

Isoprenoids are found widely in nature and have remarkably diverse structures.[1] They are utilized by all living organisms to fulfill a variety of biological roles, including serving as structural components of cell membranes, key constituents of electron transport chains, and hormones to regulate various physiological processes.[2] Many isoprenoids, produced as secondary metabolites, function as defense agents for the producers and have been one of the rich sources for human medicines.[2–3]
Successive condensation of isopentenyl diphosphate (IPP, 1, Scheme 1) and dimethylallyl diphosphate (DMAPP, 2) to construct isoprenyl backbone of desired length is a common step in the biosynthesis of all isoprenoids.[1a, 4] For decades, it was believed that the mevalonic acid (MVA) pathway is the sole source of IPP and DMAPP in all organisms.[5] Only recently, a second pathway, the deoxyxylulose phosphate (DXP) pathway (also known as methyl erythritol phosphate (MEP) pathway) was discovered,[1b, 1c, 6] in which both IPP and DMAPP are co-produced from 4-hydroxyl-3-methyl-2-butenyl diphosphate (HMBPP, 3) catalyzed by IspH (Scheme 1A).[7–11] Since IspH is not present in human and isoprenoids are essential for the survival of many pathogenic microorganisms, IspH has become an attractive target for new anti-microbial drug development.[12]
Scheme 1.
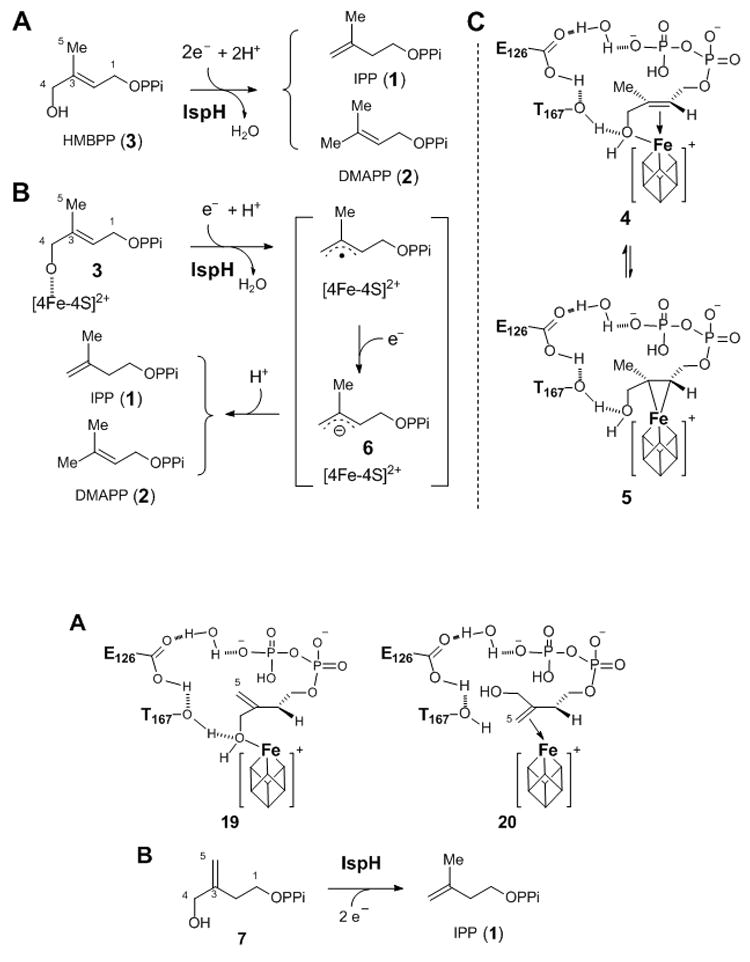
(A) The IspH-catalyzed C4 dehydroxylation reaction, (B) a possible mechanism of IspH-catalyzed reaction, and (C) two models of one-electron reduced IspH-HMBPP (3) complex.
The IspH-catalyzed conversion of 3 to 1 and 2 is an overall two-electron reductive dehydroxylation reaction. Previous biochemical, spectroscopic, and structural studies of IspH revealed the presence of a [4Fe-4S] cluster having a unique iron site to which the C4 hydroxyl group of HMBPP (3) is anchored (see 4 in Scheme 1C).[10b] This iron-sulfur cluster plays an essential role in electron transfer during IspH catalysis.[8, 13] A mechanism, resembling that of Birch reduction, has been proposed for the IspH-catalyzed reaction as shown in Scheme 1B.[8a, 8b, 9, 13] However, in view of the close proximity of the C2-C3 double bond of 3 to the unique iron site of the [4Fe-4S] cluster (~ 2.9–3.0 Å) in the crystal structure of the IspH-HMBPP complex (Figure 1) and the results of ENDOR studies of an IspH E126A mutant,[10] an alternative mechanism involving the formation of a η2-alkenyl intermediate between the C2-C3 double bond and the reduced [4Fe-4S]+ cluster (see 5 in Scheme 1C) was also proposed.[8c] To further investigate the mechanism of this intriguing reaction, we prepared a substrate analogue, 3-(hydroxymethyl)but-3-en-1-yl diphosphate (7, Scheme 2), which is expected to bind to [4Fe-4S]+ cluster in two different orientations (see 19 and 20 in Scheme 3) depending on whether formation of an metallacycle intermediate is part of catalysis. Reported herein are the experimental details and the mechanistic implications of these studies. The evaluation of the competence of 7 as an IspH substrate and analysis of the protonation of the allylic anion intermediate (6) shed new light on the mode of action of IspH.
Figure 1.

The active site of IspH with the 4-OH group of HMBPP (3) bound to the [4Fe-4S] cluster. The distances between OB to C2 and Oc to C4 are ~3.4 and 3.5 Å, respectively (pdb code: 3KE8).
Scheme 2.
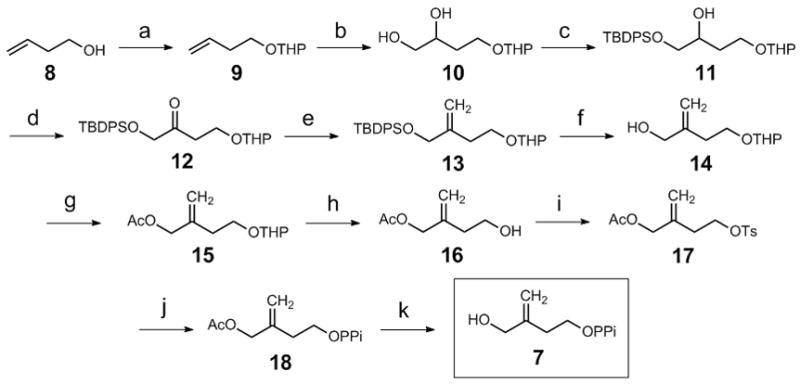
Reagents and conditions: a) DHP 1.20 eq, CH2Cl2, 0 °C, 2 hr, 95%; b) OsO4 cat., NMO 1.50 eq, acetone/KPi buffer (100 mM, pH 7.40)/THF = 2/2/1, RT, 3 hr, 90%; c) TBDPSCl 1.10 eq, Imidazole 2.00 eq, DMAP cat., CH2Cl2, RT, 12 hr, 83%; d) DMSO 3.00 eq, (COCl)2 1.50 eq, Et3N 5.00 eq, CH2Cl2, −78 °C to RT, 1 hr, 85%; e) PPh3CH3I 2.00 eq, n-BuLi 1.90 eq, THF, 0 °C to RT, 2 hr, 80%; f) TBAF 2.00 eq, THF, 90%; g) Ac2O 4.00 eq, pyridine, RT, 14 hr, 91%; h) AcOH/H2O/THF= 3/3/1, RT to 50 °C, 5 hr, 80%; i) TsCl 2.00 eq, pyridine, 0 °C, 12 hr, 90%; j) [N(n-Bu)4]3P2O7H 1.30 eq, MeCN, RT, 5 hr, 40%; k) NaOH 2.50 eq, 0°C to RT, 48 hr, 60%.
Scheme 3.
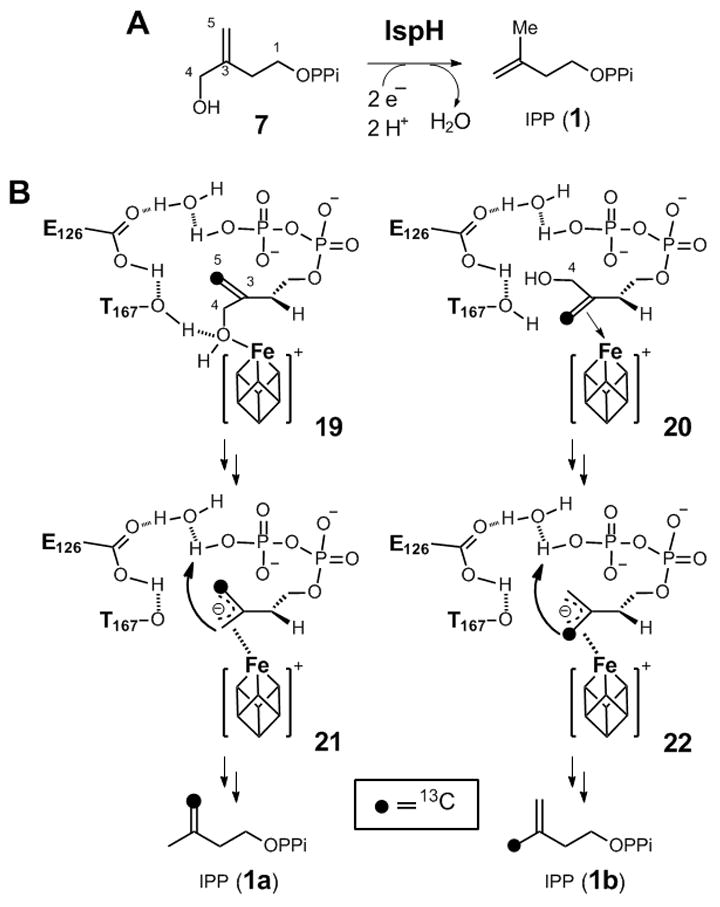
(A) The conversion of 3-(hydroxymethyl)but-3-en-1-yl diphosphate (7) to IPP (1) by IspH. (B) Two possible binding modes of [5-13C]-7 in the active site of IspH, and the anticipated respective outcomes of the pronotation step.
Synthesis of 7 followed the reaction sequence delineated in Scheme 2 (see supporting information for details). The capability of IspH to process 7 as a substrate was determined by monitoring the progress of the reaction with 1H-NMR spectroscopy.[8b, 9b] The only turnover product found in the incubation is IPP (1, Scheme 3A), which was isolated and verified by 1H-NMR spectroscopy and high-resolution mass spectrometry. The kinetic parameters for the conversion of 7 to 1 by IspH were measured using the methyl viologen assay.[8b] The analysis yielded a kcat of 484 ± 6.5 min−1 and a Km for 7 of 694 ± 79 μM. The kcat value is comparable to that of 604 ± 17 min−1 determined for the native substrate HMBPP (3) under similar conditions. However, the Km of 7 is nearly 35-fold higher than that of 3, resulting in a 44-fold reduction of the catalytic efficiency (kcat/Km) relative to that of HMBPP (3). Although 7 is a poor substrate, this result nevertheless demonstrates that the substrate of IspH does not necessarily have to have a double bond in the middle of its carbon skeleton as in 3. This finding challenges the proposed metallacycle model since the olefin moiety in 7 is further away from the apical iron atom of the [4Fe-4S] cluster (see 19 in Scheme 3) if the binding mode observed in the recent IspH-HMBPP complex is followed.[10b]
The fact that IPP is the sole product of the reaction of 7 and IspH is clearly different from the reductive dehydroxylation of 3 by IspH in which both IPP (1) and DMAPP (2) are produced in a ratio of ~5:1.[7d, 9b] This ratio is different from the ~1:3 distribution of IPP and DMAPP at thermodynamic equilibrium.[8a] The production of both IPP and DMAPP from HMBPP by IspH may be explained by the specific binding mode of 3 in the active-site of IspH.[10b] As shown by the crystal structure in Figure 1, HMBPP (3) binds to IspH in a bent conformation with its 4-OH group coordinated to the apical iron of the [4Fe-4S] cluster and its five-carbon backbone sandwiched between the [4Fe-4S] cluster and the C1 pyrophosphate group in the enzyme active site. With such geometric constraints and the lack of a nearby proton source, it was proposed that the terminal phosphate group of HMBPP serves as the proton donor in the final step (6→1 and 2, in Scheme 1B) of the dehydroxylation reaction,[8a, 9b, 10b] where the negative charge of the proposed allylic anion intermediate (6) is delocalized through C2, C3, and C4. Because OC and OB (see Figure 1) are within ~3.4–3.5 Å from the C2 and C4 position of HMBPP, they are likely involved in the protonation at C2 and C4 to yield IPP and DMAPP, respectively. This hypothesis is consistent with the pro-S stereochemistry observed for the C2 protonation step (to form IPP from HMBPP).[14] Unlike OC, OB forms a hydrogen-bond to a water molecule, which is also in H-bonding distance to E126. Thus, the ratio of IPP and DMAPP may simply reflect the different protonation state of OB and OC in the enzyme-substrate complex. Although the water molecule generated in the dehydroxylation step may serve as an alternative proton source at C4, the fact that incubation with HMBPP and its monoflouro analogue afforded IPP and DMAPP in the same ratio (~5:1)[9b] is most consistent with having the pyrophosphate (or the water molecule between OB and E126) as the proton source (see 21/22).
When compound 7 is used as the substrate, the negative charge of the proposed allylic anion intermediate will be delocalized among C3, C4, and C5 (21/22 in Scheme 3B) instead of C2, C3, and C4 (6), as seen in HMBPP (3). Hence, due to the proximal location of OB to C3, C4, and C5, OB is most likely the proton donor and protonation at either C4 or C5 will yield IPP (1) as the sole product, consistent with the experimental observations. However, since OB is located closer to C4(~3.4 Å) than to C5 (~4.6 Å), protonation is expected to occur largely at C4. Taking advantage of the anticipated preferential protonation at the site closer to OB, we probed this process using [5–13C]-7. We anticipated that if coordination of the 4-OH of 7 to the [4Fe-4S] cluster is the anchor that positions the substrate in the enzyme active site (shown as 19 in Scheme 3B), protonation at C4 of the allylic anion intermediate (21) would yield 1a when labeled 7 is used as the substrate. In contrast, if the reaction proceeds via a η2-alkenyl intermediate, as proposed by the metallocycle mechanism, coordination of the double bond of 7 to the iron-sulfur cluster may be a prerequisite to substrate orientation in the active site (Scheme 1C). Consequently, the [5-13C]-7 would bind to IspH in a conformation represented by 20. Subsequent protonation of 22 at the carbon closer to OB (now C5) should afford 1b as the product.
[5-13C]-7 was synthesized according to the reaction sequence shown in Scheme 2, except [13C]-PPh3CH3I was used in the conversion of 12 to 13 (see supporting information for details). The [5-13C]-labeled product was incubated with IspH, and the reaction was quenched at appropriate time intervals (60% and 100% conversion). After IspH was removed, the incubation mixture was analyzed by 13C-NMR spectroscopy. As shown in Figure 2A, [13C]-7 by itself gives one enriched 13C signal at 111.6 ppm. When the reaction was run to completion (Figure 2B), only one product was obtained. The sole signal that appears at 111.4 ppm can be assigned to the resonance of the terminal methylene carbon of the [13C]-labeled IPP product (1a). When the reaction was quenched at 60% conversion (Figure 2C), signals for both labeled 7 and 1a were present. Interestingly, there were no signals corresponding to [13C]-1b, which should have an enriched signal in the region of ~25 ppm (i.e., the chemical shift for the IPP methyl group). These results are consistent with the proposal that coordination of the 4-OH to the apical iron site is important to position the substrate for reaction with the [4Fe-4S] cluster, and C4 of 7 is the protonation site in IspH-catalyzed dehydroxylation of 7 to 1a.
Figure 2.

13C-NMR analysis of the incubation of [5-13C]-labeled 7 (5.0 mM) with IspH in 100 mM NaPi, pH 8.0 at 37°C. (A) in the absence of enzyme; (B) reaction was run with 5.0 μM IspH to completion (quenched after incubation for 1 h); (C) reaction was run to 60% completion with 1.0 μM IspH (quenched after incubation for 30 min).
These results are significant for two reasons. First, the outcome of the protonation experiments with [5-13C]-7 (i.e., only 1a is produced from 7) provide evidence supporting a catalytic role for the terminal phosphate group of the substrate in the final protonation step of the IspH reaction. Second, our data also shed new light on the interaction between the substrate double bond and the [4Fe-4S] cluster, which has been proposed to play an important role in IspH catalysis.[8c] However, the precise nature of this interaction has been controversial: it may be a transannular effect contributing to substrate binding as suggested by Shanmugam et al.,[15] or the driving force to form a metallacycle intermediate as proposed by Wang et al. (Scheme 1C).[8c] By comparing the incubation results with 3 and 7, it is now clear that while the olefin moiety is important for substrate binding and turnover, metallacycle formation between the double bond and the unique iron site of the [4Fe-4S] cluster is not a prerequisite for catalysis. Since the key coordinating ligand to the iron-sulfur cluster has now been shown to be the 4-OH group rather than the olefinic π-system of substrate 7 (Scheme 3B), the proposed metallacycle mechanism is less likely than the Birth reduction type mechanism (at least in the conversion of 7 to 1).[8c] Clearly, more studies are required to further delineate the catalytic mechanism of IspH. Additional experiments are also needed to determine how the reaction flux (IPP versus DMAPP) is controlled in the IspH reaction because this distribution is crucial for cellular survival. Efforts on both fronts are in progress.
Acknowledgments
Financial support for this work was partially provided by grants from the National Science Foundation (CAREER, CHE-0748504 to P.L.), the National Institutes of Health (GM 093903 to P.L.), and the Welch Foundation (F-1511 to H.-w. L.).
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org.
Contributor Information
Wei-chen Chang, Division of Medicinal Chemistry, College of Pharmacy, and Department of Chemistry and Biochemistry, University of Texas at Austin, Austin, Texas 78712 (USA), Fax: (+1)512-471-2746.
Youli Xiao, Department of Chemistry, Boston University Boston, MA 02215 (USA), Fax: (+1)617-353-6466.
Prof. Dr. Hung-wen Liu, Email: h.w.liu@mail.utexas.edu, Division of Medicinal Chemistry, College of Pharmacy, and Department of Chemistry and Biochemistry, University of Texas at Austin, Austin, Texas 78712 (USA), Fax: (+1)512-471-2746.
Prof. Dr. Pinghua Liu, Email: pinghua@bu.edu, Department of Chemistry, Boston University Boston, MA 02215 (USA), Fax: (+1)617-353-6466.
References
- 1.a) Sacchettini JC, Poulter CD. Science. 1997;277:1788–1789. doi: 10.1126/science.277.5333.1788. [DOI] [PubMed] [Google Scholar]; b) Eisenreich W, Bacher A, Arigoni D, Rohdich F. Cell Mol Life Sci. 2004;61:1401–1426. doi: 10.1007/s00018-004-3381-z. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Dairi T, Kuzuyama T, Nishiyama M, Fujii I. Nat Prod Rep. 2011;28:1054–1086. doi: 10.1039/c0np00047g. [DOI] [PubMed] [Google Scholar]
- 2.Bouvier F, Rahier A, Camara B. Prog Lipid Res. 2005;44:357–429. doi: 10.1016/j.plipres.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 3.a) Khachik F, Beecher GR, Smith JC., Jr J Cell Biochem. 1995;22:236–246. doi: 10.1002/jcb.240590830. [DOI] [PubMed] [Google Scholar]; b) Demmig-Adams B, Adams WW. Science. 2002;298:2149–2153. doi: 10.1126/science.1078002. [DOI] [PubMed] [Google Scholar]
- 4.Ruzicka L. Experientia. 1994;50:395–405. [PubMed] [Google Scholar]
- 5.a) McGarvey DJ, Croteau R. Plant Cell. 1995;7:1015–1026. doi: 10.1105/tpc.7.7.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bach TJ. Lipids. 1995;30:191–202. doi: 10.1007/BF02537822. [DOI] [PubMed] [Google Scholar]; c) Bloch K. Steroids. 1992;57:378–383. doi: 10.1016/0039-128x(92)90081-j. [DOI] [PubMed] [Google Scholar]; d) Kuzuyama T, Hemmi H, Takahashi S. Comprehensive Natural Products II. In: Mander L, Liu Hw, editors. Chemistry and Biology. I. Elsevier; Amsterdam: 2010. pp. 493–516. [Google Scholar]
- 6.a) Rohmer M. Prog Drug Res. 1998;50:135–154. doi: 10.1007/978-3-0348-8833-2_3. [DOI] [PubMed] [Google Scholar]; b) Rohmer M, Knani M, Simonin P, Sutter B, Sahm H. Biochem J. 1993;295(Pt 2):517–524. doi: 10.1042/bj2950517. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Rohmer M. Comprehensive Natural Products II. In: Mander L, Liu Hw, editors. Chemistry and Biology. I. Elsevier; Amsterdam: 2010. pp. 517–556. [Google Scholar]
- 7.a) Adam P, Hecht S, Eisenreich W, Kaiser J, Gräwert T, Arigoni D, Bacher A, Rohdich F. Proc Natl Acad Sci USA. 2002;99:12108–12113. doi: 10.1073/pnas.182412599. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rohdich F, Hecht S, Gartner K, Adam P, Krieger C, Amslinger S, Arigoni D, Bacher A, Eisenreich W. Proc Natl Acad Sci USA. 2002;99:1158–1163. doi: 10.1073/pnas.032658999. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Altincicek B, Duin EC, Reichenberg A, Hedderich R, Kollas AK, Hintz M, Wagner S, Wiesner J, Beck E, Jomaa H. FEBS Lett. 2002;532:437–440. doi: 10.1016/s0014-5793(02)03726-2. [DOI] [PubMed] [Google Scholar]; d) Gräwert T, Kaiser J, Zepeck F, Laupitz R, Hecht S, Amslinger S, Schramek N, Schleicher E, Weber S, Haslbeck M, Buchner J, Rieder C, Arigoni D, Bacher A, Eisenreich W, Rohdich F. J Am Chem Soc. 2004;126:12847–12855. doi: 10.1021/ja0471727. [DOI] [PubMed] [Google Scholar]; e) Wolff M, Seemann M, Grosdemange-Billiard C, Tritsch D, Campos N, Rodríguez-Concepción M, Boronat A, Rohmer M. Tetrahedron Lett. 2002;43:2555–2559. [Google Scholar]; f) Rekittke I, Wiesner J, Rohrich R, Demmer U, Warkentin E, Xu WY, Troschke K, Hintz M, No JH, Duin EC, Oldfield E, Jomaa H, Ermler U. J Am Chem Soc. 2008;130:17206–17207. doi: 10.1021/ja806668q. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Wang K, Wang W, No JH, Zhang Y, Oldfield E. J Am Chem Soc. 2010;132:6719–6727. doi: 10.1021/ja909664j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Gräwert T, Span I, Bacher A, Groll M. Angew Chem Int Ed. 2010;49:8802–8809. doi: 10.1002/anie.201000833. [DOI] [PubMed] [Google Scholar]; b) Xiao Y, Chu L, Sanakis Y, Liu P. J Am Chem Soc. 2009;131:9931–9933. doi: 10.1021/ja903778d. [DOI] [PubMed] [Google Scholar]; c) Wang W, Wang K, Liu YL, No JH, Li J, Nilges MJ, Oldfield E. Proc Natl Acad Sci USA. 2010;107:4522–4527. doi: 10.1073/pnas.0911087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Rohdich F, Zepeck F, Adam P, Hecht S, Kaiser J, Laupitz R, Gräwert T, Amslinger S, Eisenreich W, Bacher A, Arigoni D. Proc Natl Acad Sci USA. 2003;100:1586–1591. doi: 10.1073/pnas.0337742100. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xiao Y, Zhao ZK, Liu P. J Am Chem Soc. 2008;130:2164–2165. doi: 10.1021/ja710245d. [DOI] [PubMed] [Google Scholar]
- 10.a) Gräwert T, Rohdich F, Span I, Bacher A, Eisenreich W, Eppinger J, Groll M. Angew Chem Int Ed. 2009;48:5756–5759. doi: 10.1002/anie.200900548. [DOI] [PubMed] [Google Scholar]; b) Gräwert T, Span I, Eisenreich W, Rohdich F, Eppinger J, Bacher A, Groll M. Proc Natl Acad Sci USA. 2010;107:1077–1081. doi: 10.1073/pnas.0913045107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiao Y, Liu P. Angew Chem Int Ed. 2008;47:9722–9725. doi: 10.1002/anie.200803452. [DOI] [PubMed] [Google Scholar]
- 12.a) Oldfield E. Acc Chem Res. 2010;43:1216–1226. doi: 10.1021/ar100026v. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rodríguez-Concepción M. Curr Pharm Design. 2004;10:2391–2400. doi: 10.2174/1381612043384006. [DOI] [PubMed] [Google Scholar]
- 13.Seemann M, Janthawornpong K, Schweizer J, Böttger LH, Janoschka A, Ahrens-Botzong A, Tambou MN, Rotthaus O, Trautwein AX, Rohmer M, Schunemann V. J Am Chem Soc. 2009;131:13184–13185. doi: 10.1021/ja9012408. [DOI] [PubMed] [Google Scholar]
- 14.Laupitz R, Gräwert T, Rieder C, Zepeck F, Bacher A, Arigoni D, Rohdich F, Eisenreich W. Chem Biodiver. 2004;1:1367–1376. doi: 10.1002/cbdv.200490099. [DOI] [PubMed] [Google Scholar]
- 15.Shanmugam M, Zhang B, McNaughton RL, Kinney RA, Hille R, Hoffman BM. J Am Chem Soc. 2010;132:14015–14017. doi: 10.1021/ja106432h. [DOI] [PMC free article] [PubMed] [Google Scholar]