

Abstract
Hunter syndrome, or Mucopolysaccharidosis type II (MPS II), is a rare X-linked recessive disorder caused by a deficiency of the lysosomal enzyme iduronate-2-sulfatase (IDS). The phenotypic spectrum varies from severe to attenuated clinical forms. We report a large Brazilian family with 16 affected individuals exhibiting a very attenuated form of MPS II. Fourteen female carriers were also identified. Twelve affected male patients, whose ages ranged from 1 to 35 years, were examined. Molecular analysis showed a novel missense mutation (p.A77D) in the IDS gene, confirming the diagnosis. Nine of the family members presented some degree of heart damage, though only the proband became symptomatic and required heart transplantation. One 19-year-old adult and 1-year-old twin boys each had a normal echocardiogram. Short stature was found in two adults while macrocephaly was found in one; the remaining adults had anthropometric measures within normal range. All affected adults had normal cognitive development and were able to perform normal daily activities, except one who had mild learning disability. Two patients died due to natural causes beyond 70 years of age. The female carriers did not present any signs of disease. In this large family with a mild form of MPS II and variable degree of clinical manifestations, it is noteworthy that several affected individuals have remained asymptomatic even at advanced age and even without enzyme replacement therapy.
Introduction
Hunter syndrome, or Mucopolysaccharidosis type II (MPS II, OMIM 607014), is a rare X-linked recessive disorder caused by a deficiency of the lysosomal enzyme iduronate-2-sulfatase (IDS), leading to the progressive accumulation of glycosaminoglycans (GAGs) in several organs. It affects approximately 1 in 170,000 male newborns, whereas female carriers rarely manifest symptoms. The chronic deposition of undegraded GAGs in the connective tissue of multiple organs contributes to the progressive clinical course of the syndrome with a variable degree of severity and life expectancy. Increased urinary excretion of dermatan and heparan sulfate and low or absent activity of IDS confirms the diagnosis. It is noteworthy that neither the amount of IDS nor its activity correlates with clinical severity in MPS II patients (Martin et al. 2008; Scarpa 2011).
The phenotypic spectrum of MPS II varies, with two typical clinical forms. The severe form affects approximately 75% of all MPS II patients, with the clinical picture emerging between 18 months and 4 years of age. It is characterized by facial dysmorphism, short stature, hepatosplenomegaly, hernias, stiff joints and contractures, cardiac valve disease and obstructive respiratory complications, chronic diarrhea, hearing loss, and communicating hydrocephalus. Corneal opacities are usually absent, but progressive cognitive impairment and mental retardation become prominent. Death by progressive airway obstruction or cardiac dysfunction usually occurs between the ages of 10 and 15 years. In contrast, the attenuated form exhibits a slower progression of the disease, usually marked by fairly normal intelligence, conductive and sensorineural hearing loss, hepatosplenomegaly, and joint contractures. Retinal degeneration may develop. These patients generally survive into adulthood, although death may occur during late adolescence or later in life by airway obstruction and heart failure due to valvar defects (Martin et al. 2008; Muenzer et al. 2009; Scarpa 2011).
Over 300 mutations in the gene IDS have been described. Most of these are missense mutations and lead to impaired degradation of GAGs by lysosomes and progressive accumulation of these metabolites. Molecular analysis is essential to identify asymptomatic female carriers (Martin et al. 2008).
We report a large, nonconsanguineous Brazilian family with 16 individuals affected by a very attenuated form of MPS II, caused by a previously undescribed mutation.
Case Series
The previously asymptomatic proband of this family had an ordinary life until 23 years of age. He had normal cognitive development, allowing him to graduate from university. His clinical precedent was uneventful, except for a surgically corrected umbilical hernia during childhood. He was admitted at the Cardiologic Intensive Care Unit for acute decompensated heart failure progressing within 1 month. The physical exam at that time showed that the patient had normal height (176 cm), weight (73 kg) and occipital frontal circumference (OFC: 56 cm), as well as a mild coarse face and hepatomegaly. No other major MPS II clinical features, including lumbar gibbus or other physical deformities, were noted except for joint stiffness of the shoulders. Biochemical assays showed an increased excretion of GAGs in the urine and a deficiency of IDS activity in leukocytes, confirming the diagnosis of MPS type II.
Echocardiography demonstrated thickened valves with severe mitral regurgitation, severe left ventricular dilatation, and severe ventricle dysfunction. Once the patient did not tolerate inotropic drug withdrawal, a heart transplant was indicated for him. Unfortunately, the patient died of primary graft failure 2 days after the surgery. The postmortem histopathological analysis of the myocardium by electron microscopy revealed that GAGs accumulated in the interstitium of the myocytes.
Fifteen other affected individuals, including two deceased elders, and 14 female carriers, were identified in the family pedigree study (Fig. 1). Clinical, biochemical, and molecular tests were conducted among the 19 living family members. Twelve additional affected male patients were examined, among whom nine were adults over 18 years of age, an eight-year-old child, and one-year-old twin males. The ages of the male patients varied from 1 to 35 years (with a mean of 20 years). Anthropometric measurements were within normal centiles for the majority of them, excluding two individuals with heights of 150 and 158.5 cm and another with macrocephaly (OFC: 60.5 cm). Mild coarse facies was a universal finding, whereas hepatomegaly was observed only in the proband, probably secondary to acute decompensated heart failure. No one presented lumbar gibbus, claw hands, or deformities other than mild stiffness of shoulders (Fig. 2).
Fig. 1.
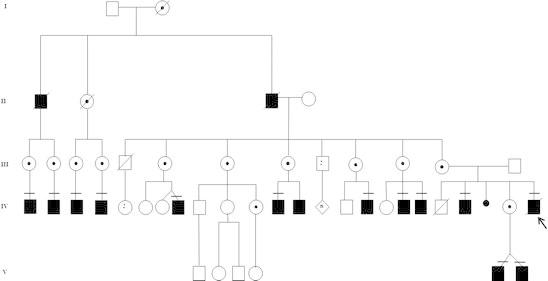
Family pedigree
Fig. 2.

Affected relatives: (a) proband; (b) proband’s brother; (c) proband’s cousin; (d) proband’s twin nephews
In this series, nine men presented some degree of heart damage, although only the proband became symptomatic. The aortic valve was involved in seven individuals, the mitral valve in five, and a ventricular septal defect was seen in one individual.
Surgical correction of hypertrophy of the adenoid tissue was necessary in five individuals and inguinal or umbilical hernia intervention in eight. Hypermetropia was present in three patients while retinal detachment secondary to trauma was present in one. Only one 32-year-old male presented mild mental retardation and hearing loss. All of the remaining adults had normal cognitive development, making it possible for them to graduate from normal school and perform satisfactorily in their ordinary jobs. The 14 female carriers have not presented any signs of disease activity; two of them died of natural causes at advanced ages.
Urinary levels of GAGs varied from 108 to 683 μg/mg creatinine (normal: 13–45), with a mean of 249.3 μg/mg. IDS activity in leukocytes ranged from undetectable to 4.3 nmol/4 h mg protein (normal: 31–110). Molecular analysis revealed a novel missense mutation (p.A77D) in the IDS gene in all of the nine affected patients and seven female carriers that were tested.
Discussion
This large Brazilian family with 16 individuals displays a very attenuated form of MPS II, with normal cognitive development and preserved quality of life. The diagnosis of this condition in otherwise asymptomatic people was only possible because of the proband´s acute decompensated heart failure.
This disease has been traditionally classified into attenuated and severe subtypes, although variations of severity between these two extremes have been observed.
Cardiovascular involvement was the most common serious complication of the disease in this family, although the proband was the only person to present cardiovascular symptoms. The fast progression of his heart failure at the young age of 23 years was conspicuous. The failure of the clinical management plus the severity of heart involvement determined his need for a heart transplantation, which was performed for the first time in the context of MPS II. Unfortunately, the surgical approach also failed in its objective, and the patient died of primary graft failure.
A remarkable variability of heart involvement was observed among affected individuals. The proband’s 28-year-old brother had only mild aortic regurgitation and his 19-year-old cousin had a normal echocardiogram. Interestingly, despite the fact that several other family members had severe valvar lesion in echocardiography studies, they had no complaints of symptoms of heart failure. In the literature, symptomatic individuals constitute more than 80% of all MPS II patients, whereas valvular disease may be found in up to 60% (Wraith et al. 2008a, b). Considering the clinical importance of cardiovascular involvement in MPS II, clinicians must always be aware of periodically monitoring heart complications during the follow-up of these patients through comprehensive clinical examination and routine echocardiography.
Life expectancy can be near normal in less severely affected individuals and such patients may reproduce, as reported previously (DiFerrante and Nichols 1972). We have identified two affected seniors that died of natural causes at advanced ages, beyond 70 years of age. The presentation of MPS II in these two individuals must have been mild enough to allow them to procreate and have long and normal lives.
Five affected males in this family needed resection of adenoid or tonsil tissues for the improvement of their upper airway obstruction, another common and potentially life-threatening complication that requires special attention (Sasaki et al. 1987). Joint restriction affecting the shoulders was noteworthy in nine individuals. In the mild form of MPS II, joint contractures usually affect the upper more prominently than the lower extremities. None of the individuals presented corneal opacities, as expected for MPS II patients.
Considering the known marked variability in the progression and the clinical phenotype of MPS II, we could expect that even individuals of the same family presenting the same mutation might manifest the attenuated form of the disease differently, as observed in this family.
The p.A77D missense mutation of IDS gene identified in the presented family has not been published before (Leiden Open Variant Database – LOVD) and probably allowed residual enzyme activity enough to prevent the severe form of MPS II and sparing from neurological impairment. Missense mutations resulting in reduced expression of IDS enzyme activity comprise the majority of gene mutations in MPS II, which have been described in patients with a whole spectrum of features, ranging from severe to attenuated forms. Those mutations that result in complete absence of the enzyme activity, such as total, partial deletions or rearrangements seem to be associated with the severe form of the disease in up to 25% of MPS II cases. Despite these associations, genotype–phenotype correlations are not predictable in all cases (Martin et al. 2008; Scarpa 2011).
Some years ago, there was no effective therapy for MPS II, and management was focused on a palliative approach. Now, the advent of enzyme replacement therapy (ERT) has changed this clinical approach, adding the possibility of improving the degradation and excretion of GAGs and changing the natural history of MPS II. Although ERT is the standard of care for MPS II, the age at which this therapy should be initiated and whether asymptomatic individuals presenting the mild form of MPS II should be treated remain uncertain (Muenzer et al. 2011; Wraith et al. 2008a, b).
In conclusion, in this large family with a very attenuated form of MPS II and a variable degree of clinical manifestations, it is noteworthy that several affected individuals have remained asymptomatic even at advanced ages without ERT.
Conflict of Interest
There are no conflicts to disclose.
Synopsis
Report of a large Brazilian family with a novel missense mutation on IDS gene, displaying a very attenuated form of MPS II, with 16 affected individuals, most of them asymptomatic.
Footnotes
Communicated by: Ed Wraith
References
- DiFerrante N, Nichols BL. A case of the Hunter syndrome with progeny. Johns Hopkins Med J. 1972;130(5):325–328. [PubMed] [Google Scholar]
- Martin R, Beck M, Eng C, Giugliani R, Harmatz P, Muñoz V, Muenzer J. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome) Pediatrics. 2008;121(2):e377–e386. doi: 10.1542/peds.2007-1350. [DOI] [PubMed] [Google Scholar]
- Muenzer J, Beck M, Eng CM, Escolar ML, Giugliani R, Guffon NH, Harmatz P, Kamin W, Kampmann C, Koseoglu ST, Link B, Martin RA, Molter DW, Muñoz Rojas MV, Ogilvie JW, Parini R, Ramaswami U, Scarpa M, Schwartz IV, Wood RE, Wraith E. Multidisciplinary management of Hunter syndrome. Pediatrics. 2009;124(6):e1228–e1239. doi: 10.1542/peds.2008-0999. [DOI] [PubMed] [Google Scholar]
- Muenzer J, Beck M, Eng CM, Giugliani R, Harmatz P, Martin R, Ramaswami U, Vellodi A, Wraith JE, Cleary M, Gucsavas-Calikoglu M, Puga AC, Shinawi M, Ulbrich B, Vijayaraghavan S, Wendt S, Conway AM, Rossi A, Whiteman DA, Kimura A. Long-term, open-labeled extension study of idursulfase in the treatment of Hunter syndrome. Genet Med. 2011;13(2):95–101. doi: 10.1097/GIM.0b013e3181fea459. [DOI] [PubMed] [Google Scholar]
- Sasaki CT, Ruiz R, Gaito R, Jr, Kirchner JA, Seshi B. Hunter’s syndrome: a study in airway obstruction. Laryngoscope. 1987;97(3 Pt 1):280–285. [PubMed] [Google Scholar]
- Scarpa M (2011) Mucopolysaccharidosis Type II. In: GeneReviews at GeneTests – Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1997–2011. Available at http://www.genetests.org. Accessed 24 May 2011
- Wraith JE, Beck M, Giugliani R, Clarke J, Martin R, Muenzer J. HOS Investigators Initial report from the Hunter Outcome Survey. Genet Med. 2008;10:508–516. doi: 10.1097/GIM.0b013e31817701e6. [DOI] [PubMed] [Google Scholar]
- Wraith JE, Scarpa M, Beck M, Bodamer OA, De Meirleir L, Guffon N, Lund AM, Malm G, Van der Ploeg AT, Zeman J. Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediatr. 2008;167:267–277. doi: 10.1007/s00431-007-0635-4. [DOI] [PMC free article] [PubMed] [Google Scholar]