

Abstract
Context:
Uterine remodeling is highly dependent on the glycosylated transmembrane protein extracellular matrix metalloproteinase (MMP) inducer (EMMPRIN). Previous studies indicate estradiol can increase EMMPRIN expression in uterine cells and promote subsequent induction of MMP production.
Objective:
The aim of this study was to investigate the role of G protein-coupled receptor 30 (GPR30) stimulation on EMMPRIN microvesicle release in the human uterine epithelial cell line hTERT-EEC (EECs).
Design:
We examined EMMPRIN release by human EECs in response to GPR30 stimulation by microvesicle isolation, Western blot, and immunocytochemistry. We employed a pharmacological approach using the GPR30-selective agonist G1 and the antagonist G15 to determine the receptor specificity of this response.
Results:
We demonstrated GPR30 expression in EECs and release of EMMPRIN in microvesicles in response to stimulation of GPR30. G1, estradiol, and cholera toxin stimulated EMMPRIN release in microvesicles as detected by Western blot and immunocytochemistry, indicating that stimulation of GPR30 can induce EMMPRIN microvesicle release.
Conclusions:
These data indicate that EMMPRIN release in microvesicles can be mediated by stimulation of GPR30 in human EECs, suggesting that inappropriate stimulation or expression of this receptor may be significant in uterine pathology.
Estradiol is required for and modulates many normal physiological processes in reproductive, immune, and vascular function. However, estradiol is also involved in pathological disease states including cancer and the reproductive disorder endometriosis (1, 2). Among the estradiol-dependent processes that play an important role in both normal and pathological uterine physiology are endometrial proliferation, angiogenesis, and remodeling as accomplished via matrix metalloproteinases (MMPs). Numerous studies have shown that MMP secretion is induced in both normal and tumor tissues by the transmembrane glycosylated protein extracellular MMP inducer (EMMPRIN) encoded by the BSG gene (3). EMMPRIN was initially identified as a tumor cell surface molecule stimulator of MMP release from fibroblasts and has been shown to be released in microvesicles in epithelial cells of the ovary and eye (4, 5). Since its discovery, abnormal expression of EMMPRIN and MMPs has been observed in a number of reproductive pathologies, including ovarian (6) and cervical cancer (7) as well as endometriosis (8).
In the uterus, EMMPRIN is strongly expressed by epithelial cells under the influence of estradiol (8, 9), although the mechanism of estradiol actions on EMMPRIN expression in these cells remains unknown. Conventionally, estradiol action involves binding to either estrogen receptor-α (ERα) or estrogen receptor-β (ERβ), encoded by ESR1 and ESR2, respectively, nuclear translocation, and alteration of transcription. However, estradiol can also mediate rapid, nongenomic signaling events through localization of nuclear ERs at the plasma membrane (10–12) or activation of the seven-pass transmembrane receptor G protein-coupled receptor 30 (GPR30), encoded by the GPER gene, by estradiol (13, 14).
Materials and Methods
Materials
G1 and G15 were obtained from Tocris (Ellisville, MO). All other chemicals including estradiol and cholera toxin (CTx) were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise specified. Stock solutions of G1 and G15 [20 and 200 μm, respectively, in dimethyl sulfoxide (DMSO)] were stored frozen. Estradiol stock solution was made in ethanol (20 μm) and stored at 4 C in the dark. After final dilution, DMSO or ethanol was less than 0.1% (vol/vol). A parallel solvent-only control was performed in all experiments with DMSO or ethanol.
Cell culture and treatment
Human uterine epithelial cells (EECs) (15) were obtained from Dr. Sabine Klonisch, University of Manitoba (Winnipeg, Canada), and grown in DMEM/F12 (Invitrogen, Carlsbad, CA) containing 10% (vol/vol) fetal bovine serum, 1% (wt/vol) penicillin/streptomycin, and 0.016% Arg/Ins at 37 C. Cells were grown to 80–90% confluency and placed in serum-free medium for 24 h before treatment. Cells were treated with 20 nm estradiol, 20 nm G1, and 200 nm G15 or parallel solvent controls for 0.1, 24, or 48 h as indicated. After treatment, conditioned medium was collected by aspiration and centrifuged for 15 min at 1500 × g at 4 C to remove cellular debris and then concentrated 50- to 80-fold by centrifugation (4000 × g for 10 min at 4 C) using Amicon ultracentrifuge 10-kDa filters (Millipore, Billerica, MA). To isolate microvesicles conditioned medium was first centrifuged to remove cellular debris as described above. Supernatant was gently pipetted into a new tube and centrifuged to pellet microvesicles (40,000 × g for 60 min at 4 C). Supernatant was aspirated. Microvesicle pellets were resuspended in 200 μl lysis buffer [10% glycerol, 62.5 mm Tris (pH 6.8), 2% sodium dodecyl sulfate] to solubilize microvesicle proteins and protein concentration was determined using the bicinchoninic acid (BCA) protein assay reagent (Thermo Scientific, Pittsburgh, PA). Equal volumes of microvesicle lysate were used for Western blotting. β1-Integrin blotting was used to verify microvesicle isolation. Cell counts were determined by hemocytometer.
Cell lysate extraction
Cells were trypsinized, pelleted, and washed twice with PBS (Cellgro, Manassas, VA) by centrifugation. Pelleted cells were extracted with Tris NP-40 EDTA buffer [10 mm Tris (pH 8.0), 1 mm EDTA, 0.5% Nonidet P-40, 1× protease inhibitor] for 45 min on ice with vortexing every 5 min. Samples were clarified by centrifugation (10,000 × g for 5 min).
RNA extraction and gene expression
Cells were trypsinized, pelleted, and washed three times with PBS. Cell pellets were then resuspended in RLT buffer (QIAGEN, Germantown, MD) supplemented with 10 μl/ml β-mercaptoethanol. RNA was isolated using the RNAeasy kit (QIAGEN) according to the manufacturer's instructions. One microgram of RNA was used in 20-μl reverse transcription reactions using a high-capacity reverse transcription kit (4368814; Applied Biosystems, Atlanta, GA), quantitative PCR (qPCR) was performed, and relative fold induction was determined using the comparative cycle threshold method as described in previous work (16). For nonquantitative PCR, cDNA was amplified using Platinum PCRSuperMix (Invitrogen) and the following gene and transcript-specific primers according to manufacturer's instructions: BSGi2 (forward 5′-GCGAGGAATAGGAATCATGG-3′, reverse 5′-TACTCTCCCCACTGGTCGTC-3′), BSGi3 (forward 5′-TTAGTCTGCGGTCCTCTTGC-3′, reverse 5′-TACTCTCCCCACTGGTCGTC-3′), BSGi4 (forward 5′-TTAGTCTGCGGTCCTCTTGC-3′, reverse 5′-TACTCTCCCCACTGGTCGTC-3′), GPER (forward 5′-ATCATCGGCCTGTGCTACTC-3′, reverse 5′-GGTTTAGGGAGCTGTTGGAG-3′), ESR1 (forward 5′-GTGCCTGGCTAGAGATCCTG-3′, reverse 5′-AGAGACTTCAGGGTGCTGGA-3′), and ESR2 (forward 5′-TCCAGGTTCAAAGAGGGATG-3′, reverse 5′-GTCGGCACTTCTCTGTCTCC-3′).
Electrophoresis and Western blotting
SDS-PAGE was performed using 4–12% gradient gels (Thermo Scientific) with HEPES running buffer (Thermo Scientific), and proteins were electrophoretically transferred to polyvinylidene difluoride membranes in Towbin buffer [25 mm Tris, 193 mm glycine, 0.1% sodium dodecyl sulfate (pH 8.4), 20% methanol, vol/vol] for Western blotting. Nonspecific membrane binding sites were blocked with a 5% solution of nonfat milk powder in Tris-buffered saline with Tween (12.5 mm Tris, 140 mm NaCl, 0.1% vol/vol Tween, pH adjusted to 7.6), and membranes were probed in blocking solution with specific antibodies: 1:500 dilution of anti-N-terminal EMMPRIN (N19; Santa Cruz Biotechnology, Santa Cruz, CA), 1:1000 dilution of anti-C-terminal EMMPRIN (Cell Signaling Technology, Danvers, MA), or 1:1000 dilution of anti-GPR30 (sc-48524-R; Santa Cruz Biotechnology). Species-appropriate anti-IgG antibodies (Cell Signaling) conjugated to alkaline phosphatase were then used to probe primary antibodies. SuperSignal West Pico chemiluminescent substrate (Thermo Scientific) was used to image for phosphatase activity. All anti-EMMPRIN Western blots were performed with the anti-C-terminal antibody (Cell Signaling) unless otherwise specified.
Immunofluorescence
Cells were cultured on μ-dishes (Ibidi, München, Germany) and treated as described above. After treatment, cells were washed twice with PBS and fixed in cold 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, MA) in PBS for 1 h at room temperature, washed in PBS (five times for 3 min each), permeabilized with 0.05% Triton X-100 in PBS for 15 min, washed in PBS (five times for 3 min each), blocked overnight in a 5% solution of nonfat milk powder in Tris-buffered saline with Tween, and washed in PBS (five times for 3 min each). Fixed, permeabilized samples were immunolabeled with a 1:100 dilution of anti-EMMPRIN C-terminal antibodies (Cell Signaling), washed, incubated in a 1:200 dilution of rabbit antimouse IgG conjugated to Alexa 488 (Invitrogen), and washed; all washes were in PBS (five times for 3 min each), and all antibodies were diluted in blocking solution. PBS was then removed, and 200 μl Prolong Gold (Invitrogen) was added to each μ-dish. Samples were cured overnight at 4 C. Images were collected using a 1.4 NA ×63 objective on a Zeiss LSM 700 confocal microscope at the Keck Imaging Facility at the University of Illinois Champaign-Urbana (Champaign, IL).
Statistical analysis
An ANOVA model to evaluate experimental variability between individual experiments was used. For qPCR, the difference between the threshold cycle (Ct) of the target gene and housekeeping gene (GAPDH) was determined using the 2−ΔΔCt method. The threshold cycle was defined as the cycle number where all transcripts were in the linear phase of amplification. The difference between the target gene and GAPDH was then normalized to control treatment (no treatment) expression and expressed as a relative fold difference. Statistical significance was measured to identify treatment effects within each gene evaluated by post hoc orthogonal contrast statements.
For immunoblots, densitometric analysis was performed using Image J software (National Institutes of Health, Bethesda, MD). An ANOVA test was used to determine statistical significance of the differences between treatment and control groups for each given treatment. The least significant difference test indicated differences between groups (P < 0.05 was considered significant).
Quantification of pixel intensity for immunofluorescence images was performed using Image J software (National Institutes of Health). Line scans across eight cells in each treatment group were measured, and average intensity was plotted against percent width of the cell. The Riemann sum method was used to calculate the total pixel intensity for each scan.
Results
Expression of BSG isoforms and full-length EMMPRIN protein in EECs
Our past work indicated that EMMPRIN expression in endometrial epithelial cells in vivo was increased during the proliferative phase of the cycle. Thus, we examined whether the BSG gene (encoding EMMPRIN protein) and EMMPRIN protein were expressed in the human EEC line. RT-PCR of EEC mRNA (Fig. 1A) showed robust expression of BSG isoform 2 and weaker expression of isoforms 3 and 4. Isoform 1 is retinal specific (17) and was not assayed. We also examined EMMPRIN protein expression in EEC lysates and conditioned medium. Western blotting revealed the presence of mature glycosylated (50–70 kDa) EMMPRIN (black arrowhead) and unprocessed (26 kDa) EMMPRIN (white arrowhead) in the cellular lysate (Fig 1B); alternate probing with antibodies for the N terminus and C terminus of EMMPRIN confirmed the presence of full-length EMMPRIN protein. Western blotting of concentrated conditioned medium from cells revealed the presence of only mature full-length EMMPRIN protein as shown in Fig. 1C (black arrowhead), indicating the presence of mature, glycosylated full-length EMMPRIN in conditioned medium.
Fig. 1.
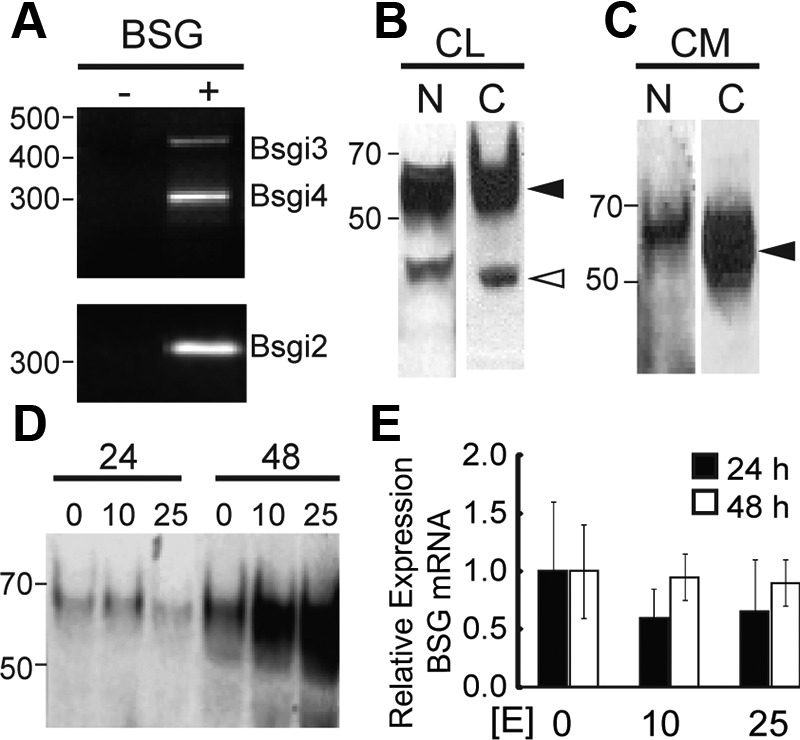
BSG and EMMPRIN expression in the presence and absence of estradiol. Panel A, Expression of BSG isoforms 2 (310 bp), 3 (454 bp), and 4 (298 bp) reported by RT-PCR of EEC mRNA with transcript-specific primers. Panel B, Immunoblot detection of the N terminus (N) and C terminus (C) of glycosylated (black arrowhead) and unmodified (white arrowhead) EMMPRIN in EEC lysate (CL). Panel C, Immunoblot detection of the N terminus (N) or C terminus (C) of glycosylated EMMPRIN (black arrowhead) in concentrated EEC-conditioned medium (CM). Panel D, Immunoblot detection of EMMPRIN in concentrated conditioned medium after treatment with 0, 10, or 25 nm estradiol for 24 or 48 h. BSG expression was not significantly different between treatments or time points. Panel E, Relative expression of BSG by RT-qPCR of EEC mRNA after treatment with 0, 10, or 25 nm estradiol for 24 h (black bars) or 48 h (white bars).
Regulation of EMMPRIN release by estradiol
Next we tested whether estradiol could alter release of EMMPRIN into the medium. As shown in Fig. 1D, EMMPRIN release into conditioned medium increased with time between 24 and 48 h of treatment (lanes 1–3 compared with lanes 4–6). More notably, increasing the dose of estrogen from 0 to 25 nm during the 48-h period increased EMMPRIN release (lane 4 compared with lane 6). We hypothesized that this was due to an increase in BSG gene expression or alternatively to an increased release of EMMPRIN protein already present in the cell. We used qPCR to assay for BSG transcript levels after treatment with estradiol and found no significant change in BSG transcript levels after treatment with 0, 10, or 25 nm estrogen for either 24 or 48 h (Fig. 1E). This suggested that the change in EMMPRIN protein release in response to estrogen treatment was not due to altered BSG gene transcription.
The role of GPR30 in estradiol-mediated EMMPRIN release
To determine which ERs mediate estradiol-stimulated EMMPRIN release, we examined ER gene expression in EECs. RT-PCR of EEC mRNA demonstrated that all three receptors, GPR30, ERα, and ERβ, encoded by GPER, ESR1, and ESR2, respectively, were expressed in this EEC line (Fig. 2A). Furthermore, the presence of GPR30 protein in EECs was demonstrated by Western blot (Fig. 2B) of EEC cell lysate. Because GPR30 is known to mediate nongenomic estradiol responses in other tissues but has not been studied in EECs, we used a pharmacological approach to study its possible role in EMMPRIN release using two well-characterized selective modifiers of GPR30: the selective agonist G1 and the selective antagonist G15. For this approach, conditioned medium from cells was collected after 48 h of pharmacological treatment, concentrated, and assayed by Western blot for EMMPRIN. Treatment with 20 nm G1, 20 nm estradiol, or 10 μm CTx increased EMMPRIN release into medium dramatically compared with vehicle controls (Fig. 2C). In contrast, treatment with the GPR30 antagonist G15 (200 nm) showed little or no EMMPRIN release as seen with vehicle controls (ethanol and DMSO). Significantly, 200 nm G15 was able to block both estradiol- and G1-stimulated release of EMMPRIN into the medium. These results suggest that estradiol-stimulated EMMPRIN release is mediated by GPR30. Previous work in other tissues has demonstrated GPR30 activation of Gαs leading to increased production of cAMP (14). If Gαs proteins stimulate EMMPRIN release, then constitutive activation of Gαs by CTx should also promote EMMPRIN release. Indeed, we found that treatment with CTx (Fig. 2C) increased EMMPRIN release, suggesting that release can be accomplished by activation of Gαs. These data are consistent with the hypothesis that stimulation of GPR30 and activation of Gαs results in EMMPRIN release.
Fig. 2.
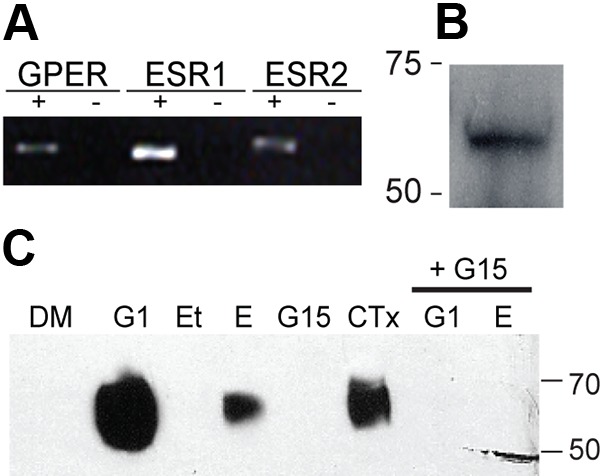
GPER, ESR1, ESR2, and GPR30 expression and effect of G1, estradiol, and CTx on EMMPRIN level in conditioned medium. A, Expression of GPER, ESR1, and ESR2, which encode GPR30, ERα, and ERβ, respectively, reported by RT-PCR of EEC mRNA with gene-specific primers. B, Immunoblot detection of GPR30 in EEC lysate. C, Immunoblot detection of EMMPRIN in concentrated conditioned medium after treatment with 0.1% DMSO (DM), 20 nm G1, 0.1% ethanol (Et), 20 nm estradiol (E), 10 μm CTx, or 200 nm G15 alone (G15) or supplemented with either 20 nm G1 (G1+G15) or 20 nm estradiol (G15+E) for 48 h.
EMMPRIN release occurs via microvesicle shedding
Next we examined whether EMMPRIN release by EECs occurs via microvesicle shedding. Cells were treated with 0 or 20 nm estradiol for 24 or 48 h or 10 μm CTx for 24 h. Microvesicles were isolated from the conditioned medium and solubilized for Western blotting and probed for EMMPRIN and β1-integrin. Consistent with our findings in Fig. 2C, EMMPRIN release increased with estradiol and CTx treatment (Fig. 3A). β1-Integrin also increased with estradiol and CTx treatment as seen in Fig. 3B. Densitometric analysis of immunoblots indicated a significant increase in EMMPRIN in solubilized microvesicles in the presence of estradiol and CTx (Fig. 3C). A concomitant increase in β1-integrin in solubilized microvesicles was also seen with estradiol and CTx treatment (Fig. 3D). Increased EMMPRIN and β1-integrin release were accompanied by an increase in total protein in the microvesicular fraction, suggesting an increase in the number of microvesicles shed rather than increased incorporation of EMMPRIN into microvesicles (Fig. 3E). This interpretation was also supported by a parallel increase in β1-integrin, another transmembrane protein found in microvesicles (18), in the microvesicular fraction (Fig. 3B).
Fig. 3.

Microvesicle release of EMMPRIN in response to estradiol and CTx treatment. Solubilized microvesicles recovered from conditioned medium after treatment with either 0 or 20 nm estradiol for 24 or 48 h or 10 μm CTx for 24 h were assayed by immunoblot, densitometric analysis, and BCA assay. All densitometric analysis results are given as mean ± sd of triplicate densitometric analyses. A, Immunoblot detection of EMMPRIN in solubilized microvesicles recovered from conditioned medium after treatment. B, Immunoblot detection of β1-integrin in solubilized microvesicles recovered from conditioned medium after treatment. C, Mean EMMPRIN pixel intensity for samples from cells treated for 48 h with estradiol or treated with CTx were significantly (indicated by a) higher than samples from cells treated for 24 h without estradiol. Estradiol treatment for 48 h significantly increased mean EMMPRIN pixel intensity (indicated by b). EMMPRIN pixel intensity after CTx treatment was not significantly different from 24 h estradiol treatment (indicated by c). D, Mean β1-integrin intensity for samples from cells treated for 48 h, treated with estradiol, or treated with CTx were significantly (indicated by a) higher than samples from cells treated for 24 h without estradiol. Treatment for 48 h with estradiol significantly increased mean β1-integrin pixel intensity (indicated by b). Mean β1-integrin pixel intensity after treatment with CTx was not significantly different from 24 h estradiol treatment (indicated by c). E, Protein concentration of solubilized microvesicles recovered from conditioned media after treatment was determined by BCA assay. Results are given as mean ± sd. Mean protein concentration for samples from cells treated for 48 h, treated with estradiol, or treated with CTx were significantly (indicated by a) higher than samples from cells treated for 24 h without estradiol. Treatment for 48 h with estradiol significantly increased mean protein concentration (indicated by b). Mean protein concentration after treatment with CTx was not significantly different from 24 h estradiol treatment (indicated by c).
EMMPRIN microvesicle release was also stimulated after treatment with the GPR30 agonist G1, and estradiol- or G1-induced microvesicle release was blocked by the GPR30 antagonist G15 (Fig. 4A). This was accomplished by an increase in number of microvesicles released as indicated by concomitant increases in β1-integrin and total protein (Fig. 4, B and E) rather than a selective incorporation of EMMPRIN into microvesicles, in which β1-integrin release would presumably remain constant. These results concurred with our observations of EMMPRIN release into concentrated conditioned medium and confirmed that EMMPRIN release is accomplished by microvesicle shedding. Densitometric analysis demonstrated that EMMPRIN and β1-integrin both significantly increased in response to G1 or estradiol. In contrast, the addition of the GPR30 receptor antagonist G15 prevented G1- and estradiol-induced increases in EMMPRIN and β1-integrin in the microvesicular fraction.
Fig. 4.

Microvesicle release of EMMPRIN and cell proliferation with G1, estradiol, and G15 treatment. All cells were treated with either 0.1% DMSO (DM), 20 nm G1, 0.1% ethanol (Et), 20 nm estradiol (E), or 200 nm G15 alone or supplemented with either 20 nm G1 (G1+G15) or 20 nm estradiol (G15+E) for 48 h unless otherwise indicated. All densitometric analysis results are given as mean ± sd of triplicate densitometric analysis. Panel A, Immunoblot detection of EMMPRIN in solubilized microvesicles recovered from conditioned medium after treatment. Panel B, Immunoblot detection of β1-integrin in solubilized microvesicles recovered from conditioned medium after treatment. Panel C, Mean EMMPRIN pixel intensity was not significantly different for samples indicated by a. Treatment with estradiol or G1 significantly increased mean EMMPRIN pixel intensity compared with vehicle controls indicated by b. Treatment in the presence of G15 with G1 or estradiol significantly reduced mean EMMPRIN pixel intensity compared with treatment with G1 or estradiol alone as indicated by c. Panel D, Mean β1 pixel intensity for samples from treated cells were not significantly different as indicated by a. Treatment with estradiol or G1 significantly increased mean β1-integrin pixel intensity compared with treatment with vehicle controls as indicated by b. Treatment in the presence of G15 with either G1 or estradiol significantly reduced mean β1-integrin pixel intensity compared with in the absence of G15 as indicated by c. Panel E, Protein concentration of solubilized microvesicles recovered from conditioned media after treatment was determined by BCA assay. Results are given as mean ± sd. Mean protein concentration for samples from treated cells were not significantly different as indicated by a. Treatment with estradiol or G1 significantly increased mean protein concentration compared with treatment with vehicle as indicated by b. Treatment with G1 or E in the presence of G15 significantly reduced mean protein concentration compared with the absence of G15 as indicated by c. Panel F, Cells were counted after treatment for 0.1 (black bars) or 48 (white bars) hours. Results are given as mean ± sd from five experiments. At 0.1 h after treatment, there was no significant difference between treatments as indicated by a. At 48 h of treatment, no significant difference in cell number is seen between treatment with vehicle or G1 as indicated by b. Forty-eight hours of treatment with estradiol in the presence or absence of G15 significantly increased cell number compared with vehicle control, media, or G1 as indicated by c. Treatment with CTx significantly decreased cell number compared with medium alone as indicated by d.
Increased release of EMMPRIN-containing microvesicles in response to estradiol could be due in part to cell proliferation because estradiol is known to stimulate epithelial cell proliferation. To examine the effect of estradiol on EEC proliferation and investigate whether proliferation might involve signaling through GPR30, we treated cells with vehicle controls, estradiol (with and without G15), G1 (with and without G15), and CTx and performed cell counts after 0.1 or 48 h of treatment. Estradiol stimulated cell proliferation as indicated by a significant 47% increase in cell numbers compared with vehicle controls (see Fig. 4D). Interestingly, G1 failed to mimic this result. Cell proliferation in the presence of G1 was not significantly different from that of medium and vehicle controls (Fig. 4D). Treatment with G15 failed to diminish the proliferative effect of estradiol although EMMPRIN release was prevented (Fig. 4A). Cell proliferation was also inhibited by CTx (Fig. 4F) while simultaneously stimulating EMMPRIN microvesicle release (Fig. 3A).
To further investigate cellular release of EMMPRIN, we performed immunocytochemistry for EMMPRIN after treatment with estradiol, G1, or CTx. Immunolabeling for EMMPRIN at 24-h intervals showed treatment-specific alteration in EMMPRIN protein localization (Fig 5). Before treatment (Fig. 5, top row, 0.1 h, no treatment), EMMPRIN labeling was low intensity and diffuse throughout the cells. Treatment with estradiol, G1, or CTx resulted in a higher intensity of EMMPRIN at the plasma membrane within minutes (Fig. 5, second row, 0.1 h, with treatment). A magnified view of this intensity change at 0.1 h in the presence or absence of treatment is shown in Fig. 6A. Line-scan analysis of pixel intensity before (Fig. 6B) and after (Fig. 6C) treatment indicated an increase in membrane intensity with estradiol, G1, or CTx treatment. Integral calculation of line scans indicated no change in total cell pixel intensity (Fig. 6D). This membrane localization was maintained at high intensity for 24 h and diminished after 48 h (Fig. 5, fourth and sixth rows). This timeline was consistent with our previous results indicating increased EMMPRIN release into conditioned medium via microvesicles after 48 h of treatment. Control cells treated with vehicle for 24 or 48 h showed EMMPRIN labeling comparable to that of 0.1 h of vehicle treatment (third and fifth rows).
Fig. 5.

EMMPRIN localization in response to estradiol, G1, or CTx treatment. Cells were treated with either 20 nm estradiol (first column), 20 nm G1 (second column), or 10 μm CTx (third column). Rows indicate the presence (+) or absence (−) of treatment and the duration (0.1, 24, or 48 h) of treatment. After treatment, cells were fixed, permeabilized, and immunolabeled for EMMPRIN protein and visualized using confocal microscopy. Insets show labeling in no-primary controls. Scale bars, 20 μm.
Fig. 6.

Cellular distribution of EMMPRIN with estradiol, G1, and CTx treatment. A, Cells were treated with either 20 nm estradiol (first column), 20 nm G1 (second column), or 10 μm CTx (third column). Rows indicate the presence (+) or absence (−) of treatment; all treatments were performed for 0.1 h. After treatment, cells were fixed, permeabilized, and immunolabeled for EMMPRIN protein and visualized using confocal microscopy. Scale bars, 10 μm. B, Line scans were performed on cells treated with vehicle controls as indicated, and mean pixel intensity ± sd of triplicate analysis and sem was calculated (n = 3). C, Line scans were performed on cells treated with estradiol, G1, or CTx as indicated, and mean pixel intensity ± sd of triplicate analysis and sem was calculated (n = 8 cells for each treatment). D, Total pixel intensity for each scan was determined by Riemann sum integration for vehicle controls (white bars) and treatments (black bars) as indicated. Average total pixel intensity ± sd of triplicate analysis and sem was calculated (n = 8 cells for each treatment).
Discussion
In this report, we studied the potential role of GPR30 in mediating EMMRPIN release. Currently, the normal function of GPR30 in vivo remains unclear because GPR30-null animals exhibit no reported reproductive deficits (19). Interestingly, GPR30 expression and activation have been implicated in pathological states including proliferation of breast, uterine, and thyroid cancer cells and endometriotic cells (20–22). Here we characterize GPR30 signaling in response to estradiol in human endometrial cells. EMMPRIN is released by human endometrial cells (EECs) in response to treatment with either estradiol or the GPR30-selective agonist G1. Both estradiol- and GPR30-induced release of EMMPRIN occurs via microvesicle shedding. Other studies have correlated overexpression of GPR30 with increased myometrial invasion and poor prognosis in uterine carcinomas (23). GPR30 expression is also strongly associated with HER2 expression and tumor progression in breast carcinomas (24). These reports suggest that cell invasion, potentially mediated by EMMPRIN, may be linked to GPR30 expression and subsequent activation. Our results reveal a putative mechanism by which estradiol can stimulate release of EMMPRIN-containing microvesicles as stimulation of GPR30 in EECs resulted in rapid, increased microvesicle release via activation of stimulatory G proteins.
Previous studies suggest estrogen regulation of EMMPRIN release in EECs. Here we report that BSG and EMMPRIN are expressed in cultured human EECs and that treatment with estrogen increased the release of EMMPRIN into the surrounding medium. We found that estrogen stimulation does not alter BSG transcript levels, and instead, we observed a rapid change in EMMPRIN intensity intracellularly consistent with a nongenomic signaling event. Additional experiments with the GPR30-selective agonist G1 and the GPR30-selective antagonist G15 showed EMMPRIN release can be mediated by a change in microvesicle release. Additionally, treatment with CTx constitutively activating Gαs resulted in microvesicle release, indicating that microvesicle shedding can be mediated by Gαs, the Gα subunit previously identified as coupled to GPR30. These findings were supported by a concomitant increase in β1-integrin release.
Numerous breast (13, 25–28), endometrial (20, 29, 30), ovarian (1, 31–33), and prostate cancer (34) cell lines are sensitive to the proliferative and invasive effects of estrogen. GPER is expressed in many of these cell types, and stimulation of GPR30 by estrogen and estrogenic compounds can initiate proliferation and migration in vitro (20, 21, 28, 32). Clinically, high levels of GPER expression in endometrial and ovarian cancer correlate with increased cell invasion, poor prognosis, and decreased survival (23, 24, 28, 35). Adverse effects of increased GPER expression have also been observed in breast cancer. High GPR30 expression is associated with increased breast tumor volume and metastasis (24). Here we link estrogen signaling and increased cell invasion through estrogen-stimulated activation of GPR30 and subsequent EMMPRIN release.
EMMPRIN is a critical mediator of cell invasion and metastasis. Shedding of EMMPRIN-containing microvesicles occurs both in normal and pathological tissue during tissue remodeling (36). In normal tissues, limited quantities of microvesicles are released in response to specific stimulation (37); in contrast, tumor cells shed microvesicles constitutively in a largely unregulated fashion (4). Our results showed stimulation of EMMPRIN-containing microvesicle release in response to GPR30 activation by estrogen and the selective GPR30 agonist G1 in normal EECs, suggesting a role for GPR30 in normal uterine remodeling. Interestingly, this signaling pathway may be perturbed in endometrial pathologies including endometriosis. The highly invasive human endometriotic cell line H-38 has been shown to overexpress GPR30 and lacks expression of ERα and ERβ, suggesting that increasing GPR30 levels may permit inappropriate EMMPRIN release and subsequent invasion (22).
Reports in many estrogen-responsive cancer cell lines demonstrate proliferation in response to estrogen (20, 21, 32); however, treatment with G1 failed to stimulate cell proliferation in EECs even though EMMPRIN microvesicle release was markedly increased. Estradiol alone successfully elicited both an increase in microvesicle release and an increase in cell number. Taken together, these results indicate EMMPRIN microvesicle release, but not proliferation, is mediated through activation of GPR30. The effect of GPR30 on cell proliferation is varied. In ER-negative endometrial carcinoma cells (38) and endometriotic H-38 cells (22), GPR30 mediates proliferation via phosphoinositide 3-kinase activation. In contrast, G1 inhibited estradiol-mediated cell proliferation in the mouse uterus but was dependent on stromal ERK1/2 activation (39). GPR30-mediated phosphoinositide 3-kinase activation and subsequent cell proliferation may be indicative of uterine pathology because this response was not seen in our studies using normal EECs or in normal mouse uterus but is present in carcinoma and endometriotic cell lines.
In addition to proliferation, estrogen can also mediate carcinogenesis. Anti-estrogen therapies include selective ER degraders, selective ER modulators, and aromatase inhibitors. These therapies have complex physiological actions and many have been shown to lack selectivity for ERα and ERβ and have effects on GPR30. Two commonly used ER antagonists, tamoxifen and fulvestrant, have well-documented agonist effects on GPR30 (40). Increased GPER expression has been correlated with tamoxifen-induced uterine bleeding and endometrial thickening and associated with uterine cancer. Moreover, a decreased survival rate was seen in breast cancer patients with GPR30-positive tumors treated only with tamoxifen, and an increase in GPR30 is seen with tamoxifen-only treatment (41).
The correlation of reproductive carcinogenesis with increased GPER expression indicates that modulation of this receptor may be significant for therapeutic management of estrogen-responsive cancers. Our studies demonstrated that pharmacological inhibition of GPR30 signaling using G15 prevented EMMPRIN microvesicle release stimulated by both estrogen and G1. GPR30-selective antagonists including G15 and the recently synthesized G36 (42) may represent a novel class of anti-estrogen therapeutics for modifying the actions of GPR30 in cancer. Additional studies demonstrating the specific actions of GPR30 stimulation on cell invasion, proliferation, and migration are warranted to understand the putative role of novel selective therapeutic agents in cancer treatment and to increase understanding of the physiological interplay between ER signaling in different pathological states.
Acknowledgments
This work was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, through cooperative agreement U54 HD 40093 (to R.A.N.) as part of the Specialized Cooperative Centers Program in Reproductive Research and by the American Medical Association Foundation Seed Grant (to L.A.B.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- BCA
- Bicinchoninic acid
- CTx
- cholera toxin
- DMSO
- dimethyl sulfoxide
- EEC
- human uterine epithelial cell line hTERT-EEC
- EMMPRIN
- extracellular MMP inducer
- ERα
- estrogen receptor-α
- ERβ
- estrogen receptor-β
- GPR
- G protein-coupled receptor 30
- MMP
- matrix metalloproteinase
- qPCR
- quantitative PCR.
References
- 1. Edwards DP. 2005. Regulation of signal transduction pathways by estrogen and progesterone. Annu Rev Physiol 67:335–376 [DOI] [PubMed] [Google Scholar]
- 2. Lange CA. 2008. Challenges to defining a role for progesterone in breast cancer. Steroids 73:914–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gabison EE, Hoang-Xuan T, Mauviel A, Menashi S. 2005. EMMPRIN/CD147, an MMP modulator in cancer, development and tissue repair. Biochimie (Paris) 87:361–368 [DOI] [PubMed] [Google Scholar]
- 4. Millimaggi D, Mari M, D'Ascenzo S, Carosa E, Jannini EA, Zucker S, Carta G, Pavan A, Dolo V. 2007. Tumor vesicle-associated CD147 modulates the angiogenic capability of endothelial cells. Neoplasia 9:349–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alcazar O, Hawkridge AM, Collier TS, Cousins SW, Bhattacharya SK, Muddiman DC, Marin-Castano ME. 2009. Proteomics characterization of cell membrane blebs in human retinal pigment epithelium cells. Mol Cell Proteomics 8:2201–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sillanpää S, Anttila M, Suhonen K, Hämäläinen K, Turpeenniemi-Hujanen T, Puistola U, Tammi M, Sironen R, Saarikoski S, Kosma VM. 2007. Prognostic significance of extracellular matrix metalloproteinase inducer and matrix metalloproteinase 2 in epithelial ovarian cancer. Tumour Biol 28:280–289 [DOI] [PubMed] [Google Scholar]
- 7. Wu Y, Zhou X, Zheng PS. 2011. Involvement of CD147 isoform4 in the proliferation of SiHa cells: a possible molecular mechanism of cervical cancer. Oncol Rep 26:717–724 [DOI] [PubMed] [Google Scholar]
- 8. Braundmeier AG, Fazleabas AT, Lessey BA, Guo H, Toole BP, Nowak RA. 2006. Extracellular matrix metalloproteinase inducer regulates metalloproteinases in human uterine endometrium. J Clin Endocrinol Metab 91:2358–2365 [DOI] [PubMed] [Google Scholar]
- 9. Noguchi Y, Sato T, Hirata M, Hara T, Ohama K, Ito A. 2003. Identification and characterization of extracellular matrix metalloproteinase inducer in human endometrium during the menstrual cycle in vivo and in vitro. J Clin Endocrinol Metab 88:6063–6072 [DOI] [PubMed] [Google Scholar]
- 10. Pedram A, Razandi M, Levin ER. 2006. Nature of functional estrogen receptors at the plasma membrane. Mol Endocrinol 20:1996–2009 [DOI] [PubMed] [Google Scholar]
- 11. Levin ER. 2005. Integration of the extranuclear and nuclear actions of estrogen. Mol Endocrinol 19:1951–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harrington WR, Kim SH, Funk CC, Madak-Erdogan Z, Schiff R, Katzenellenbogen JA, Katzenellenbogen BS. 2006. Estrogen dendrimer conjugates that preferentially activate extranuclear, nongenomic versus genomic pathways of estrogen action. Mol Endocrinol 20:491–502 [DOI] [PubMed] [Google Scholar]
- 13. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. 2005. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 307:1625–1630 [DOI] [PubMed] [Google Scholar]
- 14. Filardo EJ, Quinn JA, Frackelton AR, Jr, Bland KI. 2002. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol 16:70–84 [DOI] [PubMed] [Google Scholar]
- 15. Hombach-Klonisch S, Kehlen A, Fowler PA, Huppertz B, Jugert JF, Bischoff G, Schlüter E, Buchmann J, Klonisch T. 2005. Regulation of functional steroid receptors and ligand-induced responses in telomerase-immortalized human endometrial epithelial cells. Journal of molecular endocrinology 34:517–534 [DOI] [PubMed] [Google Scholar]
- 16. Braundmeier AG, Nowak RA. 2006. Cytokines regulate matrix metalloproteinases in human uterine endometrial fibroblast cells through a mechanism that does not involve increases in extracellular matrix metalloproteinase inducer. Am J Reprod Immunol 56:201–214 [DOI] [PubMed] [Google Scholar]
- 17. Redzic JS, Armstrong GS, Isern NG, Jones DN, Kieft JS, Eisenmesser EZ. 2011. The retinal specific CD147 Ig0 domain: from molecular structure to biological activity. J Mol Biol 411:68–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Atay S, Gercel-Taylor C, Kesimer M, Taylor DD. 2011. Morphologic and proteomic characterization of exosomes released by cultured extravillous trophoblast cells. Exp Cell Res 317:1192–1202 [DOI] [PubMed] [Google Scholar]
- 19. Otto C, Fuchs I, Kauselmann G, Kern H, Zevnik B, Andreasen P, Schwarz G, Altmann H, Klewer M, Schoor M, Vonk R, Fritzemeier KH. 2009. GPR30 does not mediate estrogenic responses in reproductive organs in mice. Biol Reprod 80:34–41 [DOI] [PubMed] [Google Scholar]
- 20. Vivacqua A, Bonofiglio D, Albanito L, Madeo A, Rago V, Carpino A, Musti AM, Picard D, Andò S, Maggiolini M. 2006. 17β-Estradiol, genistein, and 4-hydroxytamoxifen induce the proliferation of thyroid cancer cells through the g protein-coupled receptor GPR30. Mol Pharmacol 70:1414–1423 [DOI] [PubMed] [Google Scholar]
- 21. Vivacqua A, Bonofiglio D, Recchia AG, Musti AM, Picard D, Andò S, Maggiolini M. 2006. The G protein-coupled receptor GPR30 mediates the proliferative effects induced by 17β-estradiol and hydroxytamoxifen in endometrial cancer cells. Mol Endocrinol 20:631–646 [DOI] [PubMed] [Google Scholar]
- 22. Lin BC, Suzawa M, Blind RD, Tobias SC, Bulun SE, Scanlan TS, Ingraham HA. 2009. Stimulating the GPR30 estrogen receptor with a novel tamoxifen analogue activates SF-1 and promotes endometrial cell proliferation. Cancer Res 69:5415–5423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Smith HO, Leslie KK, Singh M, Qualls CR, Revankar CM, Joste NE, Prossnitz ER. 2007. GPR30: a novel indicator of poor survival for endometrial carcinoma. Am J Obstet Gynecol 196:386.e1–e9; discussion 386.e9–e11 [DOI] [PubMed] [Google Scholar]
- 24. Filardo EJ, Graeber CT, Quinn JA, Resnick MB, Giri D, DeLellis RA, Steinhoff MM, Sabo E. 2006. Distribution of GPR30, a seven membrane-spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin Cancer Res 12:6359–6366 [DOI] [PubMed] [Google Scholar]
- 25. Filardo EJ, Quinn JA, Bland KI, Frackelton AR., Jr 2000. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol 14:1649–1660 [DOI] [PubMed] [Google Scholar]
- 26. Carmeci C, Thompson DA, Ring HZ, Francke U, Weigel RJ. 1997. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics 45:607–617 [DOI] [PubMed] [Google Scholar]
- 27. Albanito L, Sisci D, Aquila S, Brunelli E, Vivacqua A, Madeo A, Lappano R, Pandey DP, Picard D, Mauro L, Andò S, Maggiolini M. 2008. Epidermal growth factor induces G protein-coupled receptor 30 expression in estrogen receptor-negative breast cancer cells. Endocrinology 149:3799–3808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang D, Hu L, Zhang G, Zhang L, Chen C. 2010. G protein-coupled receptor 30 in tumor development. Endocrine 38:29–37 [DOI] [PubMed] [Google Scholar]
- 29. He YY, Cai B, Yang YX, Liu XL, Wan XP. 2009. Estrogenic G protein-coupled receptor 30 signaling is involved in regulation of endometrial carcinoma by promoting proliferation, invasion potential, and interleukin-6 secretion via the MEK/ERK mitogen-activated protein kinase pathway. Cancer Sci 100:1051–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Leblanc K, Sexton E, Parent S, Bélanger G, Déry MC, Boucher V, Asselin E. 2007. Effects of 4-hydroxytamoxifen, raloxifene and ICI 182 780 on survival of uterine cancer cell lines in the presence and absence of exogenous estrogens. Int J Oncol 30:477–487 [PubMed] [Google Scholar]
- 31. Soloff MS, Szego CM. 1969. Purification of estradiol receptor from rat uterus and blockade of its estrogen-binding function by specific antibody. Biochem Biophys Res Commun 34:141–147 [DOI] [PubMed] [Google Scholar]
- 32. Albanito L, Madeo A, Lappano R, Vivacqua A, Rago V, Carpino A, Oprea TI, Prossnitz ER, Musti AM, Andò S, Maggiolini M. 2007. G protein-coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17β-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer Res 67:1859–1866 [DOI] [PubMed] [Google Scholar]
- 33. Henic E, Noskova V, Hoyer-Hansen G, Hansson S, Casslen B. 2009. Estradiol attenuates EGF-induced rapid uPAR mobilization and cell migration via the G-protein-coupled receptor 30 in ovarian cancer cells. Int J Gynecol Cancer 19:214–222 [DOI] [PubMed] [Google Scholar]
- 34. Chan QK, Lam HM, Ng CF, Lee AY, Chan ES, Ng HK, Ho SM, Lau KM. 2010. Activation of GPR30 inhibits the growth of prostate cancer cells through sustained activation of Erk1/2, c-jun/c-fos-dependent upregulation of p21, and induction of G2 cell-cycle arrest. Cell Death Differ 17:1511–1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Smith HO, Arias-Pulido H, Kuo DY, Howard T, Qualls CR, Lee SJ, Verschraegen CF, Hathaway HJ, Joste NE, Prossnitz ER. 2009. GPR30 predicts poor survival for ovarian cancer. Gynecol Oncol 114:465–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sidhu SS, Mengistab AT, Tauscher AN, LaVail J, Basbaum C. 2004. The microvesicle as a vehicle for EMMPRIN in tumor-stromal interactions. Oncogene 23:956–963 [DOI] [PubMed] [Google Scholar]
- 37. Taraboletti G, D'Ascenzo S, Borsotti P, Giavazzi R, Pavan A, Dolo V. 2002. Shedding of the matrix metalloproteinases MMP-2, MMP-9, and MT1-MMP as membrane vesicle-associated components by endothelial cells. Am J Pathol 160:673–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wei Y, Zhang Z, Liao H, Wu L, Wu X, Zhou D, Xi X, Zhu Y, Feng Y. 2012. Nuclear estrogen receptor-mediated Notch signaling and GPR30-mediated PI3K/AKT signaling in the regulation of endometrial cancer cell proliferation. Oncol Rep 27:504–510 [DOI] [PubMed] [Google Scholar]
- 39. Gao F, Ma X, Ostmann AB, Das SK. 2011. GPR30 activation opposes estrogen-dependent uterine growth via inhibition of stromal ERK1/2 and estrogen receptor α (ERα) phosphorylation signals. Endocrinology 152:1434–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Prossnitz ER, Barton M. 2011. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol 7:715–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pandey DP, Lappano R, Albanito L, Madeo A, Maggiolini M, Picard D. 2009. Estrogenic GPR30 signalling induces proliferation and migration of breast cancer cells through CTGF. EMBO J 28:523–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dennis MK, Field AS, Burai R, Ramesh C, Petrie WK, Bologa CG, Oprea TI, Yamaguchi Y, Hayashi S, Sklar LA, Hathaway HJ, Arterburn JB, Prossnitz ER. 2011. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J Steroid Biochem Mol Biol 127:358–366 [DOI] [PMC free article] [PubMed] [Google Scholar]