

Abstract
Equilibrium constants, such as the dissociation constant (Kd), are a key measurement of noncovalent interactions that are of importance to the proper functioning of molecules in living systems. Frontal analysis (FA) is a simple and accurate capillary electrophoresis (CE) method for the determination of Kd. Microchip CE coupled with laser-induced fluorescence (LIF) detection was used to determine Kd of protein-DNA interactions using the FA method. A model system of immunoglobulin E (IgE) and the IgE-binding aptamer was selected to demonstrate the capability of FA in microchip CE. Because the fluorescence emission was dependent on the dye migration velocity, the velocity of the free aptamer was adjusted to be the same as that of the aptamer-IgE complex by setting up individual separation voltage configurations for the free and bound aptamers. The ratio of the free and bound aptamers in the equilibrium mixture was directly measured from the heights of their plateaus detected at 1.0 cm from the intersection of the microchip, and no internal standard was needed. The Kd of the IgE-aptamer pair was determined as 6 ± 2 nM which is consistent with the reported results (8 nM).
Keywords: Capillary electrophoresis, Dissociation constant, Frontal analysis, Laser-induced fluorescence, Microchip
1 Introduction
Microchip capillary electrophoresis (CE) coupled with laser-induced fluorescence (LIF) detection offers CE separations with speed, sensitivity, ease of automation and low consumption of reagents [1–3]. Many techniques developed in traditional CE have been transferred onto microchip CE with appropriate modifications [3–7]. Multilane microchips have been developed to obtain high throughput analysis and were applied in DNA sequencing, genotyping, pharmaceuticals and other areas [8–12].
Noncovalent interactions between molecules, such as protein-DNA and protein-drug, are of great importance to their proper functions in living systems [13]. Binding constants usually are the appropriate measurement of the interactions. In the development of new drugs, the binding constant is one of the fundamental parameters for the evaluation of drug performance. Methods to determine binding constants include spectroscopy, microdialysis, calorimetry and separation techniques such as CE, etc. [13–17]. All methods have advantages and disadvantages. The newly-developed CE methods have advantages, such as accuracy and small reagent consumption. Several CE methods are used for binding constant determination, including affinity CE [18], partial filling technique [19, 20], frontal analysis (FA) [13], vacancy peak method [21] and Hummel-Dreyer method [22]. Among these FA is relatively simple to perform and produces robust binding constant values.
The principle of FA in CE is based on the separation of ligand from protein and protein-ligand complex [13, 23]. A larger volume of sample solution than normal is introduced into the capillary. Under an applied voltage, ligand, protein and protein-ligand complex migrate at their individual velocities since they have different electrophoretic mobilities. The ligand may appear as a flat-topped peak without dispersion due to diffusion during separations. The plateau height is an exact measurement of free ligand concentration in the incubated protein-ligand mixture. The peak height of the ligand at the initial concentration can be obtained when the ligand at the same initial concentration as in the mixture is run with the same injection volume. Therefore, binding constants can be obtained by plotting the binding curve of the number of bound ligand molecules per protein (r) versus the initial ligand concentration.
The prerequisite of FA in CE is enough electrophoretic mobility difference between ligand and protein or protein-ligand complex to give adequate separation when UV detection is used. However, when LIF detection is used, only the fluorescent-labeled molecules and the resulting complex can be detected. Therefore, the primary prerequisite for FA using LIF detection is the electrophoretic mobility difference between the fluorescently-labeled ligand and the ligand-involved complex. The binding target, such as non-labeled protein, does not affect the detection of the free and bound fluorescently-labeled ligand. In addition, the effect of complex formation on fluorescence emission may be negligible [24]. Therefore, the ratio of free ligand to the bound ligand can be obtained in a single experiment.
In this paper, FA in microchip CE coupled with LIF detection was developed to determine the dissociation constant of a protein-aptamer pair. The IgE-aptamer pair was chosen as a model system, and the aptamer was labeled with hexachlorofluorescein, while the protein was label-free. The large difference in mobilities between the free aptamer and the IgE-aptamer complex produced good resolution. The running condition was optimized and the dissociation constant was determined.
2 Materials and methods
2.1 Instrumentation
A Microfluidic Tool Kit (μTK) (Micralyne, Edmonton, AB, Canada) consisting of a high voltage power supply and a LIF detection system with excitation at 532 nm from a frequency-doubled Nd:YAG diode-pumped solid-state laser (4 mW) was used for microchip separations. The voltage program was controlled by LabView software (National Instruments, Austin, TX, USA). The excitation laser beam was focused on the separation channel through a 20× microscope objective, and the emission was collected with the same objective and was filtered by a 550 nm long-pass filter and a 568.2 nm bandpass filter before arriving at a PMT detector. Data were recorded at 20 Hz with Turbochrome software (Version 6.2.1) from Perkin-Elmer Corp. (Cupertino, CA, USA). The single-T borofloat glass microchip was purchased from Micralyne (Edmonton, AB, Canada). The long channel (8.5 cm) bisects the short channel (0.8 cm) at 0.5 cm. Four plastic reservoirs cut from 200 μL pipette tips (5.0 mm I.D.) were attached around the access holes using two-part epoxy.
2.2 Chemicals and Reagents
Tris, glycine and potassium dibasic phosphate were obtained from Fisher Scientific Company (Fair Lawn, NJ, USA). Human myeloma IgE (1.63 mg/mL or 8.36 μM, assuming a molecular weight of 195 kDa) was obtained from Athens Research and Technology (Athens, GA, USA). IgE-binding aptamer (5′-/5HEX/GGG GCA CGT TTA TCC GTC CCT CCT AGT GGC GTG CCC C-3′) was synthesized and labeled with 6-carboxy-2′,4,4′,5′,7,7′-hexachlorofluorescein, succinimidyl ester (6-HEX, SE) at the 5′ end, and was HPLC-purified by Integrated DNA Technologies (Coralville, IA, USA).
The stock solutions of Tris, glycine and phosphate were prepared with distilled-deionized water (dd-water) from a Milli-Q System (Millipore Corp., Bedford, MA, USA) at concentrations of 250, 1920, 50 mM, respectively. The separation buffer of Tris/glycine/K2HPO4 (TGK) was prepared by mixing appropriate aliquots of the Tris, glycine and phosphate stocks buffers and diluting with dd-water to give final concentrations of 25 mM Tris, 192 mM glycine and 5.0 mM K2HPO4. IgE and aptamer stock solutions were prepared in dd-water at concentrations of 2.0 μM and 50.0 μM, respectively. Aliquots of the IgE and aptamer stocks were placed in several centrifuge tubes and were stored in a −20 °C freezer for short term use. The remaining IgE and aptamer stocks were stored in a −70 °C freezer. For sample preparation, the stock solutions of IgE and aptamer were diluted with dd-water to give concentrations of 200 nM each.
2.3 Procedure
All liquid loaded into the microchip reservoirs were filtered through 0.22 μm filters. The TGK separation buffer was degassed by applying a vacuum in a syringe. All samples were prepared in the TGK separation buffer. An aliquot of the TGK separation buffer (30 μL) was pipetted into a 0.6-mL centrifuge tube, followed by an appropriate volume of dd-water, a volume of aptamer stock solution (200 nM), and finally 10 μL of the IgE stock solution (200 nM). The final total volume of each sample was 100 μL. After being vortexed for several seconds, the sample mixture was incubated for 30 min at room temperature.
The injection method was a modified gated injection [25] using the voltage configuration shown in Figure 1 where the voltage unit is kilovolts (kV). The loading voltage configuration (Figure 1a, 5 s) confined the sample solution flow from the sample reservoir (SR) to the sample waste reservoir (SW). Injection was carried out by setting an electric field between SR and the buffer waste reservoir (BW) while the buffer reservoir (BR) and SW were floated (Figure 1b). During the separation process, the SR was floated to protect the sample solution from the voltage impact, while the sample leaking from the channel connected with the SR was flushed to the SW (Figures 1c and 1d). Note that the voltage values were changed as described in the corresponding text.
Figure 1.

Voltage configuration. (a) Sample loading (5 s); (b) injection (3 s, or more); (c) dispensing to get plateau height for free aptamer; (d) dispensing to get plateau height for bound aptamer. SR, sample reservoir; SW, sample waste reservoir; BR, buffer reservoir; BW, buffer waste reservoir; F, floating; and G, ground. Voltages are in the unit of kilovolts (kV). See text for details.
Prior to analysis, the glass microchip was conditioned by sequentially adding 1.0 M NaOH, dd-water and TGK separation buffer into SR, SW and BR while applying a vacuum on BW for 5 min each. After NaOH conditioning, all the reservoirs were flushed with water several times. Following the condition steps, aliquots (50 μL) of the TGK separation buffer were pipetted into BR and BW while SW was filled with 46 μL of the TGK separation buffer. The SR was washed once with water, sucked for 2 s with a vacuum, and then was loaded with 50 μL of the sample solution. After focusing the laser beam onto the separation channel, the voltage configuration and Turbochrome software were manually switched on simultaneously. The detection point was at 1.0 cm from the intersection of the microchip.
3 Results and discussion
3.1 Principle of Kd determination using FA
In the IgE-aptamer pair, aptamer is the binding ligand, and IgE protein is the binding target. IgE-aptamer complex dissociates according to the following equation.
| (1) |
where PA represents protein-aptamer complex, P and A are the protein and the fluorescently labeled aptamer, respectively. The dissociation constant of complex PA is expressed in Equation 2
| (2) |
where [P]f, [A]f and [PA] are concentrations of the free protein, the free aptamer and the complex, respectively, in the equilibrium mixture; P0 and A0 are the original concentrations of the protein and the aptamer, respectively.
The ratio (r) between the free and bound aptamers in the equilibrium mixture is expressed in Equation 3.
| (3) |
With the combination of Equations 2 and 3, Kd is determined by Equation 4 [26].
| (4) |
where the values of P0 and A0 are known. Therefore, Kd is determined by the ratio r. In the model system, only the aptamer is fluorescently labeled. In the sample incubation buffer and the separation buffer (pH ~8.0), the incorporation of an aptamer and a protein produces a complex entity with a large difference in electrophoretic mobilities relative to the free aptamer. A large injection volume may produce two or three plateaus, the bound aptamer, the free aptamer and the overlap of the two. Therefore, the ratio r can be determined by the two plateau heights of the free (Hf) and the bound (Hb) aptamers with Equation 5.
| (5) |
The complex formation does not affect the fluorescence emission [24]. However, the fluorescence intensity is dependent upon the apparent velocity of dyes which flow through the laser beam detection window. This effect is discussed in the following section.
3.2 Fluorescence intensity dependence on dye migration velocity
In experiments, we noticed that the magnitude of the fluorescence signal depended on the electric field strength which produced different migration velocities for fluorescently-labeled aptamer and the complex. To further study this effect, a detailed experiment was designed to examine the fluorescence intensity as a function of the applied voltage between the SR and BW (Grounded) with the BR and SW floated. A dye-containing sample (IgE aptamer 500 nM) plug was pumped (SR 6.0 kV) through the detection window. The voltage on SR was then dropped immediately to 0.0 kV increased from there in 1 kV intervals up to 6.0 kV and then decreased from 6.0 kV to 0.0 kV in 1 kV intervals, with each interval being 10 s in length. The signal magnitude changed with the changes of the applied voltage as shown in Figure 2. As can be seen, at the SR voltage of 0, the fluorescence signal rapidly decreased and continued to exponentially decrease until 1.0 kV on the SR was turned on. With each increase in voltage from 0 to 6.0 kV the fluorescence magnitude increased. Similarly, the signal decreased with successive drops in applied voltage from 6 to 0 kV.
Figure 2.

Migration velocity dependence of fluorescence emission. Sample solution (500 nM IgE aptamer) was first pumped through the detection window at a voltage of 6.0 kV (SR), and then voltage on the SR was varied from 0.0 kV to 6.0 kV and from 6.0 kV to 0.0 kV in intervals of 1.0 kV for a duration of 10 s each. The last signal was produced by dispensing step (Figure 1c). The numbers on the plateaus are the voltages applied on the SR with the unit of kV.
This voltage dependence of fluorescence emission is attributed to the photobleaching effect of dyes when exposed to high-power laser irradiation [27]. Photobleaching is a phenomenon that occurs when a fluorophore loses the ability to fluoresce due to photon-induced chemical degradation.[28] The average number of excitation and emission cycles that occur for a particular fluorophore before photobleaching is dependent upon the molecular structure and the local environment. Some fluorophores bleach quickly after emitting only a few photons, while others can undergo thousands or millions of cycles before being bleached. Photobleaching of dyes is irradiating-time dependent, the longer the irradiation on the dyes, the more significant the bleaching [28]. In LIF detection for CE, the excitation laser beam has a specific size which can be adjusted by tuning the focusing on the channel; and the dyes flow through the detection window at their individual apparent velocities, which produce a transmission time for a specific dye molecule. When the bulk dye in a plug continuously flows through the laser beam, some of the dye may be photobleached depending on the robustness and the residence time of the dye in the laser beam. Therefore, the bulk dye exposed in the laser beam may form a concentration gradient with the highest concentration at the incoming boundary of dye and laser beam and lowest at the other outgoing side. The detected fluorescence emission is produced by the dye gradient in the laser beam, and the total emission intensity will be lower than without photobleaching. The sharp rise and fall of the fluorescence signal after voltage switching as shown in Figure 2 may be due to the rapid photobleaching of the fluorescently labeled aptamer relative to the slow data collecting rate (20 Hz). The apparent velocity of dyes can be controlled by varying the electric field strength produced by applied voltages. Therefore, this photobleaching effect in FA experiments can be adjusted by appropriately setting individual separation voltage configurations for each component so as to provide the same migration velocities for each component.
3.3 Injection volume effect on plateaus
The injection volume was controlled by varying the injection time at a constant electric field. During sample injection, a voltage of 5800 V was applied between SR and BW, while BR and SW were floated. This voltage configuration produced an electric field strength of 682 V/cm for electrokinetic sample injection. After injection, the dispensing step was immediately switched on and the large sample plug was pumped through the detection window. The electric field across the separation channel was adjusted to be the same as that during sample injection (682 V/cm). Figure 3 shows the injection volume effect on the shape of plateaus in terms of injection time. As can be seen, when the injection volume was small, free aptamer and IgE-aptamer complex were separated completely, but no plateaus were formed. With the increase in the injection volume, the two peaks became broadened and leveled off, and finally three plateaus were formed, corresponding to the complex (Plateau 1), the overlap of the complex and the free aptamer (Plateau 3) and free aptamer (Plateau 2) when 10-s injection was carried out. It was also noted that two small decreasing plateaus appeared at the right side of plateau 3. The two plateaus may be attributed to the fluorescently-labeled impurities in the IgE aptamer or the resulting complex the impurities formed with IgE. The complex front reached the center of the detection beam at 3.25 s, and free aptamer arrived at 7.15 s. Therefore, the apparent velocities of IgE-aptamer complex and the free aptamer were 0.31 and 0.14 cm/s (detection length 1.0 cm), respectively, under the running conditions.
Figure 3.

Peak shapes produced at different injection volumes. Injection times were 0.5, 1.5, 3 and 10 s as shown in the figure. The incubated sample consisted of IgE and aptamer at 20 and 40 nM, respectively. Signals 1, 2 and 3 are bound aptamer, free aptamer and the overlap of the free and bound aptamers, respectively.
3.4 Determination of Kd
According to the discussions above, the voltage configuration was designed as shown in Figure 1. The gated injection method was used [25]. The loading step (Figure 1a) pumped the sample solution through the cross section (5 s). During the injection step (Figure 1b), the BR and SW were floated, and the sample solution was pumped into the separation channel with a time duration of 3 s. Two dispensing processes (Figure 1c and Figure 1d) were carried out alternately following sample injection. The voltage configuration in Figure 1c produced an electric field strength of 683 V/cm (E1), which was used to obtain the free aptamer plateau; and the voltage setup in Figure 1d produced 311 V/cm (E2), which was to get the bound aptamer plateau. This experimental design came from the fact that the migration time ratio of the free and bound aptamers was 2.2. Therefore, the electric field strength ratio of E1 and E2 should also be 2.2 to adjust the velocity effect of fluorescence emission of the free aptamer. Figure 4 shows the plateau results obtained at the two dispensing voltage configurations. As can be seen, the pair of the free and bound aptamers obtained at the higher electric field had a larger fluorescence response than the other pair at the lower electric field strength, respectively. The plateau ratio r of the free and bound aptamers was obtained by Equation 5, where Hf was the free aptamer plateau height obtained at E1, and Hb was the bound aptamer plateau obtained at E2. The plateau height was the average of 3 injections. Table 1 shows the Kd values determined at different aptamer concentrations in the incubation buffer while the IgE concentration was kept constant (20.0 nM). As can be seen, the obtained Kd is independent of the aptamer concentrations used, and the average value is 6 ± 2 nM, which is consistent with the reported results of 8 nM [29] and 9 nM [30].
Figure 4.
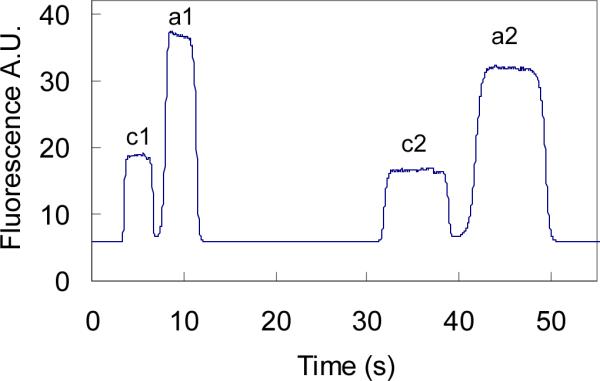
Two sets of plateaus obtained at two voltage configurations. The sample solution consisted of 20 nM IgE and 70 nM aptamer. The set of c1 and a1 was obtained with the voltage setup in Figure 1c. The other set of c2 and a2 was obtained with the voltage in Figure 1d. Plateaus c (c1 and c2) and a (a1 and a2) are the bound and free aptamers, respectively.
Table 1.
Determined Kda) at different aptamer concentrations
| Aptamer (nM) | 20 | 30 | 40 | 50 | 60 | 70 | ave |
|---|---|---|---|---|---|---|---|
| r = Hf/Hb | 0.53 | 0.93 | 1.67 | 2.02 | 2.38 | 2.83 | - |
| Kd (nM) | 3.7 | 4.1 | 8.4 | 6.9 | 5.3 | 4.8 | 6± 2 |
Calculated with Equation 4.
4 Concluding remarks
Microchip CE is a powerful tool for binding studies of protein-DNA systems. The short detection length (1.0 cm) used here reduced the analysis time and might reduce protein loss due to adsorption on the channel walls during separation. FA is an accurate and dependable method to determine the dissociation constants of some slow-off binding systems. LIF detection used in FA is advantageous for high sensitivity and no internal standard requirement.
Although the migration velocity dependence of fluorescence emission initially caused some trouble in experiments, this effect was conveniently adjusted by setting up a compensating voltage configuration according to the relative migration velocities of the free and bound aptamers. To reduce possible error due to fluctuations of fluorescence emission with time, multiple injections and separations can be alternatively carried out for both voltage configurations. One surely can use an internal standard for the binding experiments and the blank aptamer (without IgE added) to determine the bound aptamer ratio, but the right internal standard often remains hard to choose for FA.
In FA, care must be taken to insure the separation buffer and the sample buffer have the same conductivities to avoid the sample stacking effect. The same sample and separation buffers are usually used, but their conductivities may still have a little difference when the sample solution contains some proteins and aptamers which probably increase the viscosity of the sample solution and decrease its conductivity. In addition, stacking may occur due to ionic transport number mismatch at the boundary of the sample plug and the separation buffer [31]. The two effects may be negligible when low concentrations of proteins and aptamers are used.
The fluorescence linearity with concentration may be another concern. Therefore, the free and bound aptamers should produce plateaus with similar heights, which can be controlled by choosing the approximate original concentrations of proteins and aptamers.
Finally, it should be noted that the method developed here may only be useful for slow-off binding systems with Kd < 10 nM.
Acknowledgements
We appreciate Professor Carl J. Seliskar and Aigars Piruska (Department of Chemistry, University of Cincinnati) for a discussion of photobleaching phenomenon. This project was supported by NIH with grant number GM 69547.
Abbreviations
- CE
capillary electrophoresis
- FA
frontal analysis
- LIF
laser-induced fluorescence
- TGK
Tris/Glycine/K2HPO4
- BR
buffer reservoir
- SR
sample reservoir
- BW
buffer waste reservoir
- SW
sample waste reservoir
References
- [1].Dolnik V, Liu S. J. Sep. Sci. 2005;28:1994–2009. doi: 10.1002/jssc.200500243. [DOI] [PubMed] [Google Scholar]
- [2].Lacher NA, Garrison KE, Martin RS, Lunte SM. Electrophoresis. 2001;22:2526–2536. doi: 10.1002/1522-2683(200107)22:12<2526::AID-ELPS2526>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- [3].Dolník V, Liu S, Jovanovich S. Electrophoresis. 2000;21:41–54. doi: 10.1002/(SICI)1522-2683(20000101)21:1<41::AID-ELPS41>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- [4].Gong M, Wehmeyer KR, Limbach PA, Arias F, Heineman WR. Anal. Chem. 2006;78:3730–3737. doi: 10.1021/ac0521798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Currie CA, Heineman WR, Halsall HB, Seliskar CJ, et al. J. Chromatogr. B. 2005;824:201–205. doi: 10.1016/j.jchromb.2005.07.035. [DOI] [PubMed] [Google Scholar]
- [6].Koutny LB, Schmalzing D, Taylor TA, Fuchs M. Anal. Chem. 1996;68:18–22. doi: 10.1021/ac9508311. [DOI] [PubMed] [Google Scholar]
- [7].Tu J, Halsall HB, Seliskar CJ, Limbach PA, et al. J. Pharmaceut. Biomed. 2005;38:1–7. doi: 10.1016/j.jpba.2004.11.035. [DOI] [PubMed] [Google Scholar]
- [8].Gao Q, Shi Y, Liu S. Fresen. J. Anal. Chem. 2001;371:137–145. doi: 10.1007/s002160100957. [DOI] [PubMed] [Google Scholar]
- [9].Shi Y, Simpson PC, Scherer JR, Wexler D, et al. Anal. Chem. 1999;71:5354–5361. doi: 10.1021/ac990518p. [DOI] [PubMed] [Google Scholar]
- [10].Woolley AT, Sensabaugh GF, Mathies RA. Anal. Chem. 1997;69:2181–2186. doi: 10.1021/ac961237+. [DOI] [PubMed] [Google Scholar]
- [11].Carrilho E. Electrophoresis. 2000;21:55–65. doi: 10.1002/(SICI)1522-2683(20000101)21:1<55::AID-ELPS55>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- [12].Emrich CA, Tian H, Medintz IL, Mathies RA. Anal. Chem. 2002;74:5076–5083. doi: 10.1021/ac020236g. [DOI] [PubMed] [Google Scholar]
- [13].Østergaard J, Heegaard NHH. Electrophoresis. 2003;24:2903–2913. doi: 10.1002/elps.200305526. [DOI] [PubMed] [Google Scholar]
- [14].Rundlett KL, Armstrong DW. Electrophoresis. 2001;22:1419–1427. doi: 10.1002/1522-2683(200105)22:7<1419::AID-ELPS1419>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- [15].Oravcova J, Bohs B, Lindner W. J. Chromatogr. B. 1996;677:1–28. doi: 10.1016/0378-4347(95)00425-4. [DOI] [PubMed] [Google Scholar]
- [16].Busch MHA, Kraak JC, Poppe H. J. Chromatogr. A. 1997;777:329–353. doi: 10.1016/s0021-9673(97)00369-5. [DOI] [PubMed] [Google Scholar]
- [17].Sebille B, Zini R, Madjar C-V, Thuaud N, Tillement J-P. J. Chromatogr. B. 1990;531:51–77. doi: 10.1016/s0378-4347(00)82280-x. [DOI] [PubMed] [Google Scholar]
- [18].Shimura K, Kasai K.-i. Anal. Biochem. 1997;251:1–16. doi: 10.1006/abio.1997.2212. [DOI] [PubMed] [Google Scholar]
- [19].Xu H, Yu X-D, Li X-D, Chen H-Y. Chromatographia. 2005;61:419–422. [Google Scholar]
- [20].Nilsson M, Johansson G, Isaksson R. Electrophoresis. 2004;25:1022–1027. doi: 10.1002/elps.200305786. [DOI] [PubMed] [Google Scholar]
- [21].Busch MHA, Boelens HFM, Kraak JC, Poppe H. J. Chromatogr. A. 1997;775:313–326. [Google Scholar]
- [22].Rudnev AV, Aleksenko SS, Semenova O, Hartinger CG, et al. J. Sep. Sci. 2005;28:121–127. doi: 10.1002/jssc.200401930. [DOI] [PubMed] [Google Scholar]
- [23].Kraak JC, Busch S, Poppe H. J. Chromatogr. A. 1992;608:257–263. doi: 10.1016/0021-9673(92)87132-r. [DOI] [PubMed] [Google Scholar]
- [24].Krylov SN, Berezovski M. Analyst. 2003;128:571–575. doi: 10.1039/b212913b. [DOI] [PubMed] [Google Scholar]
- [25].Jacobson SC, Koutny LB, Hergenroeder R, Moore AW, Jr., Ramsey JM. Anal. Chem. 1994;66:3472–3476. [Google Scholar]
- [26].Huang CC, Cao Z, Chang HT, Tan W. Anal. Chem. 2004;76:6973–6981. doi: 10.1021/ac049158i. [DOI] [PubMed] [Google Scholar]
- [27].Wang GR. Lab Chip. 2005;5:450–456. doi: 10.1039/b416209a. [DOI] [PubMed] [Google Scholar]
- [28].Wang GR, Fiedler HE. Exp. Fluids. 2000;29:257–264. [Google Scholar]
- [29].German I, Buchanan DD, Kennedy RT. Anal. Chem. 1998;70:4540–4545. doi: 10.1021/ac980638h. [DOI] [PubMed] [Google Scholar]
- [30].Wiegand TW, Williams PB, Dreskin SC, Jouvin MH, et al. J. Immunol. 1996;157:221–230. [PubMed] [Google Scholar]
- [31].Liu Y, Foote RS, Jacobson SC, Ramsey JM. Lab Chip. 2005;5:457–465. doi: 10.1039/b416414h. [DOI] [PubMed] [Google Scholar]