

Abstract
Lower respiratory tract virus infections are the major cause of asthma exacerbations. Severity of infection and age at initial encounter with virus appear to be major determinants of the risk for allergic asthma later in life. In animal models, reinfection of mice initially infected as neonates leads to markedly enhanced alterations in airway function and inflammation, unlike reinfection of older mice. Both innate and adaptive immune responses contribute to this susceptibility with lung dendritic cells showing marked differences in phenotype and function in young compared to older mice, and these differences are further enhanced following virus infection. These findings have implications for therapeutic targeting, for example, of RSV G and F surface proteins at different stages of the response to infection.
Introduction
The importance of virus infections in asthma requires little debate. There is overwhelming evidence for the importance of infection with respiratory viruses in the exacerbation of asthma in both children and adults. Using molecular diagnostics, a viral pathogen has been identified in the majority of the wheezing episodes which occur in the first years of life [1]. Indeed, all children in early life have been infected with a respiratory virus with more than half experiencing lower respiratory tract disease together with a wheezing episode before school age [2]. The most common viruses associated with these early onset wheezing episodes are respiratory syncytial virus (RSV), human rhinovirus (HRV), and metapneumovirus. The ties between respiratory virus infections in early life and development of asthma in childhood have been the subject of many papers and reviews [3–5]. Although respiratory viruses are associated with asthma exacerbations, the underlying mechanisms are not entirely clear. Added to the complexities of the associations are genetic factors that govern the immune response to these viruses, attributes of the viruses themselves and potentially important differences among the strains of virus. The aim of this article is not to continue the debate about viruses triggering asthma but to review two important aspects pertinent to the association of respiratory viruses and asthma in humans and in experimental asthma mice.
Importance of Age at First Infection and Response to Reinfection
Although the debate about a causal link between virus infection and reactive airway disease remains unresolved, there is increasing evidence to suggest that the age at initial infection is an important factor. Several studies have demonstrated that children hospitalized for RSV bronchiolitis during infancy were more likely to have subsequent episodes of wheezing and asthma compared to non-hospitalized children [6, 7]. In infants, young age at first RSV infection has been associated with T helper (Th) 2 responses [8]. This was demonstrated by higher levels of IL-4 in RSV-positive infants younger than 3 months of age compared to those older than 3 months of age. This may suggest that for RSV infection, the earlier the first infection, the more skewed the immune response may be. Less consistent were results in a cohort of infants with a family history of allergy and asthma where peripheral blood mononuclear cell responses to activation by the mitogenic lectin phytohemagglutinin or to RSV and rhinovirus were examined [9]. Children who wheezed with RSV infection had reduced PHA-induced IL-13 production at birth, which diminished marginally after the first year of life. Somewhat in contrast, the wheezing infant had lower PHA- and RV-induced IFN-γ responses unlike those with detectable IFN responses at birth who were less likely to wheeze in the first year. These studies cannot answer the critical question of whether RSV, HRV, or other early respiratory virus infection does indeed lead to asthma or the infection, especially severe bronchiolitis, is simply a marker or predictor. In addition, it is unclear in these infant studies whether the immune response is altered by the virus infection or whether the nature of the response is related to a developmental stage of the immune system at the time of initial infection.
Other approaches have addressed the issue of whether early RSV infection alters the host to increase the subsequent risk of asthma or is merely a marker of an underlying predisposition for asthma. The Tennessee Asthma Bronchiolitis study reported that infants with a healthcare visit for bronchiolitis during RSV-dominant months (December to February) were more likely to develop asthma between 4 and 5.5 years of age [10]. Thus, infants born approximately 4 months prior to the first winter viral peak encounter following birth were at the highest risk of developing childhood asthma. In this cohort, it appeared that the more severe the infant bronchiolitis event, the greater the odds of developing asthma [11]. This extended to severity defined by hospitalization, emergency department visits for asthma, or need for rescue corticosteroids. These results have been confirmed to a large extent in similar although not identical cohorts [7].
It also follows that initial host response to viral infection, crucial to asthma pathogenesis, is in part genetically programmed. Genetic polymorphisms relevant to atopy, asthma, and infection have been described for both innate and adaptive immunity [12]. In fact, there are many polymorphisms among the toll-like receptors that are associated with response to infection and infection severity, and to atopy and asthma, indicating common genetic determinants for virus infection and asthma [13–16]. As these data accumulate, there is increasing evidence to support the notion that genetic polymorphisms control susceptibility to severe respiratory virus infection and, as a result, the risk for subsequent development of asthma.
There is now evidence that different RSV strains impact the immune system and airway function differently, at least in mice and in primary human bronchial epithelial cells [17, 18]. Thus, the interplay between age at initial infection, genetic susceptibility polymorphisms and virus strain will likely dictate the outcome of respiratory virus infection and airway disease.
Impact of Age at Initial Infection in Animal Models of RSV Disease
Because of the many limitations to establish causality of virus infection and asthma in humans, many species have been used to develop animal models of human disease, with studies in mice and RSV infection predominating [5]. Most of these studies utilized intranasal inoculation of human RSV with viral replication peaking 3–4 days after infection. In mice, RSV can induce the production of a wide variety of cytokines (IFN-γ, IL-4, IL-5, IL-10, IL-12, and IL-13) and the chemokines CCL2, CCL3, CCL5, CXCL10, and KC [19], as well as pro-inflammatory lipid mediators [20, 21]. RSV infection can induce airway hyperresponsiveness to inhaled methacholine (MCh) and neutrophilic and eosinophilic inflammation [5]. Epithelial cell damage, inflammation, and neural pathways are also involved in RSV-induced AHR in mice [22, 23].
One of the important limitations in many of the human epidemiological studies has been the ability to distinguish the impact of primary versus secondary RSV infection. Repeated infection is common at all ages, indicating the possibility of important interactions and outcomes in RSV-reinfected hosts. It is well documented that prior exposure to allergen in a sensitized host leads to enhanced Th2-dominant immune responses against RSV [24–26]. In turn, when the immune system was Th2-primed by allergen, infection with RSV enhanced the allergic response [27]. Taken together, the results suggest that the response to allergen may depend on the host’s immune setting induced at the time of initial virus encounter.
Re-infection with RSV has been examined in mice. Primary infection of neonatal mice (during the first week of life) with RSV was associated with reduced and delayed IFN-γ responses [28]. Upon re-infection, these mice developed more severe weight loss with a Th2-biased lymphocyte response and airway eosinophilia compared with mice initially infected with RSV at an older age. We have examined the consequences of reinfection with RSV and determined how the responses differed if initial infection was in neonates or weanling animals, in particular examining the consequences on airway responsiveness [29]. Both age groups developed significant AHR after primary RSV infection at 1 or 3 weeks of age. When re-infected with RSV 5 weeks later, the two age groups developed very different responses. Initial RSV infection at 3 weeks of age elicited a protective airway response upon re-infection characterized by an increased airway inflammatory response consisting primarily of lymphocytes, but without development of AHR, eosinophilia, or mucus hyperproduction. In contrast, initial neonatal infection failed to protect the airways on re-infection and resulted in enhanced AHR, increased IL-13 production associated with mucus hyperproduction, and airway eosinophilia (Fig. 1) [29]. Moreover, IL-13 was shown to be critical to the development of the asthma-like phenotype after re-infection of mice initially infected as newborns.
Figure 1.
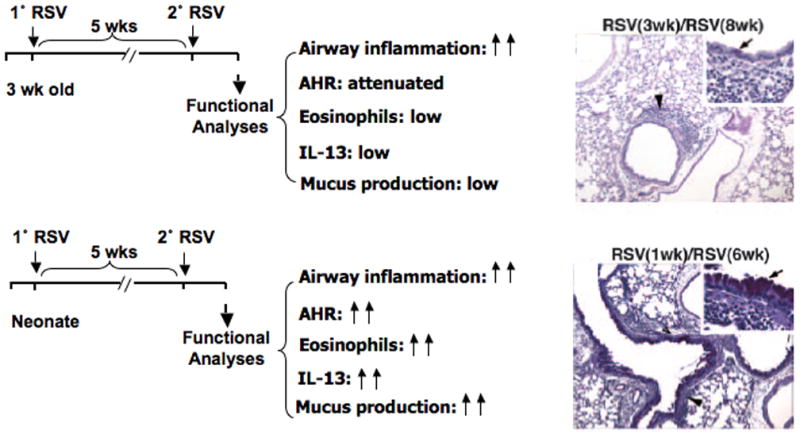
Reinfection of mice initially infected with RSV as neonates leads to enhanced lung responses. In addition to alterations in airway function, inflammation, and eosinophilia, levels of IL-13 in BAL fluid were increased and goblet cell metaplasia mucus production were markedly increased as demonstrated in the PAS-stained photomicrographs (arrow inset).
The underlying mechanisms responsible for these significant age differences are slowly being unraveled and have clinical implications. An association between severity of RSV bronchiolitis and deficient IFN-γ production has been demonstrated in infants [30–33]. Infants hospitalized for severe lower respiratory tract illness due to RSV infection had lower IFN-γ production from blood mononuclear cells compared to those with milder illnesses [30, 31]. Moreover, the levels of IFN-γ measured in nasopharyngeal aspirates were lower in infants hospitalized for severe RSV bronchiolitis compared to those exhibiting milder disease. These clinical studies suggested that IFN-γ plays an important role in determining the outcome of RSV-mediated disease. Similarly, in animal models and humans, we and others found that neonates demonstrated lower IFN-γ responses to initial RSV infection compared with weanling or adult mice [29, 30, 32–34]. The protective role of IFN-γ was confirmed in the RSV re-infection model using IFN-γ deficient mice, which developed significantly greater AHR, airway eosinophilia, and mucus hyperproduction following re-infection compared to the WT mice, regardless of the absence of significant differences in the response to initial infection [34]. Provision of IFN-γ during primary neonatal infection prevented the development of enhanced AHR and lung histopathology upon re-infection with RSV. These results indicated a critical role for IFN-γ during the initial infection stage that dictated the subsequent outcomes of re-infection with RSV. This may also be true in humans so that provision of IFN-γ in infancy may interfere with the development of altered airway responses on re-infection at a later age.
Specific IgE antibodies against viral pathogens have been identified in clinical studies following different viral infections, including RSV [35]. Development of virus-specific IgE has been associated with a more severe disease outcome [35, 36]. In the mouse model, primary RSV infection can also lead to the production of RSV-specific IgE, which may contribute to the development of exaggerated airway responses upon reinfection [37]. Thus, there is a certain parallelism between RSV-mediated wheezing and allergen-triggered asthma. In the mouse re-infection model, RSV-specific IgE enhanced the development of Th2-biased airway responsiveness on re-infection of mice initially infected as neonates [37]. Studies using mouse Sendai virus (SeV) infection pointed to a potential mechanism [38]. Infection with mouse SeV resulted in increased expression of the high affinity IgE receptor (FcεRI) on lung dendritic cells (DCs), and was type I interferon receptor-dependent. Crosslinking of FcεRI on DCs resulted in CCL28 production, recruitment of IL-13-producing CD4+ T cells and development of mucus metaplasia. In this way, the development of virus-specific IgE could play a critical role in re-infection-induced airway inflammation and AHR.
To define the mechanism underlying the enhanced responsiveness in neonatally-infected mice re-infected with RSV, potential differences in lung DCs were examined given their central role as APCs in T cell polarization. Lung DCs from neonatal mice differed from those obtained from 5 week old mice [39]. Neonatal lung DCs expressed higher baseline levels of OX40L and lower cytoplasmic levels of IL-12, a known inducer of IFN-γ production, and these differences were amplified following RSV infection with further increases in OX40L expression in infected neonates and higher IL-12 expression in infected, older mice. In parallel with increased expression of OX40L during neonatal infection, administration of anti-OX40L during primary but not secondary infection dramatically reduced the consequences of re-infection, preventing the enhancement of AHR, airway eosinophilia, mucus hyperproduction, and increases in the Th2 cytokines, IL-5 and IL-13. Together, these data positioned the OX40L-OX40 pathway as a major regulator of the skewed response of re-infected mice initially exposed to RSV in the neonatal period.
Expression levels of thymic stromal lymphopoietin (TSLP) in the lungs at baseline were higher in neonates than in adult mice and the levels increased following RSV infection [39]. Administration of anti-TSLP before neonatal RSV infection reduced the accumulation of lung DCs, decreased OX40L expression on lung DCs, and attenuated the enhancement of airway responses following re-infection, including AHR, eosinophilic airway inflammation, mucus hyperproduction, and IL-13. Taken together, the data identified the involvement of a TSLP-OX40L-OX40 axis in the enhanced development of RSV-induced AHR and inflammation when mice initially infected as neonates were later re-infected with RSV (Fig. 2).
Figure 2.
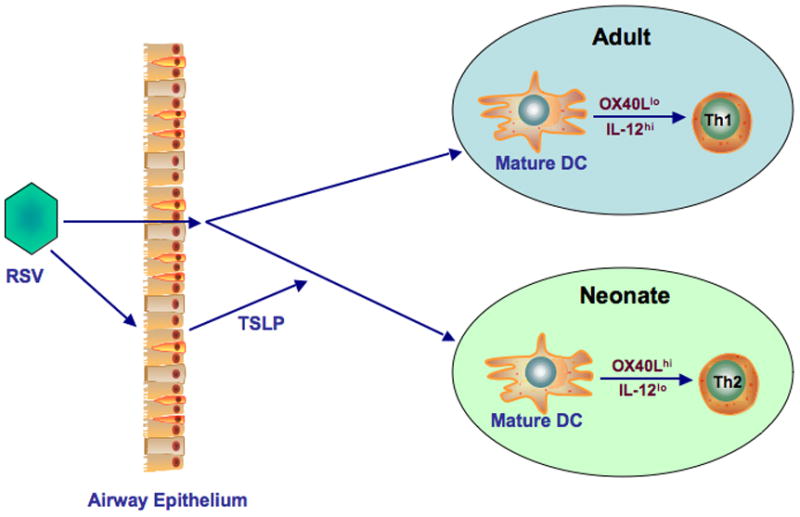
Phenotypic and functional differences between lung DCs in neonatal and adult mice. At baseline, neonatal DCs express higher levels of OX40 ligand and lower levels of IL-12 in contrast to adult lung DCs. Infection with RSV enhances these differences, mediated at least in part, by RSV-induced TSLP.
Influence of RSV Surface Proteins on Host Response to RSV
There are three virally encoded surface transmembrane proteins on RSV, G, F, and SH, all associated with modifying aspects of the host response to infection. The G or attachment protein is a type II glycoprotein with a single hydrophobic region that serves as a signal peptide and membrane anchor. An extracellular cysteine rich ectodomain contains a CX3C chemokine motif that may facilitate virus attachment to cells expressing the CX3C chemokine receptor (CX3CR1) and alter CX3CL1 (fractalkine)-mediated immune responses, a potential immune evasion mechanism [40]. The G-protein appears to be necessary for efficient virus replication in vivo [41]. The F-protein is implicated in fusion of the virion to cells and its deletion results in a marked reduction in infectivity. The SH-protein is a minor surface protein shown to form cation-selective ion channels [42] and interact with G-protein [43].
RSV infection leads to lung epithelium damage and release of a number of immunomodulatory factors [44]. Following RSV replication, soluble G-protein is released which may also alter the immune response [40, 45]. The immunomodulatory effects of RSV are exhibited at many levels including innate immunity, T-cell response to infection, and cell trafficking [44, 46–48]. One potentially important activity for RSV G-protein is the suppression of TLR4 signaling. F-protein interacts with TLR4 whereas G-protein reduces TLR4 activity [48]. Downstream effects of TLR signaling may also be seen in that RSV G-protein modulates suppression of cytokine signaling (SOCS1 and SOCS3) expression to inhibit type I IFN and interferon-stimulated gene (ISG)-15 expression [47]. SOCS1 and SOCS3 appear to be the most efficient downregulators of type I IFN production [49]. This would indicate that RSV G-protein signaling through TLR may be important in reducing type I IFN production, enhancing virus replication.
In a similar way, fractalkine (CX3CL1) is a chemokine involved in the attraction of antigen-specific killer T cells and NK cells [50]. The RSV G-protein competes with fractalkine for binding to its receptor, CX3CR1 [44]. CX3CR1 T cells specific for RSV and NK cells were markedly increased in the lungs of mice infected with a G-deficient mutant [51]. In humans, there are at least two observations supporting the hypothesis that G-protein immune modulation, even suppression, plays a role in dictating clinical severity. A mutation in CX3CR1, which reduces the affinity for fractalkine, was found in 38% of children hospitalized with severe RSV infection. This was compared to 21% in a control group [52]. As well, mutations in TLR4 that reduce receptor activity were over-represented in infants hospitalized with RSV in whom there was a 20% versus 5% frequency in infants with mild or no disease [53].
Therapeutic Implications
Studies in mice vaccinated with different recombinant vaccinia virus constructs expressing G- or F-proteins showed differential priming effects on CD4 T cell subsets [54]. F-protein primed both CD4 and CD8 T cells towards a Th1-like cytokine response while G-protein primed CD4 T cells towards Th2-like responses [55]. Efficient virus clearance appeared to require Th1 responses characterized by IFN-γ, IL-2, and IL-12 expression whereas Th2 responses, IL-4, IL-10, and IL-13, were ineffective and led to allergic disease and asthma. Th2 cytokine responses have also been linked to RSV vaccine-enhanced disease in mice and cotton rats immunized with formalin-inactivated RSV vaccine or vaccinia vectors expressing RSV F- or G- proteins [45]. Despite much effort, alternative vaccination strategies have not materialized to date.
An alternative but only prophylactic approach has been the administration of palivizumab, a humanized anti-F monoclonal antibody. This has been effective in reducing RSV hospitalizations and disease severity in high-risk infants and young children [56]. Post-infection treatment with paliviuzumab was ineffective [57] and may have had some deleterious effects if administered late [58].
There is a long history of the benefits using antibodies targeting the central conserved motif of RSV G-protein in rodents. Anti-G antibodies that interfere with G-protein binding to CX3CR1 were effective in a post-infection model using murine [59, 60] or high affinity human monoclonal antibodies [61]. In these studies, anti-G antibodies were more effective than anti-F antibodies in reducing both lung inflammation and pro-inflammatory cytokine production [59]. The potential superiority of anti-G could be the result of direct antiviral activity dependent on the intact antibody including the Fc sequence or is dependent on immunomodulatory activity mediated through the antigen combining site. In fact, intact antibody was active as both an anti-viral and anti-inflammatory agent whereas the F(ab′)2 fragment reduced the influx of inflammatory cells to the lung, but exhibited minimal anti-viral activity [60].
Conclusion
There is now conclusive evidence that asthma is a chronic inflammatory condition that if not initiated by respiratory virus infection is triggered and often sustained by respiratory viruses such as RSV, HRV, and others. For RSV and likely the other asthma-associated viruses, the age at initial infection is a critical factor in determining the host response to this or a repeated infection. Together with genetics, atopy, and unique virus strain characteristics, age shapes the immune response and severity of the disease. Given the apparent lack of new vaccination strategies, at least in the near future, administration of anti-RSV antibodies is the only approved prophylactic therapy. However, to be maximally effective, this approach must take advantage of the apparent differences seen when targeting F versus G surface protein. Whereas anti-F antibodies have been reasonably well suited for prophylaxis, targeting the G-protein may show added benefit including therapeutic applications. Studies need to examine potential differences between the two strategies in very young infants where RSV re-infection can impact disease severity and potentially the inception of asthma as demonstrated in the mouse.
Highlights.
Age and severity of first infection with virus impacts both innate and adaptive immune responses.
Age at initial encounter with virus is an important determinant of later susceptibility to asthma development.
Phenotypic and functional differences among lung dendritic cells are seen in neonatal compared to older mice.
Targeting of G or F RSV surface proteins has different immunomodulatory effects that likely impact therapeutic outcomes.
Acknowledgments
This work was supported by NIH grants HL-61005, AI-77609, and HL-36577. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NHLBI or the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jackson DJ, Gangnon RE, Evans MD, Roberg KA, Anderson EL, Pappas TE, Printz MC, Lee WM, Shult PA, Reisdorf E, Carlson-Dakes KT, Salazar LP, DaSilva DF, Tisler CJ, Gern JE, Lemanske RF., Jr Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am J Resp Crit Care Med. 2008;178:667–672. doi: 10.1164/rccm.200802-309OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2*.Martinez FD, Wright AL, Taussig LM, Holberg CJ, Halonen M, Morgan WJ. Asthma and wheezing in the first six years of life. New Engl J Med. 1995;332:133–1338. doi: 10.1056/NEJM199501193320301. Original article defining the relationship of early virus-induced wheezing on the natural history of the disease. [DOI] [PubMed] [Google Scholar]
- 3.Jackson DJ, Lemanske RF., Jr The role of respiratory virus infections in childhood asthma inception. Immunol Allergy Clin N Am. 2010;30:513–522. doi: 10.1016/j.iac.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu P, Hartert TV. Evidence for a causal relationship between respiratory syncytial virus infection and asthma. Expert Rev Anti Infect Ther. 2011;9:731–745. doi: 10.1586/eri.11.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Han J, Takeda K, Gelfand EW. The role of RSV infection in asthma initiation and progression: Findings in a mouse model. Pulm Med. 2011;2011:748038. doi: 10.1155/2011/748038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6**.Sigurs N, Aljassim F, Kjellman B, Robinson PD, Sigurbergsson F, Bjarnason R, Gustafsson PM. Asthma and allergy patterns over 18 years after severe RSV bronchiolitis in the first year of life. Thorax. 2010;65:1045–1052. doi: 10.1136/thx.2009.121582. Identifies the importance of severity of virus-induced disease as a predictor of asthma. [DOI] [PubMed] [Google Scholar]
- 7.Escobar GJ, Ragins A, Li SX, Prager L, Masaquel AS, Kipnis P. Recurrent wheezing in the third year of life among children born at 32 weeks’ gestation or later: relationship to laboratory-confirmed, medically attended infection with respiratory syncytial virus during the first year of life. Arch Pediatr Adolesc Med. 2010;164:915–922. doi: 10.1001/archpediatrics.2010.177. [DOI] [PubMed] [Google Scholar]
- 8.Kristjansson S, Bjarnarson SP, Wennergren G, Palsdottir AH, Arnadottir T, Haraldsson A, Jonsdottir I. Respiratory syncytial virus and other respiratory viruses during the first 3 months of life promote a local Th2-like response. J Allergy Clin Immunol. 2005;116:805–811. doi: 10.1016/j.jaci.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 9.Gern JE, Brooks GD, Meyer P, Chang A, Shen K, Evans MD, Tisler C, Dasilva D, Roberg KA, Mikus LD, Rosenthal LA, Kirk CJ, Shult PA, Bhattacharya A, Li Z, Gangnon R, Lemanske RF., Jr Bidirectional interactions between viral respiratory illnesses and cytokine responses in the first year of life. J Allergy Clin Immunol. 2006;117:72–78. doi: 10.1016/j.jaci.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 10.Carroll KN, Wu P, Gebretsadik T, Griffin MR, Dupont WD, Mitchel EF, Hartert TV. Season of infant bronchiolitis and estimates of subsequent risk and burden of early childhood asthma. J Allergy Clin Immunol. 2009;123:964–966. doi: 10.1016/j.jaci.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11*.Carroll KN, Wu P, Gebretsadik T, Griffin MR, Dupont WD, Mitchel EF, Hartert TV. The severity-dependent relationship of infant bronchiolitis on the risk and morbidity of early childhood asthma. J Allergy Clin Immunol. 2009;123:1055–1061. doi: 10.1016/j.jaci.2009.02.021. Linked severity of the infant bronchiolitis event to greater odds of developing childhood-onset asthma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bartlett NW, McLean GR, Chang Y-S, Johnston SL. Genetics and epidemiology: Asthma and infection. Curr Opin Allergy Clin Immunol. 2009;9:395–400. doi: 10.1097/ACI.0b013e32833066fa. [DOI] [PubMed] [Google Scholar]
- 13.Moller-Larsen S, Nyegaard M, Haagerup A, Vestbo J, Kruse TA, Børglum AD. Association analysis identifies TLR7 and TLR8 as novel risk genes in asthma and related disorders. Thorax. 2008;63:1064–1069. doi: 10.1136/thx.2007.094128. [DOI] [PubMed] [Google Scholar]
- 14.Lachheb J, Dhifallah IB, Cheibi H, Hamzaoui K, Hamzaoui A. Toll-like receptors and CD14 genes polymorphisms and susceptibility to asthma in Tunisian children. Tissue Antigens. 2008;71:417–425. doi: 10.1111/j.1399-0039.2008.01011.x. [DOI] [PubMed] [Google Scholar]
- 15.Smit LA, Siroux V, Bouzigon E, Oryszczyn MP, Lathrop M, Demenais F, Kauffmann F. Epidemiological Study on the Genetics and Environment of Asthma, Bronchial Hyperresponsiveness, and Atopy (EGEA) Cooperative Group: CD14 and toll-like receptor gene polymorphisms, country living, and asthma in adults. Am J Respir Crit Care Med. 2009;179:363–368. doi: 10.1164/rccm.200810-1533OC. [DOI] [PubMed] [Google Scholar]
- 16.Smit LA, Bongers SI, Ruven HJ, Rijkers GT, Wouters IM, Heederik D, Omland O, Sigsgaard T. Atopy and new-onset asthma in young Danish farmers and CD14, TLR2, and TLR4 genetic polymorphisms: A nested case-control study. Clin Exp Allergy. 2007;37:1602–1608. doi: 10.1111/j.1365-2222.2007.02831.x. [DOI] [PubMed] [Google Scholar]
- 17*.Stokes KL, Chi MH, Sakamoto K, Newcomb DC, Currier MG, Huckabee MM, Lee S, Goleniewska K, Pretto C, Williams JV, Hotard A, Sherrill TP, Peebles RS, Jr, Moore ML. Differential pathogenesis of respiratory syncytial virus clinical isolates in BALB/c mice. J Virol. 2011;85:5782–5793. doi: 10.1128/JVI.01693-10. Demonstrated important strain differences among clinical virus isolates. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Villenave R, O’Donoghue D, Thavagnanam S, Touzelet O, Skibinski G, Heaney LG, McKaigue JP, Coyle PV, Shields MD, Power UF. Differential cytopathogenesis of respiratory syncytial virus prototypic and clinical isolates in primary pediatric bronchial epithelial cells. Virol J. 2011;8:43. doi: 10.1186/1743-422X-8-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller AL, Bowlin TL, Lukacs NW. Respiratory syncytial virus-induced chemokine production: linking viral replication to chemokine production in vitro and in vivo. J Infect Dis. 2004;189:1419–1430. doi: 10.1086/382958. [DOI] [PubMed] [Google Scholar]
- 20.Fullmer JJ, Khan AM, Elidemir O, Chiappetta C, Stark JM, Colasurdo GN. Role of cysteinyl leukotrienes in airway inflammation and responsiveness following RSV infection in BALB/c mice. Pediatr Allergy Immunol. 2005;16:593–601. doi: 10.1111/j.1399-3038.2005.00248.x. [DOI] [PubMed] [Google Scholar]
- 21.Han J, Jia Y, Takeda K, Shiraishi Y, Okamoto M, Dakhama A, Gelfand EW. Montelukast during primary infection prevents airway hyperresponsiveness and inflammation after reinfection with respiratory syncytial virus. Am J Respir Crit Care Med. 2010;182:455–463. doi: 10.1164/rccm.200912-1811OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ogra PL. Respiratory syncytial virus: The virus, the disease and the immune response. Paediatr Respir Rev. 2004;5:S119–S126. doi: 10.1016/S1526-0542(04)90023-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dakhama A, Lee YM, Gelfand EW. Virus-induced airway dysfunction: Pathogenesis and biomechanisms. Pediatr Infect Dis J. 2005;24:S159–S169. doi: 10.1097/01.inf.0000188155.46381.15. [DOI] [PubMed] [Google Scholar]
- 24.Mäkelä MJ, Tripp R, Dakhama A, Park JW, Ikemura T, Joetham A, Waris M, Anderson LJ, Gelfand EW. Prior airway exposure to allergen increases virus-induced airway hyperresponsiveness. J Allergy Clin Immunol. 2003;112:861–869. doi: 10.1016/s0091-6749(03)02020-7. [DOI] [PubMed] [Google Scholar]
- 25.Park JW, Taube C, Yang ES, Joetham A, Balhorn A, Takeda K, Miyahara N, Dakhama A, Donaldson DD, Gelfand EW. Respiratory syncytial virus-induced airway hyperrespnsiveness is independent of IL-13 compared with that induced by allergen. J Allergy Clin Immunol. 2003;112:1078–1087. doi: 10.1016/j.jaci.2003.08.046. [DOI] [PubMed] [Google Scholar]
- 26.Kalina WV, Gershwin LJ. Progress in defining the role of RSV in allergy and asthma: from clinical observations to animal models. Clin Dev Immunol. 2004;11:113–119. doi: 10.1080/10446670410001722131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barends M, Van Oosten M, De Rond CG, Dormans JA, Osterhaus AD, Neijens HJ, Kimman TG. Timing of infection and prior immunization with respiratory syncytial virus (RSV) in RSV-enhanced allergic inflammation. J Infect Dis. 2004;189:1866–1872. doi: 10.1086/386341. [DOI] [PubMed] [Google Scholar]
- 28.Culley FJ, Pollott J, Openshaw PJ. Age at first viral infection determines the pattern of T cell-mediated disease during reinfection in adulthood. J Exp Med. 2002;196:1381–1386. doi: 10.1084/jem.20020943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29**.Dakhama A, Park JW, Taube C, Joetham A, Balhorn A, Miyahara N, Takeda K, Gelfand EW. The enhancement or prevention of airway hyperresponsiveness during re-infection with respiratory syncytial virus is critically dependent on the age at first infection and interleukin-13 production. J Immunol. 2005;175:1876–1883. doi: 10.4049/jimmunol.175.3.1876. Characterized a model demonstrating the importance of age at initial infection on a full spectrum of lung responses to re-infection at a later age. [DOI] [PubMed] [Google Scholar]
- 30.Aberle JH, Aberle SW, Dworzak MN, Mandl CW, Rebhandl W, Vollnhofer G, Kundi M, Popow-Kraupp T. Reduced interferon gamma expression in peripheral blood mononuclear cells of infants with severe respiratory syncytial virus disease. Am J Respir Crit Care Med. 1999;160:1263–1268. doi: 10.1164/ajrccm.160.4.9812025. [DOI] [PubMed] [Google Scholar]
- 31.Bont L, Heijnen CJ, Kavelaars A, van Aalderen WM, Brus F, Draaisma JT, Geelen SM, van Vught HJ, Kimpen JL. Peripheral blood cytokine responses and disease severity in respiratory syncytial virus bronchiolitis. Eur Respir J. 1999;14:144–149. doi: 10.1034/j.1399-3003.1999.14a24.x. [DOI] [PubMed] [Google Scholar]
- 32.Renzi PM, Turgeon JP, Marcotte JE, Drblik SP, Berube D, Gagnon MF, Spier S. Reduced interferon-gamma production in infants with bronchiolitis and asthma. Am J Respir Crit Care Med. 1999;159:1417–1422. doi: 10.1164/ajrccm.159.5.9805080. [DOI] [PubMed] [Google Scholar]
- 33.Legg JP, Hussain IR, Warner JA, Johnston SL, Warner JO. Type 1 and type 2 cytokine imbalance in acute respiratory syncytial virus bronchiolitis. Am J Respir Crit Care Med. 2003;168:633–639. doi: 10.1164/rccm.200210-1148OC. [DOI] [PubMed] [Google Scholar]
- 34.Lee YM, Miyahara N, Takeda K, Prpich J, Oh A, Balhorn A, Joetham A, Gelfand EW, Dakhama A. IFN-gamma production during initial infection determines the outcome of reinfection with respiratory syncytial virus. Am J Respir Crit Care Med. 2008;177:208–218. doi: 10.1164/rccm.200612-1890OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Welliver RC, Wong DT, Sun M, Middleton E, Jr, Vaughan RS, Ogra PL. The development of respiratory syncytial virus-specific IgE and the release of histamine in nasopharyngeal secretions after infection. New Engl J Med. 1981;305:841–846. doi: 10.1056/NEJM198110083051501. [DOI] [PubMed] [Google Scholar]
- 36.Koraka P, Murgue B, Deparis X, Setiati TE, Suharti C, van Gorp EC, Hack CE, Osterhaus AD, Groen J. Elevated levels of total and dengue virus-specific immunoglobulin E in patient with varying disease severity. J Med Virol. 2003;70:91–98. doi: 10.1002/jmv.10358. [DOI] [PubMed] [Google Scholar]
- 37.Dakhama A, Lee YM, Ohnishi H, Jing X, Balhorn A, Takeda K, Gelfand EW. Virus-specific IgE enhances airway responsiveness on reinfection with respiratory syncytial virus in newborn mice. J Allergy Clin Immunol. 2009;123:138–145. doi: 10.1016/j.jaci.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 38*.Grayson MH, Cheung D, Rohlfing MM, Kitchens R, Spiegel DE, Tucker J, Battaile JT, Alevy Y, Yan L, Agapov E, Kim EY, Holtzman MJ. Induction of high-affinity IgE receptor on lung dendritic cells during viral infection leads to mucous cell metaplasia. J Exp Med. 2007;204:2759–2769. doi: 10.1084/jem.20070360. Elucidate a potentially important mechanisms how respiratory virus infection can induce or exacerbate atopic disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Han J, Dakhama A, Jia Y, Wang M, Zeng W, Takeda K, Shiraishi Y, Okamoto M, Ziegler SF, Gelfand EW. Responsiveness to RSV in neonates is mediated through TSLP and OX40 ligand. J Allergy Clin Immunol. 2012 doi: 10.1016/j.jaci.2012.08.033. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tripp RA, Jones KP, Haynes LM, Zheng H, Murphy PM, Anderson LJ. CX3C chemokine mimicry by respiratory syncytial virus G glycoprotein. Nat Immunol. 2001;2:732–738. doi: 10.1038/90675. [DOI] [PubMed] [Google Scholar]
- 41.Teng MN, Whitehead SS, Collins PL. Contribution of the respiratory syncytial virus G glycoprotein and its secreted and membrane-bound forms to virus replication in vitro and in vivo. Virology. 2001;289:283–296. doi: 10.1006/viro.2001.1138. [DOI] [PubMed] [Google Scholar]
- 42.Gan SW, Ng L, Lin X, Gong X, Torres J. Structure and ion channel activity of the human respiratory syncytial virus (hRSV) small hydrophobic protein transmembrane domain. Protein Sci. 2008;17:813–820. doi: 10.1110/ps.073366208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Low KW, Tan T, Ng K, Tan BH, Sugrue RJ. The RSV F and G glycoproteins interact to form a complex on the surface of infected cells. Biochem Biophy Res Commun. 2008;366:308–313. doi: 10.1016/j.bbrc.2007.11.042. [DOI] [PubMed] [Google Scholar]
- 44.Oshansky CM, Zhang W, Moore E, Tripp RA. The host response and molecular pathogenesis associated with respiratory syncytial virus infection. Future Microbiol. 2009;4:279–297. doi: 10.2217/fmb.09.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Castilow EM, Olson MR, Varga SM. Understanding respiratory syncytial virus (RSV) vaccine-enhanced disease. Immunol Res. 2007;39:225–239. doi: 10.1007/s12026-007-0071-6. [DOI] [PubMed] [Google Scholar]
- 46.Becker Y. Respiratory syncytial virus (RSV) evades the human adaptive immune system by skewing the Th1/Th2 cytokine balance toward increased levels of Th2 cytokines and IgE, markers of allergy – a review. Virus Genes. 2006;33:235–252. doi: 10.1007/s11262-006-0064-x. [DOI] [PubMed] [Google Scholar]
- 47**.Oshansky CM, Krunkosky TM, Barber J, Jones LP, Tripp RA. Respiratory syncytial virus proteins modulate suppressors of cytokine signaling 1 and 3 and the type I interferon response to infection by a toll-like receptor pathway. Viral Immunol. 2009;22:147–161. doi: 10.1089/vim.2008.0098. Findings demonstrate the impact of RSV surface proteins signal through the TLR pathway and critically regulate SOCS protein activities. [DOI] [PubMed] [Google Scholar]
- 48.Shingai M, Azuma M, Ebihara T, Sasai M, Funami K, Ayata M, Ogura H, Tsutsumi H, Matsumoto M, Seya T. Soluble G protein of respiratory syncytial virus inhibits toll-like receptor 3/4-mediated IFN-beta induction. Intl Immunol. 2008;20:1169–1180. doi: 10.1093/intimm/dxn074. [DOI] [PubMed] [Google Scholar]
- 49.Krebs DK, Hilton DJ. SOCS: Physiological suppressors of cytokine signaling. J Cell Sci. 2000;113:2813–2819. doi: 10.1242/jcs.113.16.2813. [DOI] [PubMed] [Google Scholar]
- 50.Umehara H, Bloom E, Okazaki T, Domae N, Imai T. Fractalkine and vascular injury. Trends Immunol. 2001;22:602–607. doi: 10.1016/s1471-4906(01)02051-8. [DOI] [PubMed] [Google Scholar]
- 51.Tripp RA, Oshansky C, Alvarez R. Cytokines and respiratory syncytial virus infection. Proc Am Thorac Soc. 2005;2:147–149. doi: 10.1513/pats.200502-014AW. [DOI] [PubMed] [Google Scholar]
- 52*.Amanatidou V, Sourvinos G, Apostolakis S, Tsilmigaki A, Spandios DA. T280M variation of the CX3C receptor gene is associated with increased risk for severe respiratory syncytial virus bronchiolitis. Pediatr Infect Dis. 2006;25:410–414. doi: 10.1097/01.inf.0000214998.16248.b7. Identify critical receptor polymorphisms that are associated with severity of RSV disease. [DOI] [PubMed] [Google Scholar]
- 53*.Tal G, Mandelberg A, Dalal I, Cesar K, Somekh E, Tal A, Oron A, Itskovich S, Ballin A, Houri S, Beigelman A, Lider O, Rechavi G, Amariglio N. Association between common toll-like receptor 4 mutations and severe respiratory syncytial virus disease. J Infect Dis. 2004;189:2057–2063. doi: 10.1086/420830. Identify critical receptor polymorphisms that are associated with severity of RSV disease. [DOI] [PubMed] [Google Scholar]
- 54.Alwan WH, Openshaw PJ. Distinct patterns of T- and B-cell immunity to respiratory syncytial virus induced by individual viral proteins. Vaccine. 1993;11:431–437. doi: 10.1016/0264-410x(93)90284-5. [DOI] [PubMed] [Google Scholar]
- 55.Johnson TR, Graham BS. Secreted respiratory syncytial virus G glycoprotein induces interleukin-5 (IL-5), IL-13, and eosinophilia by an IL-4-independnt mechanism. J Virol. 1999;73:8485–8495. doi: 10.1128/jvi.73.10.8485-8495.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramilo O. Evolution of prophylaxis: MoAB, siRNA, vaccine, and small molecules. Paediatr Respir Rev. 2009;10:23–25. doi: 10.1016/S1526-0542(09)70011-9. [DOI] [PubMed] [Google Scholar]
- 57.Georgescu G, Chemaly RF. Palivizumab: Where to go from here? Expert Opin Biol Ther. 2009;9:139–147. doi: 10.1517/14712590802610692. [DOI] [PubMed] [Google Scholar]
- 58.Malley R, DeVincenzo J, Ramilo O, Dennehy PH, Meissner HC, Gruber WC, Sanchez PJ, Jafri H, Balsley J, Carlin D, Buckingham S, Vernacchio L, Ambrosino DM. Reduction of respiratory syncytial virus (RSV) in tracheal aspirates in intubated infants by use of humanized monoclonal antibody to RSV F protein. J Infect Dis. 1998;178:1555–1561. doi: 10.1086/314523. [DOI] [PubMed] [Google Scholar]
- 59*.Haynes LM, Caidi H, Radu GU, Miao C, Harcourt JL, Tripp RA, Anderson LJ. Therapeutic monoclonal antibody treatment targeting respiratory syncytial virus (RSV) G protein mediates viral clearance and reduces the pathogenesis of RSV infection in BALB/c mice. J Infect Dis. 2009;200:439–447. doi: 10.1086/600108. Showed important differences in antibody targeting of G versus F surface proteins. [DOI] [PubMed] [Google Scholar]
- 60.Miao C, Radu GU, Caidi H, Tripp RA, Anderson LJ, Haynes LM. Treatment with respiratory syncytial virus G glycoprotein monoclonal antibody or F(ab′)2 components mediates reduced pulmonary inflammation in mice. J Gen Virol. 2009;90:1119–1123. doi: 10.1099/vir.0.009308-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61**.Collarini EJ, Lee FE, Foord O, Park M, Sperinde G, Wu H, Harriman WD, Carroll SF, Ellsworth SL, Anderson LJ, Tripp RA, Walsh EE, Keyt BA, Kauvar LM. Potent high-affinity antibodies for treatment and prophylaxis of respiratory syncytial virus derived from B cells of infected patients. J Immunol. 2009;183:6338–6345. doi: 10.4049/jimmunol.0901373. Important methodological advance for isolating high affinity antibodies targeting RSV surface proteins. [DOI] [PubMed] [Google Scholar]