

Abstract
Congenital disorders of glycosylation (CDG) are a group of metabolic disorders caused by deficient protein glycosylation. PMM2-CDG, the most common CDG, is caused by phosphomannomutase (PMM) deficiency. Clinical symptoms often include neurological involvement in addition to dysmorphic features, failure to thrive, cardiac failure, renal, and endocrine abnormalities. To our knowledge, lymphatic edema in CDG has not been reported. We present two cases of lymphatic edema in PMM2-CDG patients. The first patient was noted to have a larger right leg circumference at two years. Ultrasound investigations did not reveal any obvious vascular or lymphatic malformation. The swelling increased in size over time. At 12 years, lymphoscintigraphy revealed decreased lymphatic draining in both legs, which was more profound in the right leg. The second patient was treated for pulmonary stenosis at age 2 months. Postoperative, the patient suffered from protein-losing enteropathy, hypothyroidism, recurrent bacterial infections, and bilateral lymphatic edema. General condition improved after thyroxin treatment and albumin infusions; however, the bilateral pedal and leg edema remained unresolved. Contrast studies of the lymphatic system showed bilateral hypoplasia distal to the knees. Although both children had secondary factors worsening lymphatic edema in PMM2-CDG, hypoalbuminemia, recurrent infections, cardiac failure, and endocrine abnormalities could not fully explain the clinical features. The additional factors were treated successfully but the therapy did not resolve the lymphatic edema. Based on the abnormal imaging studies of the lymphatic system, we propose that lymphatic vessel hypoplasia is the major cause for lymphatic edema in our patients with PMM2-CDG.
Introduction
Congenital disorders of glycosylation (CDG) form a group of rare metabolic disorders caused by deficient glycosylation of proteins and/or lipids. A deficiency of phosphomannomutase (PMM) activity leads to the most common form of CDG, PMM2-CDG (formerly CDG-Ia; OMIM 212065). Most patients develop neurological symptoms such as hypotonia, strabismus, cerebellar hypoplasia, and psychomotor retardation. Dysmorphic features, failure to thrive, malabsorption, cardiac failure, frequent infections, endocrine, and renal anomalies are common as well (de Lonlay et al. 2001).
Although congenital lymphatic edema has not been described as a feature of PMM2-CDG, edematous fluid accumulation is common in CDG and shows variable clinical features. Generalized edema or localized edema of the extremities is frequently reported in CDG due to low oncotic pressure, cardiac failure, or abnormal endocrine balance (Truin et al. 2008). Another study on 17 patients from three CDG groups (PMM2-CDG, MPI-CDG, and ALG6-CDG) reports lower extremity lymphedema in the presence of protein-losing enteropathy in two patients (Damen et al. 2004). Pericardial effusion is relatively common, but ascites and hydrops fetalis has been reported as well (Leticee et al. 2010). The origin of localized fluid accumulation is not fully explained. Treatment of hypoalbuminemia and cardiac failure do not always lead to improvement of the pericardial and abdominal fluid accumulation (Truin et al. 2008). An inflammatory process has been suggested as a possible cause of therapy resistant fluid accumulation.
In this report, we present two patients who developed severe progressive lymphatic edema. Despite adequate treatment of all factors leading to a possible chronic swelling of the extremities, the lymphatic edema persisted and caused debilitating restrictions in free movement and recurrent infections in the patients.
Case Reports
Patient 1
The female patient was born term to healthy nonconsanguineous parents. Upon delivery, she was noted to have dysmorphic features, abnormal fat distribution, inverted nipples, long fingers, and strabismus. Neonatal screening revealed hypothyroidism. Due to feeding difficulties, she remained hospitalized and was diagnosed with a malabsorption syndrome, protein-losing enteropathy, hypertrophic cardiomyopathy, and received a gastric tube due to recurrent vomiting. MRI of the brain showed cerebral atrophy and vermis hypoplasia. Repeated thyroid screening was normal. She was also known with elevated liver function tests and coagulopathy. She was diagnosed with PMM2-CDG at the age of 2 months and found to have a common R141H/F119L PMM2-CDG mutation.
Since the age of 2 years, a larger right leg was noticed that was suspected for lymphedema. The leg circumference was increased with 2.5 cm compared to the left. Ultrasound investigation did not show a vascular or lymphatic malformation or the presence of thrombosis and no further examinations were performed at that time. Over time, the size of the leg increased and elastic stockings were measured and used for several years. Due to decreasing thyroxine (freeT4) concentrations and increasing weight gain, she was started on thyroxin supplementation. At the age of 12 years, the patient developed cellulitis of her upper right leg due to group A streptococcus infection. A concomitant inguinal abscess and further increase in the leg circumference was found due to Escherichia coli infection. The patient was treated with intravenous antibiotics and recovered well. Albumin infusions were given due to albumin levels of 2 g/dL (normal values 3.5–5.0 g/dL); liver cirrhosis was ruled out as a cause of hypoalbuminemia. An ultrasound of the right leg and abdomen were normal. An X-ray of the right showed dysplastic development of the right hip.
The size of the right leg, however, remained significantly increased due to nonpitting edema (the circumference was increased with 4 cm compared to the other leg; even after the successful treatment of the abscess; Fig. 1). A lymphoscintigraphy revealed bilateral, profoundly decreased lymphatic draining; the abnormality was more significant in the right leg. Since this episode, the patient has remained stable with the use of bilateral elastic stockings; however, she remains in a wheelchair and suffers from recurrent contact and pressure wounds of both feet.
Fig. 1.
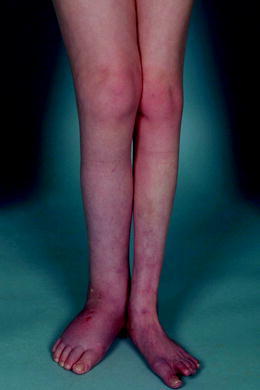
Prominent lymphatic edema of the right leg in the first patient at the age of 12 years
Patient 2
The patient was born at term with a birth weight of 2,900 g to healthy unrelated parents. At birth she had dysmorphic features, abnormal fat distribution, long fingers, and edema of both legs. She was admitted to the hospital at the age of 2 months due to tachypnea and feeding difficulties and was diagnosed with pulmonary artery stenosis. At the preoperative evaluation decreased albumin levels, elevated liver enzymes, and prolonged bleeding time were noted. Postoperative, the patient was treated for chronic protein-losing enteropathy, hypothyroidism, and recurrent bacterial infections. She demonstrated a generalized edema and bilateral nonpitting edema, most prominently on her feet and lower legs. The patient received nutritional intervention and was started on diuretics. Based on the clinical features, she was suspected for a glycosylation defect and was diagnosed with PMM2-CDG due to a homozygous R21G mutation. Upon cardiac stabilization, thyroxin treatment, and repeated albumin infusions, without signs of liver cirrhosis, the general condition improved but the bilateral pedal and leg edema remained unresolved. Contrast studies of the lymphatic system of the legs showed a significant delay in lymphatic circulation, suggesting a bilateral hypoplasia of the lymph vessels distal to the knees, but normal lymph circulation above the knees. At the age of 29 years, the patients still suffers from “heavy legs”, uses a wheel chair for longer distances and she still wears elastic stockings to minimize the bilateral lymphatic edema of her feet and both legs.
Discussion
To our knowledge, there are no other case reports on lymphatic edema based on abnormal lymph vessel development in patients with CDG. In the first patient, the right leg was increased in size from a young age, but caused a significantly progressive burden for the patient after the onset of cellulitis. The second patient developed bilateral lymphatic edema as a neonate, which did not resolve as the patient recovered from cardiac surgery, and still persists.
Several potential contributing factors to the development of edema in PMM2-CDG are present in our patients, but fail to fully explain their clinical features. First, hypoalbuminemia is common in PMM2-CDG patients (Jaeken 2010; Grünewald 2009) and is a known cause for edema as it lowers oncotic pressure. Generally, patients with low albumin levels present with generalized pitting edema; in our patients, nonpitting edema existed. Moreover, the edema did not subside after normal albumin levels were obtained as a result of albumin infusions in both patients. Second, cardiac failure was proposed as a cause for edema in both patients. The first patient was diagnosed with hypertrophic cardiomyopathy, and regular cardiac evaluation was performed by echocardiography but no signs of cardiac failure were noted. In the second patient, edema persisted after recovering from cardiac surgery. Also in this patient, there were no signs of cardiac failure by echocardiography. Finally, both patients were known with hypothyroidism, although the second patient had normal thyroxin levels with increased TSH levels when she developed the lymphatic edema. Localized (pretibial) myxedema has primarily been described in autoimmune thyroid disease (Cannavò et al. 2002; Georgala et al. 2002; Buljan-Cvijanovic et al. 1998; Forgie et al. 1994). Our patients did neither had severe hypothyroidism nor were they found to have autoimmune thyroid disease. Additionally, treatment with thyroxin, which normalized thyroid hormone levels, did not lead to any decrease of the edema.
History and physical examination were suggestive of lymphatic edema in our two patients. Moreover, as mentioned before, other causes of edema were excluded and specific studies were performed to confirm lymphatic edema (Modi et al. 2007). In the first patient, we performed a lymphoscintigraphy in which lymph transport is estimated by determining the clearance rate of a radiolabelled macromolecule from subcutis. Indirect lymphography was performed in the second patient since lymphoscintigraphy was not widely available at the time this patient was diagnosed. A large contrast molecule is injected subcutaneously and its uptake and transport is visualized by radiographic examination. This technique does not offer a quantitative result with reference values. The results of these studies differed widely from controls, confirming lymphatic edema.
In patients with PMM2-CDG with therapy resistant pericardial or ascites fluid collections, an abnormal transferrin isoelectric focusing pattern has been found in both pericardial and abdominal fluids. A regulatory dysfunction of protein and fluid balance due to glycosylated cell surface proteins, has been suggested as a possible underlying cause (Truin et al. 2008). However, this theory can only partly explain the lymphatic edema in our patients. A protein and fluid imbalance would primarily lead to fluid accumulation; a decrease in lymphatic flow velocity, which was noticed in both patients, could not be explained by this mechanism.
Although in our first patient a secondary worsening of lymph vessel structure due to infections might play an important role in the clinical course, a congenital hypoplasia of lymphatic vessels has been confirmed by lymphatic studies in the second patient. Glycoproteins play an essential role in the embryological phase, both in cell migration and organ development. Primary development of the lymphatic system can be disturbed due to hypoglycosylation of the numerous glycoproteins involved in lymphangiogenesis including podoplanin, PROX1 (prospero-related homeobox 1), LYVE1 (lymphatic vessel endothelial hyaluronan receptor 1) and VEGFC (vascular endothelial growth factor C). VEGFC, an N-glycosylated protein involved in the regulation of growth and regulation in lymphangiogenesis (Skobe and Detmar 2000), can be hypoglycosylated in PMM2-CDG patients, potentially resulting in an underdeveloped lymphatic system. Also, edema that is present during development may cause damage to the lymphatic vessels, leading to secondary hypoplasia.
In conclusion, we report on severe, progressive lymphatic edema in two patients with PMM2-CDG. Next to regulatory dysfunction of protein and fluid balance due to glycosylated cell surface proteins, we propose congenital lymph vessel hypoplasia as the most important underlying factor for the symptoms. Patients with nonpitting edema of the extremities should be screened early on for possible lymphatic anomalies, and conservative therapy should be started to prevent progression of swelling, decubitus, and infections.
Acknowledgment
The authors are thankful for the parents of the two patients of the report, for their patience, and input in the current report.
Take-Home-Message
Case report on two PMM2-CDG patients with lymphatic edema of lower extremity, primarily caused by hypoplasia of lymphatic vessels.
Footnotes
Competing interests: None declared.
References
- Buljan-Cvijanovic M, Neal JM, Zemtsov A. Euthyroid pretibial myxedema. Endocr Pract. 1998;4(6):375–377. doi: 10.4158/EP.4.6.375. [DOI] [PubMed] [Google Scholar]
- Cannavò SP, Borgia F, Vaccaro M, Guarneri F, Magliolo E, Guarneri B. Pretibial myxoedema associated with Hashimoto’s thyroiditis. J Eur Acad Dermatol Venereol. 2002;16(6):625–627. doi: 10.1046/j.1468-3083.2002.00532.x. [DOI] [PubMed] [Google Scholar]
- Damen G, de Klerk H, Huijmans J, den Hollander J, Sinaasappel M. Gastrointestinal and other clinical manifestations in 17 children with congenital disorders of glycosylation type Ia, Ib, and Ic. J Pediatr Gastroenterol Nutr. 2004;38(3):282–287. doi: 10.1097/00005176-200403000-00010. [DOI] [PubMed] [Google Scholar]
- de Lonlay P, Seta N, Barrot S, Chabrol B, Drouin V, Gabriel BM, et al. A broad spectrum of clinical presentations in congenital disorders of glycosylation I: a series of 26 cases. J Med Genet. 2001;38(1):14–19. doi: 10.1136/jmg.38.1.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forgie JC, Highet AS, Kelly SA. Myxoedematous infiltrate of the forehead in treated hypothyroidism. Clin Exp Dermatol. 1994;19(2):168–169. doi: 10.1111/j.1365-2230.1994.tb01151.x. [DOI] [PubMed] [Google Scholar]
- Georgala S, Katoulis AC, Georgala C, Katoulis EC, Hatziolou E, Stavrianeas NG. Pretibial myxedema as the initial manifestation of Graves’ disease. J Eur Acad Dermatol Venereol. 2002;16(4):380–383. doi: 10.1046/j.1468-3083.2002.00567.x. [DOI] [PubMed] [Google Scholar]
- Grünewald S. The clinical spectrum of phosphomannomutase 2 deficiency (CDG-Ia) Biochim Biophys Acta. 2009;1792(9):827–834. doi: 10.1016/j.bbadis.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Jaeken J. Congenital disorders of glycosylation. Ann N Y Acad Sci. 2010;1214:190–198. doi: 10.1111/j.1749-6632.2010.05840.x. [DOI] [PubMed] [Google Scholar]
- Leticee N, Bessieres-Grattagliano B, Dupre T, Vuillaumier-Barrot S, de Lonlay P, Razavi F, et al. Should PMM2-deficiency (CDG Ia) be searched in every case of unexplained hydrops fetalis? Mol Genet Metab. 2010;101(2–3):253–257. doi: 10.1016/j.ymgme.2010.06.009. [DOI] [PubMed] [Google Scholar]
- Modi S, Stanton AW, Mortimer PS, Levick JR. Clinical assessment of human lymph flow using removal rate constants of interstitial macromolecules: a critical review of lymphoscintigraphy. Lymphat Res Biol. 2007;5(3):183–202. doi: 10.1089/lrb.2007.5306. [DOI] [PubMed] [Google Scholar]
- Skobe M, Detmar M. Structure, function, and molecular control of the skin lymphatic system. J Investig Dermatol Symp Proc. 2000;5(1):14–19. doi: 10.1046/j.1087-0024.2000.00001.x. [DOI] [PubMed] [Google Scholar]
- Truin G, Guillard M, Lefeber DJ, Sykut-Cegielska J, Adamowicz M, Hoppenreijs E, et al. Pericardial and abdominal fluid accumulation in congenital disorder of glycosylation type Ia. Mol Genet Metab. 2008;94(4):481–484. doi: 10.1016/j.ymgme.2008.05.005. [DOI] [PubMed] [Google Scholar]