
Table 2. Summary of data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Wavelength () | 1.033 |
| Resolution () | 201.64 (1.681.64) |
| Space group | P21 |
| Unit-cell parameters (, ) | a = 56.94, b = 92.62, c = 70.10, = 106.16 |
| Total reflections | 302500 (18223) |
| Unique reflections | 84168 (6163) |
| Multiplicity | 3.6 (3.0) |
| Completeness (%) | 98.5 (97.7) |
| I/(I) | 19.7 (8.0) |
| R meas † (%) | 5.2 (17.5) |
| R merge ‡ (%) | 4.4 (14.4) |
| Wilson B (2) | 10.2 |
| Refinement | |
| R cryst | 0.148 (0.203) |
| R free § | 0.178 (0.257) |
| R.m.s.d. from ideal bond lengths () | 0.007 |
| R.m.s.d. from ideal bond angles () | 1.128 |
| Average B factors (2) | |
| All atoms | 13.3 |
| Protein | 11.1 |
| Solvent | 24.9 |
| Ramachandran plot (%) | |
| Most favoured regions | 98.74 |
| Allowed regions | 1.26 |
| Disallowed regions | 0 |
| PDB code | 4gac |
†
R meas is the redundancy-independent R factor (on intensities; Diederichs Karplus, 1997 ▶).
‡
R
merge = 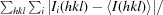
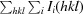 , where I(hkl) is the average value of the intensity of reflection hkl in the data set and Ii(hkl) is the intensity of the ith observation of that reflection.
, where I(hkl) is the average value of the intensity of reflection hkl in the data set and Ii(hkl) is the intensity of the ith observation of that reflection.
§
R free is calculated for a test set of reflections (5%) which were not included in the refinement.