

ABSTRACT
The study of many important intracellular bacterial pathogens requires an understanding of how specific virulence factors contribute to pathogenesis during the infection of host cells. This requires tools to dissect gene function, but unfortunately, there is a lack of such tools for research on many difficult-to-study, or understudied, intracellular pathogens. Riboswitches are RNA-based genetic control elements that directly modulate gene expression upon ligand binding. Here we report the application of theophylline-sensitive synthetic riboswitches to induce protein expression in the intracellular pathogen Francisella. We show that this system can be used to activate the bacterial expression of the reporter β-galactosidase during growth in rich medium. Furthermore, we applied this system to control the expression of green fluorescent protein during intracellular infection by the addition of theophylline directly to infected macrophages. Importantly, we could control the expression of a novel endogenous protein required for growth under nutrient-limiting conditions and replication in macrophages, FTN_0818. Riboswitch-mediated control of FTN_0818 rescued the growth of an FTN_0818 mutant in minimal medium and during macrophage infection. This is the first demonstration of the use of a synthetic riboswitch to control an endogenous gene required for a virulence trait in an intracellular bacterium. Since this system can be adapted to diverse bacteria, the ability to use riboswitches to regulate intracellular bacterial gene expression will likely facilitate the in-depth study of the virulence mechanisms of numerous difficult-to-study intracellular pathogens such as Ehrlichia chaffeensis, Anaplasma phagocytophilum, and Orientia tsutsugamushi, as well as future emerging pathogens.
IMPORTANCE
Determining how specific bacterial genes contribute to virulence during the infection of host cells is critical to understanding how pathogens cause disease. This can be especially challenging with many difficult-to-study intracellular pathogens. Riboswitches are RNA-based genetic control elements that can be used to help dissect gene function, especially since they can be used in a broad range of bacteria. We demonstrate the utility of riboswitches, and for the first time show that riboswitches can be used to functionally control a bacterial gene that is critical to the ability of a pathogen to cause disease, during intracellular infection. Since this system can be adapted to diverse bacteria, riboswitches will likely facilitate the in-depth study of the virulence mechanisms of numerous difficult-to-study intracellular pathogens, as well as future emerging pathogens.
Introduction
The detailed study of virulence proteins is critical for understanding how bacterial pathogens cause disease. The use of inducible genetic control elements has facilitated the study and understanding of gene function in many species of bacteria. Many genetic tools exist for such studies of common model organisms such as Escherichia coli and Bacillus subtilis (1–3). However, the same is not true for less genetically tractable organisms, including many intracellular pathogens such as Francisella tularensis. Riboswitches represent a potential solution, since they can be used to induce protein expression in diverse bacteria (4).
Riboswitches are RNA regulatory elements usually located in the 5′ untranslated region of mRNA. They possess an aptamer domain that binds to a specific ligand and an expression platform located downstream that undergoes a conformational change upon ligand binding, leading to an alteration of protein expression (5). Since riboswitches do not require the presence of accessory proteins (6) and can function independently of promoter identity, they can bypass pitfalls, such as promoter incompatibility or improper folding of accessory proteins, often encountered with exogenous expression of genetic control systems. In addition, riboswitches can be coupled with native promoters to drive expression (4), avoiding the incompatibility of foreign genetic regulation systems.
Unlike natural riboswitches, synthetic riboswitches do not require binding to a cellular metabolite, which allows for orthogonal genetic regulation that minimizes interruption of normal cellular function. Theophylline, a purine base structurally similar to caffeine and previously used as an antiasthmatic medication (7), has been used as a ligand for synthetic riboswitches. A number of theophylline-sensitive synthetic riboswitches have been reported as robust, broad-host-range genetic control elements (4, 8, 9). We thus anticipated that these riboswitches would be useful tools for studying the mechanisms of virulence in the Gram-negative bacterium F. tularensis.
F. tularensis is an intracellular bacterial pathogen that is the causative agent of the zoonotic disease tularemia, which is characterized by flu-like symptoms and exhibits a mortality rate of 30% (10) to 60% for the pneumonic form of the disease (11). Francisella novicida is closely related to F. tularensis, although it is rarely pathogenic to immunocompetent humans. F. novicida causes a tularemia-like disease in mice (12), possesses a genome that is 98% genetically identical to that of F. tularensis (13, 14), and has many virulence genes in common with F. tularensis. Additionally, F. novicida, like F. tularensis, exhibits the important virulence trait of replicating within host cells such as macrophages (12, 15). It is therefore often used as a model system to study F. tularensis biology.
Relatively little is known about the virulence factors required for Francisella pathogenesis, and the lack of genetic tools to modulate protein expression has hampered the investigation of protein functions (16). Many attempts to employ pre-existing tools from other organisms in Francisella have not been fruitful (17). We therefore set out to determine if riboswitches could be used to induce protein expression in Francisella and whether this system could be employed to modulate the expression of endogenous proteins during intracellular infection.
In this study, we show the efficacy of theophylline-sensitive synthetic riboswitches in inducing protein expression in F. novicida and highly virulent F. tularensis by using riboswitch-controlled β-galactosidase as a reporter. We also employed this system to control green fluorescent protein (GFP) expression during F. novicida infection of macrophages. We adapted this system to regulate the expression of an endogenous novel protein, FTN_0818, and showed that such control could functionally rescue the growth defect of an FTN_0818 mutant in minimal medium. Furthermore, riboswitch-mediated induction of FTN_0818 during macrophage infection restored the ability of the mutant strain to replicate intracellularly. This is the first demonstration that a synthetic riboswitch can be used to efficiently control an endogenous virulence factor and rescue a virulence trait in an intracellular pathogen. We anticipate that synthetic riboswitches will prove useful in numerous applications of gene expression control to study the biology of intracellular bacteria.
RESULTS
We tested a panel of eight theophylline-sensitive synthetic riboswitches that regulate gene expression in a variety of bacteria (4, 8, 9). To determine if any of these riboswitches were functional in F. novicida, each was coupled to a β-galactosidase reporter driven by a strong Francisella promoter (Pgro, the groEL promoter [18]) and cloned into the Francisella-E. coli shuttle vector pFNLTP6 (19). All reporter constructs exhibited at least a 5-fold increase in β-galactosidase activity in the presence of theophylline (see Fig. S1 in the supplemental material). Riboswitch E (E-Rs-βgal), which was rationally designed with the consensus ribosome binding site (4), exhibited a large dynamic range in F. novicida that was calculated by subtracting expression in the absence of theophylline from expression in the presence of theophylline (Fig. 1). Riboswitch F (F-Rs-βgal), selected from a library of randomized sequences (8), exhibited the lowest basal expression levels in the absence of theophylline (Fig. 1). Strains E-Rs-βgal and F-Rs-βgal were selected for further study because they exhibited the highest absolute level of gene expression in the presence of theophylline and the lowest background expression in the absence of theophylline, respectively. Each of these strains demonstrated ligand-dependent induction of β-galactosidase activity in a dose-dependent manner, even with as little as 0.1 mM theophylline (Fig. 2). These data demonstrate that riboswitches can be used effectively to modulate protein expression in F. novicida.
FIG 1.

Riboswitch-mediated control of β-galactosidase in F. novicida. Strains encoding riboswitch E (E-Rs-βgal) or F (F-Rs-βgal) upstream of lacZ were grown in the presence (●) or absence (○) of 1 mM theophylline to an OD600 between 0.7 and 0.8. β-galactosidase activity was measured in Miller units (left axis), and the activation ratio (green bar, right axis) was calculated by dividing the number of Miller units in the presence of theophylline by the number of Miller units in the absence of theophylline. The standard deviations of triplicate samples lie within the symbols. The data are representative of three independent experiments performed in triplicate.
FIG 2.
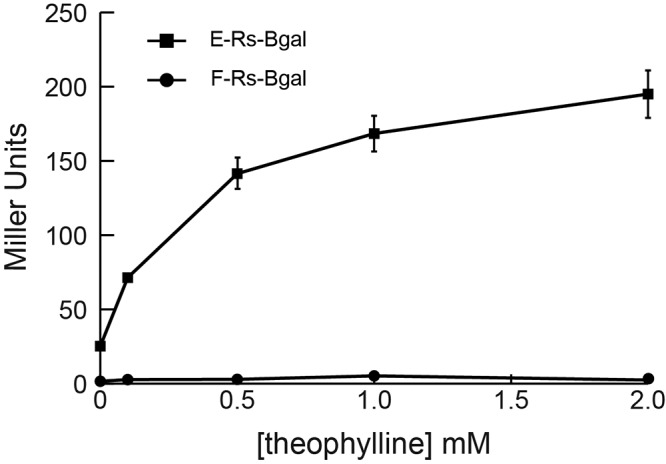
Dose-dependent riboswitch-mediated induction of β-galactosidase. Strains E-Rs-βgal and F-Rs-βgal were subcultured and grown to exponential phase (OD600 between 0.7 and 0.8) in medium containing 0, 0.1, 0.5, 1, or 2 mM theophylline. β-Galactosidase activity was measured in Miller units. The data are representative of two independent experiments performed in triplicate.
After verifying that the riboswitches function in F. novicida, we tested the feasibility of using them to control a reporter (GFP) during the bacterial infection of macrophages. We knew from previous work that strong expression levels would be required to detect fluorescence. We therefore chose riboswitch E, which induced the highest expression of β-galactosidase (Fig. 1), to drive GFP expression (strain E-Rs-GFP). As controls, we used strains constitutively expressing GFP under the control of the Pgro promoter (GFP-pos) or promoterless GFP. We did not detect GFP expression in macrophages infected with strain GFP-neg or E-Rs-GFP in the absence of theophylline (Fig. 3A and C). In contrast, GFP expression was observed in macrophages infected with GFP-pos (Fig. 3B). Importantly, the E-Rs-GFP strain expressed GFP in the presence of 1 mM theophylline (Fig. 3D). These data demonstrate the ability of a theophylline-sensitive riboswitch to regulate the expression of a bacterial reporter during the infection of host cells.
FIG 3.
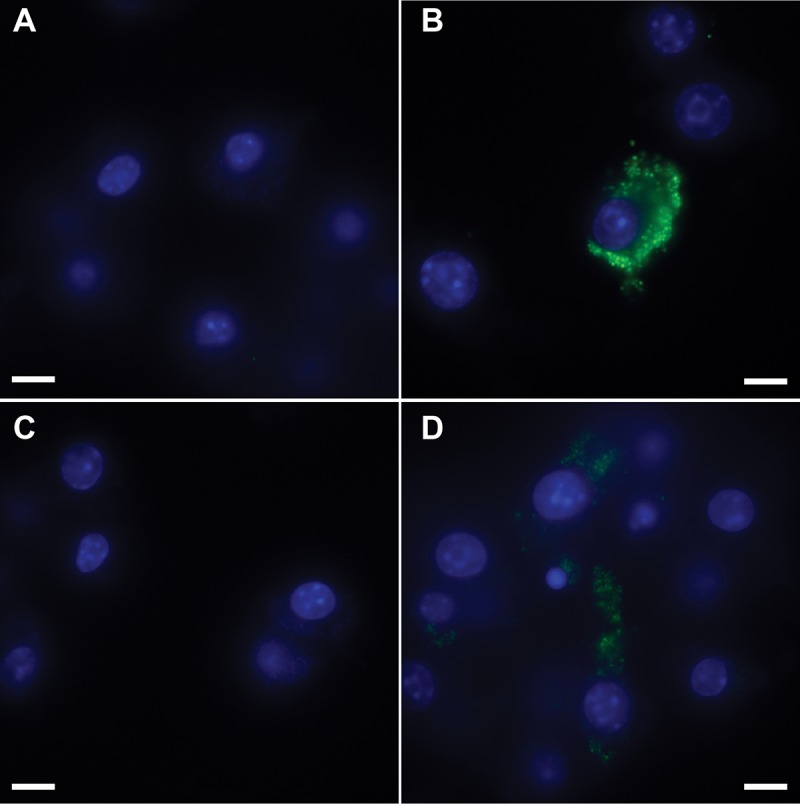
Riboswitch-mediated control of GFP (GFP-neg) in F. novicida during macrophage infection. RAW264.7 murine macrophages were infected at an MOI of 50:1 with F. novicida harboring gfp lacking a promoter (GFP-neg; panel A) or constitutively expressing gfp (GFP-pos; panel B) or the E-Rs-GFP riboswitch construct (panels C and D). At 30 min postinfection, medium without theophylline (panels A to C) or with 1 mM theophylline (panel D) was added and the macrophages were incubated at 37°C for 24 h and fixed. GFP (green) and DAPI-stained macrophage nuclei (blue) are shown. The magnification is ×100, and the scale bar represents 10 µm. The data are representative of three independent experiments in which at least 10 fields of view were analyzed for each condition.
Having demonstrated the ability to control exogenous reporter proteins (β-galactosidase and GFP) with riboswitches in F. novicida, we set out to determine if this system could be used to control an endogenous protein. FTN_0818 is a novel protein involved in biotin metabolism that is required for F. novicida replication in minimal medium (32) and within macrophages (20). We engineered a strain in which FTN_0818 is under the control of riboswitch E (E-Rs-FTN_0818) and tested its ability to grow in Chamberlain’s minimal medium. As expected, wild-type bacteria grew well while the ΔFTN_0818 mutant strain exhibited a growth defect. Even in the absence of theophylline, E-Rs-FTN_0818 replicated with wild-type kinetics (see Fig. S2 in the supplemental material), likely because of the increased background expression associated with the use of riboswitch E (Fig. 1; see Fig. S1). It should be noted that the presence of theophylline mildly slowed the growth of both the wild-type and E-Rs-FTN_0818 strains, a phenotype observed in some types of bacteria. To reduce the basal expression of FTN_0818, we placed it under the control of riboswitch F (F-Rs-FTN_0818), which exhibited much lower basal expression in the β-galactosidase assays (Fig. 1). Indeed, in the absence of theophylline, the growth rate of strain F-Rs-FTN_0818 was nearly identical to that of the ΔFTN_0818 mutant strain (Fig. 4). In contrast, in the presence of 1 mM theophylline, the growth of F-Rs-FTN_0818 was rescued to a level similar to that of the wild type (Fig. 4). These data demonstrate that a theophylline-dependent synthetic riboswitch can be used to control the expression of an endogenous bacterial protein and functionally control bacterial growth.
FIG 4.
Theophylline-dependent rescue of bacterial replication. Growth rates are represented by cell density measured as OD600 every hour for 18 h. The wild-type (WT) and ΔFTN_0818 and F-Rs-FTN_0818 mutant strains were grown overnight (~18 h) in TSB. Cultures were washed and then diluted in Chamberlain’s minimal medium to an OD600 of 0.03 in the presence or absence of 1 mM theophylline. The standard deviations of triplicate samples lie within the areas of the symbols.
The ultimate goal of this work was to modulate endogenous protein expression in the context of an intracellular infection to control and study virulence traits. To test the efficacy of this approach, we performed macrophage infections to determine whether theophylline-induced expression of FTN_0818 could rescue the intracellular growth defect of the ΔFTN_0818 mutant strain. Wild-type bacteria exhibited robust intracellular replication in the absence or presence of theophylline, demonstrating that this compound did not adversely affect this strain (Fig. 5). In contrast, the ΔFTN_0818 mutant strain was unable to replicate to significant levels. Similarly, strain F-Rs-FTN_0818 did not replicate to high levels in the presence or absence of theophylline, suggesting that the expression level of FTN_0818 induced by this riboswitch was insufficient to promote intracellular bacterial replication (Fig. 5). In contrast, the E-Rs-FTN_0818 strain exhibited very low level replication in the absence of theophylline but robust replication similar to that of the wild-type strain in the presence of 2 mM theophylline (Fig. 5). To our knowledge, these data are the first demonstration of the use of synthetic riboswitches to control the expression of a native bacterial virulence factor during the infection of host cells.
FIG 5.
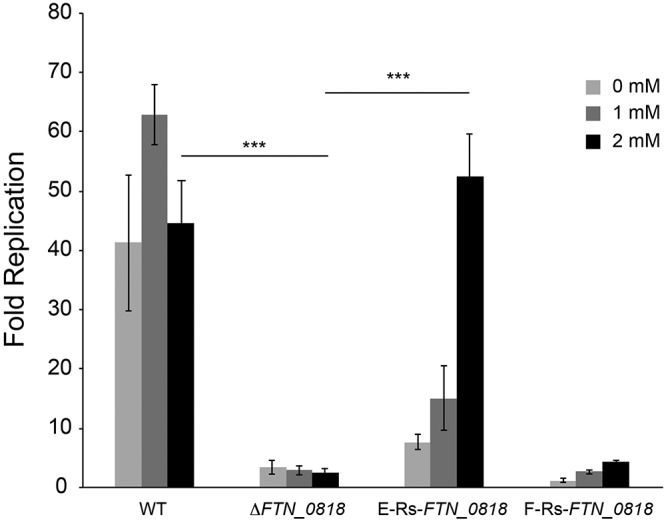
Riboswitch-mediated control of FTN_0818 facilitates intracellular replication. RAW264.7 murine macrophages were infected with the indicated F. novicida strains at an MOI of 20:1 in the presence of 0, 1, or 2 mM theophylline. Macrophages were lysed at 30 min and 24 h postinfection and plated to enumerate the intracellular CFU. Fold replication was calculated by dividing the number of CFU at 24 h postinfection by the number of CFU at 30 min. Bars represent the average fold replication of the strains, and error bars depict the standard deviations. The data are representative of four independent experiments. Asterisks indicate a significant difference in fold replication from that of the ΔFTN_0818 mutant strain in the presence of the indicated concentration of theophylline (***, P < 0.0005). WT, wild type.
To test whether theophylline-dependent synthetic riboswitches function in highly virulent species of Francisella, we tested E-Rs-βgal in F. tularensis strain SchuS4. As depicted in Fig. 6, riboswitch E induced β-galactosidase expression in a dose-dependent manner in F. tularensis to levels similar to those in F. novicida. This clearly demonstrates the utility of a theophylline-dependent synthetic riboswitch in inducing protein expression in a highly virulent Francisella strain, and taken together, these data highlight the utility of this system in controlling protein expression in intracellular pathogens.
FIG 6.
Riboswitch E controlling β-galactosidase activity in F. novicida and F. tularensis. F. novicida strain E-Rs-βgal and F. tularensis strain E-Rs-βgal were subcultured and grown to exponential phase (OD600 of ~0.7 to 0.8) in medium containing 0, 0.5, 1, or 2 mM theophylline. β-galactosidase activity was measured in Miller units. The data are representative of two independent experiments performed in triplicate.
DISCUSSION
In this study, we demonstrated the ability to induce protein expression in intracellular bacteria by using a synthetic riboswitch. Our synthetic riboswitches are induced by theophylline, a nonendogenous small molecule, and offer an orthogonal system for protein expression that minimizes interactions with natural biochemical pathways. Furthermore, the dose-dependent nature of these synthetic riboswitches allows tunable control over protein expression. This system was recently used to study a potential drug target in Streptococcus pyogenes (21) and to control gene expression in mycobacteria (22). Importantly, we have demonstrated the utility of this system in controlling proteins involved in virulence during intracellular infection. These riboswitches function in a broad range of bacteria and should therefore be easily applied, and highly useful, in studying the virulence strategies of numerous intracellular pathogens.
The riboswitch system overcomes problems encountered when adapting genetic tools from other organisms to Francisella. A possible explanation for the difficulty of transporting genetic tools from other organisms may lie partly in the unique α subunits of the RNA polymerase (RNAP) of Francisella. The α subunit serves as the initiator of RNAP assembly and binds to a supplementary promoter element (the UP element) upstream of the −35 recognition site (23). In most bacteria, the RNAP holoenzyme contains a dimer of identical α subunits (24). However, in the Francisella genus, the α subunits are encoded by two distinct genes, yielding two unique variants of the RNAP α subunit (25). Given the fundamental role of the α subunit in transcriptional regulation (26), the inefficient transcription of foreign promoters in Francisella may be attributed to the differences in RNAP. Riboswitches offer a solution to this problem, since they are readily coupled to native promoters (4).
We used this riboswitch system to control FTN_0818, an endogenous Francisella protein that is involved in biotin metabolism and is required for growth in minimal medium and intracellular replication in macrophages. When FTN_0818 expression was induced with theophylline in minimal medium, riboswitch F (F-Rs-FTN_0818) was sufficient to elicit a wild-type growth phenotype (Fig. 4). Interestingly, when stronger riboswitch E was used, even in the absence of theophylline, the strain exhibited a wild-type phenotype (see Fig. S2 in the supplemental material). This demonstrated that the background level of FTN_0818 expression induced by riboswitch E was sufficient to facilitate growth in minimal medium. However, in macrophages, riboswitch E was necessary to allow wild-type replication levels in the presence of theophylline (Fig. 5). By varying the time at which theophylline is added to infected macrophages, this system should also be useful in determining at what stage of infection a specific protein, like FTN_0818, is required.
Control of protein expression through synthetic riboswitches during the infection of mammalian cells is an important capability that will likely garner further insight into the virulence of numerous pathogenic bacteria, including Francisella. We hypothesize that it will also be feasible to regulate bacterial protein expression in in vivo systems by using synthetic riboswitches. Theophylline is an FDA-approved drug and has been tested in mouse models (27, 28). By extrapolating previous data correlating drug dosages with concentrations in blood serum (28), we have determined that it is theoretically possible to achieve concentrations in serum that are sufficient for the induction of protein expression without exceeding lethal doses of theophylline. Given the lack of tools to regulate the in vivo expression of proteins in Francisella, as well as many other intracellular pathogens, the development of riboswitches to be used in animal models is an avenue that merits further investigation. Taken together, the results presented here demonstrate the utility of synthetic riboswitches in studying virulence traits of intracellular bacteria and can be applied to a diverse set of difficult-to-study pathogens such as Ehrlichia chaffeensis, Anaplasma phagocytophilum, and Orientia tsutsugamushi, as well as future emerging pathogens.
MATERIALS AND METHODS
Bacterial strains and conditions.
The bacterial strains used in this study are listed in Table 1. Plasmid manipulations were performed by using E. coli MDS42 (Scarab Genomics, Madison, WI) transformed via electroporation. All experiments were performed with F. novicida strain U112 (15) (a gift from Denise Monack, Stanford University, Stanford, CA) or related strains and F. tularensis strain SchuS4. F. novicida overnight cultures were grown on a rolling drum at 37°C in tryptic soy broth (TSB; Difco/BD, Sparks, MD) supplemented with 0.2% l-cysteine (Sigma-Aldrich, St. Louis, MO). Growth assays were performed with Chamberlain’s minimal medium as previously described (29). For plating on solid medium, modified Mueller-Hinton (mMH; Difco/BD) plates supplemented with 0.025% ferric pyrophosphate (Sigma-Aldrich), 0.1% glucose, and 0.1% l-cysteine were used. When appropriate, kanamycin (Kan; Fisher Scientific, Fair Lawn, NJ) was added to the medium at a concentration of 30 µg ml−1.
TABLE 1 .
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Source |
|---|---|---|
| Strains | ||
| U112 | Wild-type F. novicida | D. Monack |
| ΔFTN_0818 mutant | U112 with FTN_0818 removed by allelic exchange | 32 |
| SchuS4 | Wild-type F. tularensis | |
| E-Rs-FTN_0818 | ΔFTN_0818 mutant strain complemented with FTN_0818 downstream of riboswitch E | This study |
| F-Rs-FTN_0818 | ΔFTN_0818 mutant strain complemented with FTN_0818 downstream of riboswitch F | This study |
| E-Rs-GFP | U112 containing plasmid pCKR34 | This study |
| E-Rs-βgal | U112 containing plasmid pCKR47 | This study |
| F-Rs-βgal | U112 containing plasmid pCKR43 | This study |
| GFP-neg | U112 containing plasmid pFNLTP6-NP (GFP negative control) | This study |
| GFP-pos | U112 containing plasmid pFNLTP6-gro-gfp (GFP positive control) | This study |
| Plasmids | ||
| pFNLTP6-gro-gfp | Francisella-E. coli shuttle vector with gro promoter controlling gfp | 19 |
| pFNLTP6-NP | Francisella-E. coli shuttle vector with promoter deleted | This study |
| pCKR34 | pFNLTP6 derivative with riboswitch E controlling gfp | This study |
| pCKR43 | pFNLTP6 derivative with riboswitch F controlling lacZ | This study |
| pCKR47 | pFNLTP6 derivative with riboswitch E controlling lacZ | This study |
Cloning and mutagenesis.
The plasmids used in this study are listed in Table 1. Synthetic oligonucleotide primers were purchased from Integrated DNA Technologies (Coralville, IA). DNA polymerase and restriction enzymes were purchased from New England Biolabs (Ipswich, MA). Previously published theophylline synthetic riboswitches (4, 8, 9) were adapted to F. novicida through the integration of a Francisella promoter. Riboswitch-controlled lacZ gene constructs under the control of the strong promoter Pgro were cloned into an E. coli-F. novicida shuttle vector, pFNLTP6-gro-gfp, by using the PacI and BamHI restriction sites (19) (a gift from Thomas C. Zahrt, Medical College of Wisconsin, Milwaukee, WI). Riboswitches were amplified from previously reported plasmids (4, 8, 9) and assembled by PCR downstream of the Francisella promoter Pgro and upstream of lacZ flanked by PacI and BamHI sites. After restriction enzyme digestion and ligation, constructs were transformed into E. coli MDS42. Plasmids were then extracted by using a Plasmid Midi-Prep kit (Qiagen, Germantown, MD). F. novicida U112 or F. tularensis SchuS4 was transformed as previously described (30). Plasmids for gfp were prepared as described above for lacZ. Sequences were verified by DNA sequencing (MWG Operon, Huntsville, AL). Riboswitch-mediated complement constructs of ΔFTN_0818 to be integrated into the F. novicida genome were generated by overlap PCR assembly of Pgro-driven riboswitch E or F (see sequences in Table S1 in the supplemental material) upstream of FTN_0818, followed by a Kan resistance cassette and flanked by sequences homologous to a region of the genome known to accept genomic additions without disrupting normal cell function (nucleotides 818016 to 818037 and 818649 to 818670). Transformation of plasmid DNA or allelic exchange of linear DNA was performed as previously described (30). Briefly, a 1-ml aliquot of an overnight culture was added to 50 ml of TSB supplemented with 0.2% cysteine and grown at 37°C with shaking for 2 to 4 h until the optical density at 600 nm (OD600) was ~0.8 to 1.0. Cells were then harvested by centrifugation, and the cell pellet was resuspended in 5 ml of room temperature transformation buffer (17). Plasmid or linear DNA (~1 µg) was added to 200-µl aliquots of resuspended cells and incubated at 37°C on a rolling drum for 20 min. One milliliter of TSB plus 0.2% cysteine was added, followed by another 2 h of incubation at 37°C on a rotary wheel. Cultures were then concentrated to 200 µl by centrifugation at 5,000 × g and plated on selective mMH medium.
Bacterial growth curves.
To measure bacterial growth, mutant, wild-type, and riboswitch strains (Table 1) incubated overnight in TSB plus 0.2% cysteine were centrifuged at 15,294 × g for 2 min at room temperature. Cell pellets were resuspended in Chamberlain’s minimal medium (29) and then subcultured in minimal medium with or without theophylline to an OD600 of 0.03. Subcultures were aliquoted (150 µl) into the wells of a 96-well plate and incubated overnight at 37°C with shaking in a plate reader (Bio-Tek Synergy MX, Winooski, VT). The OD600 was recorded every hour for 18 h in triplicate wells.
β-Galactosidase assay.
β-Galactosidase assays were performed as previously described (31), with the following modifications. Strains were cultured overnight at 37°C with shaking. Cells were then diluted (culture/medium) 1:30 (0 mM theophylline) or 1:25 (1 mM theophylline) into TSB containing Kan (30 µg/ml) and 0 or 1 mM theophylline to an OD600 of ~0.7 to 0.8. Theophylline and o-nitrophenyl-β-d-galactopyranoside were purchased from Sigma-Aldrich. Dose-response assays were performed as described above with the addition of various concentrations of theophylline and dilutions of 1:30 (0 and 0.25 mM theophylline), 1:27 (0.5 mM theophylline), and 1:21 (2 mM theophylline). These volumes were used to compensate for small decreases in the growth rate with increasing concentrations of theophylline.
Macrophage infections.
For infection, RAW264.7 murine macrophages (ATCC, Manassas, VA) were seeded into the wells of a chambered cover glass (CultureWell; Invitrogen, Carlsbad, CA) at 1 × 105 cells per well and incubated overnight in Dulbecco’s modified Eagle medium (high glucose, l-glutamine; DMEM; Lonza, Walkersville, MD) supplemented with 10% heat-inactivated fetal calf serum (FCS; HyClone, Logan, UT) at 37°C with 5% CO2. The medium was then removed, and the macrophages were infected with overnight cultures of various GFP-expressing strains (Table 1) that were diluted in DMEM–10% FCS to achieve a multiplicity of infection (MOI) of 50 CFU of bacteria per macrophage. The plates were centrifuged for 15 min at 233 × g at room temperature and then incubated for 30 min. The macrophages were then washed twice with DMEM and returned to incubation overnight (18 to 24 h). Next, macrophages were rinsed twice with sterile phosphate-buffered saline (PBS) and fixed by incubation in 4% paraformaldehyde (PFA) in PBS for 20 min at room temperature. The cells were again rinsed twice with PBS to wash away the PFA. To measure bacterial replication, RAW264.7 macrophages were infected as described above, with the following differences. Macrophages were seeded into the wells of 24-well tissue culture plates at 7.5 × 105 cells/well and infected at an MOI of 20 CFU per macrophage in DMEM–10% FCS containing 100 µg/ml gentamicin (Teknova, Hollister, CA) with or without 1 mM theophylline. Macrophages were then lysed with 1% saponin (Alfa Aesar, Heysham, Lancs, United Kingdom) in PBS without calcium or magnesium (Lonza, Walkersville, MD) at 30 min or 24 h. To quantify the bacterial load within macrophages at each time point, serial dilutions of the macrophage lysates were plated onto modified mMH agar plates and incubated overnight at 37°C. Finally, the n-fold replication of each strain was calculated (CFU at 24 h/CFU at 30 min).
Microscopy.
Fluorescence microscopy was performed with an Eclipse Ti microscope coupled with the Nikon Elements software package (Nikon). Image capture was performed with an Evolve EM charge-coupled device (Photometrics) by using a CFI Apo 100× (numerical aperture of 1.49) objective (Nikon) with an Intensilight epifluorescence source (Nikon). Data acquisition utilized the Nikon Perfect Focus System, which allows the capture of multipoint Z-stack data without loss of focus. Z-stack images in 10 fields of view were taken for each construct by using the fluorescein isothiocyanate (FITC) and 4',6-diamidino-2-phenylindole (DAPI) Chroma filter cubes to detect GFP and macrophage nuclei, respectively. Z-stacks probed a 5-µm section of the macrophages at 0.4 µm per slice. The FITC exposure time was 300 ms, while the DAPI exposure time was 50 ms. Images of the negative control were used as a reference to eliminate background fluorescence. Settings from the negative control were propagated to all images to allow accurate comparison. False color was applied to aid in analysis.
Statistics.
Macrophage replication data were analyzed for significance by using the unpaired Student t test with two degrees of freedom.
SUPPLEMENTAL MATERIAL
Theophylline-dependent synthetic riboswitches controlling β-galactosidase in F. novicida. Strains encoding riboswitches A to G upstream of lacZ were grown in the presence (■) or absence (□) of 1 mM theophylline to an OD600 between 0.7 and 0.8. β-galactosidase activity was measured in Miller units (y axis). The activation ratio (not shown) was calculated by dividing the number of Miller units in the presence of theophylline by the number of Miller units in the absence of theophylline and are 15, 13, 7, 19, 9, 7, 7, and 7 for riboswitches A, B, C, D, E, E*, F, and G, respectively. The standard deviations of triplicate samples are represented by error bars. The data are representative of three independent experiments performed in triplicate. Download Figure S1, TIF file, 5.5 MB.
Riboswitch E controlling FTN_0818 in minimal medium. Growth rates are represented by cell density measured as OD600 every hour for 18 h. The wild-type and ΔFTN_0818 and E-Rs-FTN_0818 mutant strains were grown overnight (~18 h) in TSB. Cultures were washed and then diluted in Chamberlain’s minimal medium to an OD600 of 0.03 in the absence or presence of 1 mM theophylline. Standard deviations of triplicate samples lie within the areas of the symbols. Download Figure S2, TIF file, 3.8 MB.
Riboswitch sequences
ACKNOWLEDGMENTS
We thank D. Stabley in the laboratory of Khalid Salaita at Emory University for assistance with microscopy images and Arturo Zychlinsky for critical reading of the manuscript.
This work was supported by NIH grants U54-AI057157 from the Southeastern Regional Center of Excellence for Emerging Infections and Biodefense to D.S.W. and GM074070 to J.P.G. and by the Camille and Henry Dreyfus Foundation. Its contents are solely our responsibility and do not necessarily represent the official views of the NIH.
ADDENDUM
During review of this manuscript, LoVullo et al. (Appl Environ Microbiol. 78:6883–6889, 2012) described a Tet-based inducible system for use in Francisella.
Footnotes
Citation Reynoso CMK, Miller MA, Bina JE, Gallivan JP, Weiss DS. 2012. Riboswitches for intracellular study of genes involved in Francisella pathogenesis. mBio 3(6):e00253-12. doi:10.1128/mBio.00253-12.
REFERENCES
- 1. Sørensen HP, Mortensen KK. 2005. Advanced genetic strategies for recombinant protein expression in Escherichia coli. J. Biotechnol. 115:113–128 [DOI] [PubMed] [Google Scholar]
- 2. Baneyx F. 1999. Recombinant protein expression in Escherichia coli. Curr. Opin. Biotechnol. 10:411–421 [DOI] [PubMed] [Google Scholar]
- 3. Scaife J, Leach DRF, Galizzi A. 1985. Genetics of bacteria. Academic Press, New York, NY. [Google Scholar]
- 4. Topp S, et al. 2010. Synthetic riboswitches that induce gene expression in diverse bacterial species. Appl. Environ. Microbiol. 76:7881–7884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Winkler WC, Breaker RR. 2005. Regulation of bacterial gene expression by riboswitches. Annu. Rev. Microbiol. 59:487–517 [DOI] [PubMed] [Google Scholar]
- 6. Barrick JE, Breaker RR. 2007. The power of riboswitches. Sci. Am. 296:50–57 [DOI] [PubMed] [Google Scholar]
- 7. Schultze-Werninghaus G, Meier-Sydow J. 1982. The clinical and pharmacological history of theophylline: first report on the bronchospasmolytic action in man by SR Hirsch in Frankfurt (Main) 1922. Clin. Exp. Allergy 12:211–215 [DOI] [PubMed] [Google Scholar]
- 8. Lynch SA, Desai SK, Sajja HK, Gallivan JP. 2007. A high-throughput screen for synthetic riboswitches reveals mechanistic insights into their function. Chem. Biol. 14:173–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Topp S, Gallivan JP. 2008. Random walks to synthetic riboswitches—a high-throughput selection based on cell motility. Chembiochem 9:210–213 [DOI] [PubMed] [Google Scholar]
- 10. Oyston PC. 2008. Francisella tularensis: unravelling the secrets of an intracellular pathogen. J. Med. Microbiol. 57:921–930 [DOI] [PubMed] [Google Scholar]
- 11. Saslaw S, Eigelsbach HT, Prior JA, Wilson HE, Carhart S. 1961. Tularemia vaccine study. II. Respiratory challenge. Arch. Intern. Med. 107:702–714 [DOI] [PubMed] [Google Scholar]
- 12. Lauriano CM, et al. 2004. MglA regulates transcription of virulence factors necessary for Francisella tularensis intraamoebae and intramacrophage survival. Proc. Natl. Acad. Sci. U. S. A. 101:4246–4249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Larsson P, et al. 2009. Molecular evolutionary consequences of niche restriction in Francisella tularensis, a facultative intracellular pathogen. PLoS Pathog. 5:e1000472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Forsman M, Sandström G, Sjöstedt A. 1994. Analysis of 16S ribosomal DNA sequences of Francisella strains and utilization for determination of the phylogeny of the genus and for identification of strains by PCR. Int. J. Syst. Bacteriol. 44:38–46 [DOI] [PubMed] [Google Scholar]
- 15. Baron GS, Nano FE. 1998. MglA and MglB are required for the intramacrophage growth of Francisella novicida. Mol. Microbiol. 29:247–259 [DOI] [PubMed] [Google Scholar]
- 16. Horzempa J, Tarwacki DM, Carlson PE, Robinson CM, Nau GJ. 2008. Characterization and application of a glucose-repressible promoter in Francisella tularensis. Appl. Environ. Microbiol. 7:2161–2270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Frank DW, Zahrt TC. 2007. Genetics and genetic manipulation in Francisella tularensis. Ann. N. Y. Acad. Sci. 1105:67–97 [DOI] [PubMed] [Google Scholar]
- 18. Ericsson M, et al. 1997. Characterization of the nucleotide sequence of the groE operon encoding heat shock proteins chaperone-60 and -10 of Francisella tularensis and determination of the T-cell response to the proteins in individuals vaccinated with F. tularensis. Infect. Immun. 65:1824–1829 PubMed; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maier TM, et al. 2004. Construction and characterization of a highly efficient Francisella shuttle plasmid. Appl. Environ. Microbiol. 70:7511–7519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Llewellyn AC, Jones CL, Napier BA, Bina JE, Weiss DS. 2011. Macrophage replication screen identifies a novel Francisella hydroperoxide resistance protein involved in virulence. PLoS One 6:e24201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bugrysheva JV, Froehlich BJ, Freiberg JA, Scott JR. 2011. The histone-like protein help is essential for growth of Streptococcus pyogenes: comparison of genetic approaches to study essential genes. Appl. Environ. Microbiol. 77:4422–4428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Seeliger JC, et al. 2012. A riboswitch-based inducible gene expression system for mycobacteria. PLoS One 7:e29266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ebright RH, Busby S. 1995. The Escherichia coli RNA polymerase alpha subunit: structure and function. Curr. Opin. Genet. Dev. 5:197–203 [DOI] [PubMed] [Google Scholar]
- 24. Charity JC, et al. 2007. Twin RNA polymerase-associated proteins control virulence gene expression in Francisella tularensis. PLoS Pathog. 3:e84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mukhamedyarov D, Makarova KS, Severinov K, Kuznedelov K. 2011. Francisella RNA polymerase contains a heterodimer of non-identical α subunits. BMC Mol. Biol. 12:50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gourse RL, Ross W, Gaal T. 2000. UPs and downs in bacterial transcription initiation: the role of the alpha subunit of RNA polymerase in promoter recognition. Mol. Microbiol. 37:687–695 [DOI] [PubMed] [Google Scholar]
- 27. McColl JD, Parker JM, Ferguson JK. 1956. A comparison of the relative toxic, emetic and convulsive actions of a series of methylated xanthine derivatives. J. Pharmacol. Exp. Ther. 116:343–350 [PubMed] [Google Scholar]
- 28. Blake KV, Massey KL, Hendeles L, Nickerson D, Neims A. 1988. Relative efficacy of phenytoin and phenobarbital for the prevention of theophylline-induced seizures in mice. Ann. Emerg. Med. 17:1024–1028 [DOI] [PubMed] [Google Scholar]
- 29. Chamberlain RE. 1965. Evaluation of live tularemia vaccine prepared in a chemically defined medium. Appl. Microbiol. 13:232–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Anthony LS, Gu MZ, Cowley SC, Leung WW, Nano FE. 1991. Transformation and allelic replacement in Francisella spp. J. Gen. Microbiol. 137:2697–2703 [DOI] [PubMed] [Google Scholar]
- 31. Miller JH. 1972. Experiments in molecular genetics, p 352–355 Cold Spring Harbor: Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 32. Napier BA, Meyer L, Bina JE, Miller MA, Sjostedt A, Weiss DS. 2012. A novel link between intraphagosomal biotin requirements and virulence in Francisella. Proc. Natl. Acad. Sci. U.S.A. 109:18084–18089 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Theophylline-dependent synthetic riboswitches controlling β-galactosidase in F. novicida. Strains encoding riboswitches A to G upstream of lacZ were grown in the presence (■) or absence (□) of 1 mM theophylline to an OD600 between 0.7 and 0.8. β-galactosidase activity was measured in Miller units (y axis). The activation ratio (not shown) was calculated by dividing the number of Miller units in the presence of theophylline by the number of Miller units in the absence of theophylline and are 15, 13, 7, 19, 9, 7, 7, and 7 for riboswitches A, B, C, D, E, E*, F, and G, respectively. The standard deviations of triplicate samples are represented by error bars. The data are representative of three independent experiments performed in triplicate. Download Figure S1, TIF file, 5.5 MB.
Riboswitch E controlling FTN_0818 in minimal medium. Growth rates are represented by cell density measured as OD600 every hour for 18 h. The wild-type and ΔFTN_0818 and E-Rs-FTN_0818 mutant strains were grown overnight (~18 h) in TSB. Cultures were washed and then diluted in Chamberlain’s minimal medium to an OD600 of 0.03 in the absence or presence of 1 mM theophylline. Standard deviations of triplicate samples lie within the areas of the symbols. Download Figure S2, TIF file, 3.8 MB.
Riboswitch sequences