acetylation by p300. J Biol Chem. 2002;277:4088–97. doi: 10.1074/jbc.M105112200. [DOI] [PubMed] [Google Scholar]
- 17.Bartusel T, Klempnauer KH. Transactivation mediated by B-Myb is dependent on TAF(II)250. Oncogene. 2003;22:2932–41. doi: 10.1038/sj.onc.1206494. [DOI] [PubMed] [Google Scholar]
- 18.Li X, McDonnell DP. The transcription factor B-Myb is maintained in an inhibited state in target cells through its interaction with the nuclear corepressors N-CoR and SMRT. Mol Cell Biol. 2002;22:3663–73. doi: 10.1128/MCB.22.11.3663-3673.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Masselink H, Vastenhouw N, Bernards R. B-myb rescues ras-induced premature senescence, which requires its transactivation domain. Cancer Lett. 2001;171:87–101. doi: 10.1016/S0304-3835(01)00631-0. [DOI] [PubMed] [Google Scholar]
- 20.Santilli G, Cervellera MN, Johnson TK, Lewis RE, Iacobelli S, Sala A. PARP co-activates B-MYB through enhanced phosphorylation at cyclin/cdk2 sites. Oncogene. 2001;20:8167–74. doi: 10.1038/sj.onc.1204943. [DOI] [PubMed] [Google Scholar]
- 21.Charrasse S, Carena I, Brondani V, Klempnauer KH, Ferrari S. Degradation of B-Myb by ubiquitin-mediated proteolysis: involvement of the Cdc34-SCF(p45Skp2) pathway. Oncogene. 2000;19:2986–95. doi: 10.1038/sj.onc.1203618. [DOI] [PubMed] [Google Scholar]
- 22.Kamano H, Burk B, Noben-Trauth K, Klempnauer KH. Differential splicing of the mouse B-myb gene. Oncogene. 1995;11:2575–82. [PubMed] [Google Scholar]
- 23.Tarasov KV, Tarasova YS, Tam WL, Riordon DR, Elliott ST, Kania G, et al. B-MYB is essential for normal cell cycle progression and chromosomal stability of embryonic stem cells. PLoS One. 2008;3:e2478. doi: 10.1371/journal.pone.0002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tanaka Y, Patestos NP, Maekawa T, Ishii S. B-myb is required for inner cell mass formation at an early stage of development. J Biol Chem. 1999;274:28067–70. doi: 10.1074/jbc.274.40.28067. [DOI] [PubMed] [Google Scholar]
- 25.Korenjak M, Taylor-Harding B, Binné UK, Satterlee JS, Stevaux O, Aasland R, et al. Native E2F/RBF complexes contain Myb-interacting proteins and repress transcription of developmentally controlled E2F target genes. Cell. 2004;119:181–93. doi: 10.1016/j.cell.2004.09.034. [DOI] [PubMed] [Google Scholar]
- 26.Lewis PW, Beall EL, Fleischer TC, Georlette D, Link AJ, Botchan MR. Identification of a Drosophila Myb-E2F2/RBF transcriptional repressor complex. Genes Dev. 2004;18:2929–40. doi: 10.1101/gad.1255204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Georlette D, Ahn S, MacAlpine DM, Cheung E, Lewis PW, Beall EL, et al. Genomic profiling and expression studies reveal both positive and negative activities for the Drosophila Myb MuvB/dREAM complex in proliferating cells. Genes Dev. 2007;21:2880–96. doi: 10.1101/gad.1600107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wen H, Andrejka L, Ashton J, Karess R, Lipsick JS. Epigenetic regulation of gene expression by Drosophila Myb and E2F2-RBF via the Myb-MuvB/dREAM complex. Genes Dev. 2008;22:601–14. doi: 10.1101/gad.1626308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Litovchick L, Sadasivam S, Florens L, Zhu X, Swanson SK, Velmurugan S, et al. Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol Cell. 2007;26:539–51. doi: 10.1016/j.molcel.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 30.Pilkinton M, Sandoval R, Colamonici OR. Mammalian Mip/LIN-9 interacts with either the p107, p130/E2F4 repressor complex or B-Myb in a cell cycle-phase-dependent context distinct from the Drosophila dREAM complex. Oncogene. 2007;26:7535–43. doi: 10.1038/sj.onc.1210562. [DOI] [PubMed] [Google Scholar]
- 31.Schmit F, Korenjak M, Mannefeld M, Schmitt K, Franke C, von Eyss B, et al. LINC, a human complex that is related to pRB-containing complexes in invertebrates regulates the expression of G2/M genes. Cell Cycle. 2007;6:1903–13. doi: 10.4161/cc.6.15.4512. [DOI] [PubMed] [Google Scholar]
- 32.Knight AS, Notaridou M, Watson RJA. A Lin-9 complex is recruited by B-Myb to activate transcription of G2/M genes in undifferentiated embryonal carcinoma cells. Oncogene. 2009;28:1737–47. doi: 10.1038/onc.2009.22. [DOI] [PubMed] [Google Scholar]
- 33.Osterloh L, von Eyss B, Schmit F, Rein L, Hübner D, Samans B, et al. The human synMuv-like protein LIN-9 is required for transcription of G2/M genes and for entry into mitosis. EMBO J. 2007;26:144–57. doi: 10.1038/sj.emboj.7601478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fung SM, Ramsay G, Katzen AL. Mutations in Drosophila myb lead to centrosome amplification and genomic instability. Development. 2002;129:347–59. doi: 10.1242/dev.129.2.347. [DOI] [PubMed] [Google Scholar]
- 35.Katzen AL, Jackson J, Harmon BP, Fung SM, Ramsay G, Bishop JM. Drosophila myb is required for the G2/M transition and maintenance of diploidy. Genes Dev. 1998;12:831–43. doi: 10.1101/gad.12.6.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manak JR, Mitiku N, Lipsick JS. Mutation of the Drosophila homologue of the Myb protooncogene causes genomic instability. Proc Natl Acad Sci USA. 2002;99:7438–43. doi: 10.1073/pnas.122231599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Manak JR, Wen H, Van T, Andrejka L, Lipsick JS. Loss of Drosophila Myb interrupts the progression of chromosome condensation. Nat Cell Biol. 2007;9:581–7. doi: 10.1038/ncb1580. [DOI] [PubMed] [Google Scholar]
- 38.Shepard JL, Amatruda JF, Stern HM, Subramanian A, Finkelstein D, Ziai J, et al. A zebrafish bmyb mutation causes genome instability and increased cancer susceptibility. Proc Natl Acad Sci USA. 2005;102:13194–9. doi: 10.1073/pnas.0506583102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahlbory D, Appl H, Lang D, Klempnauer KH. Disruption of B-myb in DT40 cells reveals novel function for B-Myb in the response to DNA-damage. Oncogene. 2005;24:7127–34. doi: 10.1038/sj.onc.1208869. [DOI] [PubMed] [Google Scholar]
- 40.García P, Frampton J. The transcription factor B-Myb is essential for S-phase progression and genomic stability in diploid and polyploid megakaryocytes. J Cell Sci. 2006;119:1483–93. doi: 10.1242/jcs.02870. [DOI] [PubMed] [Google Scholar]
- 41.Yamauchi T, Ishidao T, Nomura T, Shinagawa T, Tanaka Y, Yonemura S, et al. A B-Myb complex containing clathrin and filamin is required for mitotic spindle function. EMBO J. 2008;27:1852–62. doi: 10.1038/emboj.2008.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beall EL, Manak JR, Zhou S, Bell M, Lipsick JS, Botchan MR. Role for a Drosophila Myb-containing protein complex in site-specific DNA replication. Nature. 2002;420:833–7. doi: 10.1038/nature01228. [DOI] [PubMed] [Google Scholar]
- 43.Lorvellec M, Dumon S, Maya-Mendoza A, Jackson D, Frampton J, García P. B-Myb is critical for proper DNA duplication during an unperturbed S phase in mouse embryonic stem cells. Stem Cells. 2010;28:1751–9. doi: 10.1002/stem.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keller C, Hyrien O, Knippers R, Krude T. Site-specific and temporally controlled initiation of DNA replication in a human cell-free system. Nucleic Acids Res. 2002;30:2114–23. doi: 10.1093/nar/30.10.2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krude T. Mimosine arrests proliferating human cells before onset of DNA replication in a dose-dependent manner. Exp Cell Res. 1999;247:148–59. doi: 10.1006/excr.1998.4342. [DOI] [PubMed] [Google Scholar]
- 46.Krude T. Initiation of chromosomal DNA replication in mammalian cell-free systems. Cell Cycle. 2006;5:2115–22. doi: 10.4161/cc.5.18.3248. [DOI] [PubMed] [Google Scholar]
- 47.Dominguez-Sola D, Ying CY, Grandori C, Ruggiero L, Chen B, Li M, et al. Non-transcriptional control of DNA replication by c-Myc. Nature. 2007;448:445–51. doi: 10.1038/nature05953. [DOI] [PubMed] [Google Scholar]
- 48.McIntosh PB, Frenkiel TA, Wollborn U, McCormick JE, Klempnauer K-H, Feeney J, et al. Solution structure of the B-Myb DNA-binding domain: a possible role for conformational instability of the protein in DNA binding and control of gene expression. Biochemistry. 1998;37:9619–29. doi: 10.1021/bi972861z. [DOI] [PubMed] [Google Scholar]
- 49.Ogata K, Morikawa S, Nakamura H, Sekikawa A, Inoue T, Kanai H, et al. Solution structure of a specific DNA complex of the Myb DNA-binding domain with cooperative recognition helices. Cell. 1994;79:639–48. doi: 10.1016/0092-8674(94)90549-5. [DOI] [PubMed] [Google Scholar]
- 50.Gabrielsen OS, Sentenac A, Fromageot P. Specific DNA binding by c-Myb: evidence for a double helix-turn-helix-related motif. Science. 1991;253:1140–3. doi: 10.1126/science.1887237. [DOI] [PubMed] [Google Scholar]
- 51.Saikumar P, Murali R, Reddy EP. Role of tryptophan repeats and flanking amino acids in Myb-DNA interactions. Proc Natl Acad Sci USA. 1990;87:8452–6. doi: 10.1073/pnas.87.21.8452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cummings CM, Bentley CA, Perdue SA, Baas PW, Singer JD. The Cul3/Klhdc5 E3 ligase regulates p60/katanin and is required for normal mitosis in mammalian cells. J Biol Chem. 2009;284:11663–75. doi: 10.1074/jbc.M809374200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.He H, Tan CK, Downey KM, So AG. A tumor necrosis factor alpha- and interleukin 6-inducible protein that interacts with the small subunit of DNA polymerase delta and proliferating cell nuclear antigen. Proc Natl Acad Sci USA. 2001;98:11979–84. doi: 10.1073/pnas.221452098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lam EW, Robinson C, Watson RJ. Characterization and cell cycle-regulated expression of mouse B-myb. Oncogene. 1992;7:1885–90. [PubMed] [Google Scholar]
- 55.Zhu W, Giangrande PH, Nevins JR. E2Fs link the control of G1/S and G2/M transcription. EMBO J. 2004;23:4615–26. doi: 10.1038/sj.emboj.7600459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maga G, Hubscher U. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J Cell Sci. 2003;116:3051–60. doi: 10.1242/jcs.00653. [DOI] [PubMed] [Google Scholar]
- 57.Moldovan GL, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell. 2007;129:665–79. doi: 10.1016/j.cell.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 58.Zhou J, Hu X, Xiong X, Liu X, Liu Y, Ren K, et al. Cloning of two rat PDIP1 related genes and their interactions with proliferating cell nuclear antigen. J Exp Zool A Comp. Exp Biol. 2005;303:227–40. doi: 10.1002/jez.a.150. [DOI] [PubMed] [Google Scholar]
- 59.Davidson CJ, Tirouvanziam R, Herzenberg LA, Lipsick JS. Functional evolution of the vertebrate Myb gene family: B-Myb, but neither A-Myb nor c-Myb, complements Drosophila Myb in hemocytes. Genetics. 2005;169:215–29. doi: 10.1534/genetics.104.034132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chayka O, Kintscher J, Braas D, Klempnauer KH. v-Myb mediates cooperation of a cell-specific enhancer with the mim-1 promoter. Mol Cell Biol. 2005;25:499–511. doi: 10.1128/MCB.25.1.499-511.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.