

Abstract
Lymphocyte development occurs in a stepwise progression through distinct developmental stages. This ordered maturation ensures that cells express a single, non-autoreactive antigen receptor, which is the cornerstone of a diverse adaptive immune response. Expression of a mature antigen receptor requires assembly of the antigen receptor genes by the process of V(D)J recombination, a reaction that joins distant gene segments through DNA double-strand break (DSB) intermediates. These physiologic DSBs are generated by the recombinase-activating gene (RAG) -1 and -2 proteins, and their generation is regulated by lymphocyte and developmental stage-specific signals from cytokine receptors and antigen receptor chains. Collectively, these signals ensure that V(D)J recombination of specific antigen receptor genes occurs at discrete developmental stages. Once generated, RAG-induced DSBs activate the ataxia-telangiectasia mutated (ATM) kinase to orchestrate a multifaceted DNA damage response that ensures proper DSB repair. In response to RAG DSBs, ATM also regulates a cell type-specific transcriptional response, and here we discuss how this genetic program integrates with other cellular cues to regulate lymphocyte development.
Keywords: NFκB, Pim2, RAG, V(D)J recombination, development, interleukin-7, lymphocyte, pre-BCR, survival
Introduction
Lymphocytes proceed through discrete stages as they develop into mature B and T cells. Lymphocyte-specific, stage-specific and lineage-specific signals direct differentiation to ensure that each cell uniquely expresses the appropriate antigen receptor genes at the correct developmental stage. These signals culminate in the assembly of functional antigen receptor genes and expression of a non-autoreactive antigen receptor. The second exon of all antigen receptor genes must be assembled from variable (V), joining (J) and, at some loci, diversity (D) gene segments.1 These distinct gene segments are joined through the generation and repair of DNA breaks. This process of V(D)J recombination is initiated by the recombinase-activating genes, RAG1 and RAG2, which encode proteins that, together, form the RAG endonuclease.2-4 RAG binds to recombination signals, specific DNA sequences, at the border of the V, D and J segments and generates DNA double-strand breaks (DSBs).2-4 These RAG DSBs are processed and joined by the non-homologous end-joining (NHEJ) pathway of DNA DSB repair.5-7
In developing B lymphocytes, the heavy chain (IgH) gene is rearranged first at the pro-B cell stage and is expressed in conjunction with the surrogate light chains (VpreB and λ5) to form the pre-BCR.8 Expression of the pre-BCR signals transition to the pre-B cell developmental stage.8 Pre-B cells subsequently assemble a functional light chain (IgL) gene at either the kappa (IgLκ) or lambda (IgLλ) locus.9 The ordered rearrangement of IgH and IgL genes is regulated by diverse cellular signals.
In pre-B cells, the interleukin-7 receptor (IL-7r) and the pre-BCR are the principal regulators of cell proliferation, survival and maturation.8,10,11 These signals cooperate to regulate accessibility of the antigen receptor genes, RAG expression and transcriptional programs necessary for cell type-specific differentiation.8,10 In addition to these cell surface receptors, recent studies have shown that signals from RAG DNA breaks activate genetic programs that have important consequences for developing lymphocytes. Here we review how these DSB-dependent signals are integrated with other signals that drive lymphocyte development and differentiation.
RAG DSBs Activate Canonical DNA Damage Responses
RAG DSBs are generated in the G1 phase of the cell cycle as RAG2 is phosphorylated and degraded upon entry into S-phase.12 Similar to other DSBs generated in G1, RAG DSBs trigger canonical DNA damage responses through activation of ATM and DNA-PKcs, which are both members of the phosphatidylinositol-3-kinase (PI3K)-like family of serine-threonine kinases.5-7 Both ATM and DNA-PKcs function as transducers in the DNA damage response to phosphorylate numerous downstream effectors.13-16
ATM and DNA-PKcs have unique and redundant roles in response to RAG-mediated DSBs.5,17,18 Deficiency of either of these kinases results in errors in repair of the broken DNA ends and immune deficiencies in mice and humans.5,7,19-23 Following induction of RAG DSBs, ATM is responsible for promoting the stability of the broken DNA (coding) ends in a post-cleavage complex until they are joined.24 DNA-PKcs promotes the activity of the Artemis endonuclease, which is required to open hairpin-sealed ends generated by the RAG endonuclease.15,25,26 These kinases function in conjunction with other NHEJ proteins to ensure proper repair of RAG DSBs.5-7 Moreover, ATM phosphorylates the histone variant H2AX (γ-H2AX) for several kilobases surrounding DNA breaks, including RAG DSBs.27-29 Phosphorylated H2AX recruits proteins to the site of the DNA break, amplifying DNA damage responses and supporting normal DSB repair, including preventing the resection of RAG-induced DSBs.28,30
RAG DSBs Activate a Unique Transcriptional Program
In addition to signals important in DSB repair, RAG DSBs also activate programs that regulate broader cellular responses. Similar to genotoxic DSBs, in response to RAG DSBs, ATM phosphorylates the transcription factor p53, resulting in its stabilization and increased transcriptional activity.31-35 Once activated, p53 regulates the expression of numerous cell cycle and cell death genes, including p21, PUMA, NOXA and BAX.33,36 Deficiency of p53 results in a higher incidence of lymphoid malignancies with chromosomal translocations, consistent with an important role for this tumor suppressor in eliminating cells at risk for aberrant DNA repair and malignant transformation.33,37-41 However, mechanisms must exist to ensure that p53 does not induce the death of all cells attempting to assemble antigen receptor genes.
Indeed, RAG DSBs trigger NFκB-dependent transcription, which regulates expression of a genetic program that includes survival genes.31,42 The dual regulation of survival and cell death pathways by RAG DSBs is temporally balanced, allowing time for proper assembly of the antigen receptor genes, but ensuring elimination of cells with persistent DSBs at risk for errant repair. These opposing processes may enforce normal development and suppress malignant transformation. Moreover, RAG DSBs initiate expression of lymphocyte-specific cellular programs that integrate with other developmental cues in pre-B cells, including those initiated by the IL-7r and pre-BCR, and support lymphocyte differentiation and maturation.42
IL-7r Signaling in Pre-B Cells
IL-7 signaling is critical for early lymphocyte development, as evidenced by the block in B and T cell maturation at early stages in mice deficient in IL-7 or the IL-7r.10,43,44 In pre-B cells, the IL-7r signals through both the JAK-STAT and the PI3K-Akt pathways to support cell survival and proliferation while simultaneously suppressing IgL gene accessibility and RAG expression.10 In this manner, IL-7 induces clonal expansion of pre-B cells and prevents generation of RAG DSBs at antigen receptor loci in actively dividing cells.
Activation of the IL-7r in pre-B cells leads to phosphorylation and dimerization of STAT5, which results in nuclear translocation and initiation of gene expression.10,45,46 STAT5 mediates the upregulation of anti-apoptotic and proliferation genes, including Bcl2, Bcl-XL, Mcl-1 and Pim-1, CCND2 (cyclin-D2) and CCND3 (cyclin-D3).10,45,46 Additionally, STAT5 coordinates increased accessibility of the IgH locus and simultaneous inactivation of the IgL locus, thereby preventing light chain recombination.46-48
The IL-7r also phosphorylates and activates the Akt (PKB) kinase, which cooperates with STAT5 signaling to support pre-B cell survival at least in part through phosphorylation and inactivation of the pro-apoptotic protein BAD.10,31,49 Akt also phosphorylates the FoxO1 and FoxO3a transcription factors resulting in their cytoplasmic retention and inactivation.50-53 Attenuation of IL-7 signals disables Akt and stabilizes the FoxO transcription factors, resulting in expression of RAG and activation of IgL gene rearrangement.50-53 As such, loss of IL-7 signals is necessary for IgL chain gene assembly, but the concomitant loss of survival signals would be detrimental to pre-B cells.
Pre-BCR Signaling
Ligand-independent oligomerization of the pre-BCR promotes pathways that participate in the regulation of cellular proliferation, allelic exclusion of the IgH chain gene and activation of IgLκ chain gene rearrangement.8,54-56 The pre-BCR signals through the Igα and Igβ subunits to activate multiple downstream pathways.8,54 Activation of the RAS kinase triggers a kinase cascade that includes MEK to ERK activation, which leads to expression of the Aiolos transcriptional repressor and the E2A transcription factor.48 Aiolos inhibits expression of CCND3 (cyclin-D3) to support cell cycle arrest, while E2A activates transcription of the IgLκ chain genes.48 Additionally, pre-BCR signals support PI3K-Akt signaling, thus, augmenting IL-7r signaling through this pathway to suppress FoxO transcriptional activity and inhibit RAG expression.50,51 The pre-BCR complex also activates SLP-65 (BLNK), which further augments Aiolos expression and upregulates expression of IRF4, another important regulator of IgLκ transcription.57,58 Pre-BCR signals have also been demonstrated to promote proliferation in conditions of low IL-7 concentrations and to upregulate expression of the chemokine receptor CXCR4, which promotes migration of pre-B cells to distinct bone marrow compartments in response to its ligand CXCL12.8,58
Integration of IL-7 Receptor and Pre-BCR Signals
The IL-7r and the pre-BCR combine to provide signals necessary for ongoing B cell maturation. The integration of these signaling pathways controls pre-B cell proliferation, survival and IgL chain gene recombination.8,10,11 IL-7r signals promote cell proliferation while simultaneously suppressing RAG expression and IgLκ chain gene accessibility.10,53,58 These signals must be attenuated for continued differentiation. Pre-BCR signaling may, in part, regulate IL-7 signaling through downregulation of IL-7r expression.59,60 Moreover, pre-BCR signals upregulate CXCR4 expression, which can promote cell migration in response to CXCL12 to bone marrow microenvironments that are devoid of IL-7-producing stromal cells.58 As IL-7 diminishes, PI3K-Akt signaling ceases, resulting in activation of FoxO transcription factors and expression of the RAG machinery.53 Additionally, loss of IL-7 signals triggers increased expression of SLP-65 (BLNK) and Syk, two critical intracellular signaling components of the pre-BCR, which, in turn, activate Aiolos and IRF4 expression to suppress proliferation and activate IgLκ transcription, respectively.53,57,58 In this manner, the IL-7r and the pre-BCR cooperate to control RAG expression, IgLκ accessibility and B cell maturation.
Regulation of Lymphocyte Developmental Processes by RAG DSB Signaling
In addition to the extrinsic signals from the IL-7r and pre-BCR, pre-B cells are also influenced by intrinsic signals triggered by the DSBs generated during IgL chain gene rearrangement. Initiation of IgL chain gene assembly requires the attenuation of IL-7 signaling, which results in concomitant loss of Pim1 and Akt survival signals with rapid induction of cell death mechanisms.10,31,53,61,62 However, shortly after their generation during IgL chain gene assembly, RAG DSBs upregulate expression of Pim2, a member of a family of pro-survival kinases that includes Pim1 and Akt.31,42,63 Upregulation of Pim2 provides a cytokine-independent survival signal through maintenance of the phosphorylation and inactivation of BAD, which promotes increased levels of Bcl2.31,36 This early survival signal counters p53-associated cell death mechanisms, allowing time for proper repair of RAG DSBs and assembly of a functional IgL chain gene.
Pre-B cells frequently must undergo several successive IgL chain gene re-arrangements, each of which can take several hours to complete. Thus, pre-B cells need to survive for an extended period of time as they attempt to assemble and express a functional IgL chain gene.64 Indeed, Pim2-deficient mice have fewer IgLλ-expressing B cells, consistent with reduced pre-B cell survival and time to complete IgL chain gene rearrangement.31,65 The pro-survival functions of Pim2 replace lost IL-7 survival signals to counter cell death mechanisms and provide optimal opportunity for completion of IgL chain gene assembly. Importantly, if DSBs cannot be repaired, cell death pathways overcome the early survival signals to eliminate cells at risk of aberrant DSB repair, such as translocations. Thus, expression of Pim2 by RAG DSBs results in a temporal ordering of early survival followed by late cell death mechanisms, which promotes normal lymphocyte development while suppressing malignant transformation.
In striking contrast to the pro-proliferative properties of Pim1 and Akt downstream of IL-7, expression of Pim2 by RAG DSBs inhibits IL-7-driven proliferation.31,66,67 The mechanisms underlying the anti-proliferative properties of Pim2 are unclear. Notably, though, Pim2 is not sufficient to block proliferation but functions in conjunction with other DSB-dependent checkpoint activators to maintain cell cycle arrest.31 This unique function of Pim2 in cells undergoing V(D)J recombination may be important to help enforce the G1 to S checkpoint in the presence of low levels of IL-7. As such, the anti-proliferative function of Pim2 downstream of RAG DSBs may integrate with pre-BCR signals to inhibit IL-7r signaling and maintain cell cycle arrest. In this regard, DNA damage responses and antigen receptor signals cooperate to prevent pre-B cells with RAG DSBs from entering S-phase, where these DNA breaks could be repaired as translocations or as replicated broken chromosomes.
In pre-B cells RAG DSBs also induce expression of genes with known functions in lymphocyte migration and homing such as CD69, L-selection (CD62L) and SWAP-70.42,68-72 SWAP-70 and L-selectin regulate integrin-mediated adhesion and chemokine responses, respectively, to control lymphocyte homing to secondary lymphoid organs.69,70,73,74 CD69 mediates downregulation of the S1P1 receptor, which binds to sphingosine-1-phosphate (S1P) and promotes egress of lymphocytes out of the thymus and other secondary lymphoid organs.71,72 The S1P1 receptor is also required to promote the migration of newly generated B cells from the bone marrow.75,76 Expression of CD69 blocks S1P1 receptor expression in immature B cells and prevents their migration into the periphery.75 Thus, the induction of CD69 by RAG DSBs may prevent pre-B cells from exiting the bone marrow prior to completing IgL chain gene assembly. Moreover, expression of CD69, SWAP-70 and L-selectin by RAG DSBs may function in conjunction with signals from the pre-BCR, which promote CXCR4 expression and migration away from IL-7-producing stromal cells.58 In this regard, the combination of antigen receptor and DSB-dependent signals could promote the residence of pre-B cells in bone marrow compartments devoid of IL-7 while they complete assembly of the IgL chain genes and expression of a mature BCR.
Interestingly, regulation of developmental programs by physiologic DSBs is also observed during later stages of B cell development in the spleen. The DNA DSBs generated during class switch recombination in mature B cells promote a transcriptional program that controls differentiation of activated B cells into plasma cells.77,78 Thus, signals from physiologic DSBs regulate cell type-specific genetic programs that function at various stages of lymphocyte development to promote normal immune cell maturation.
Concluding Remarks
All lymphocytes generate DSBs as they assemble functional antigen receptor genes at different developmental stages. The specific antigen receptor genes that are rearranged and the timing of the DSBs generation are regulated by surface receptor signals, such as those from the IL-7r and the pre-BCR in developing B cells. These external cellular cues trigger pathways that control (1) accessibility of antigen receptor genes to transcriptional machinery, (2) RAG recombinase expression and (3) cell proliferation and survival. In large, pre-B cells, IL-7r and pre-BCR signals cooperate to promote survival, proliferation and clonal expansion while suppressing RAG expression and IgL chain gene accessibility (Fig. 1). To continue maturation and transition to the small pre-B cell stage, IL-7r signaling must be attenuated, which occurs, at least in part, through migration away from IL-7-producing stromal cells. Loss of IL-7 signals results in expression of RAG, IgL germline transcription and modification of pre-BCR signaling. Once generated, the RAG DSBs themselves trigger a DNA damage response that activates a unique lymphocyte-specific program, which intersects with IL-7r and antigen receptor signals to direct lymphocyte differentiation. In this regard, RAG DSBs and the pre-BCR regulate pathways to suppress the proliferative response to IL-7 and likely promote pre-B cell residence in bone marrow niches devoid of IL-7 (Fig. 1). Together, these mechanisms maintain G1 arrest in cells undergoing IgL recombination. Simultaneously, RAG DSBs activate survival mechanisms to replace loss IL-7-driven survival signals, permitting time for repair of the broken DNA ends and expression of a functional IgL chain gene. The integration of these DNA damage responses with extrinsic development cues creates a coordinated signaling network to manage DNA breaks, enforce normal lymphocyte development and suppress malignant transformation.
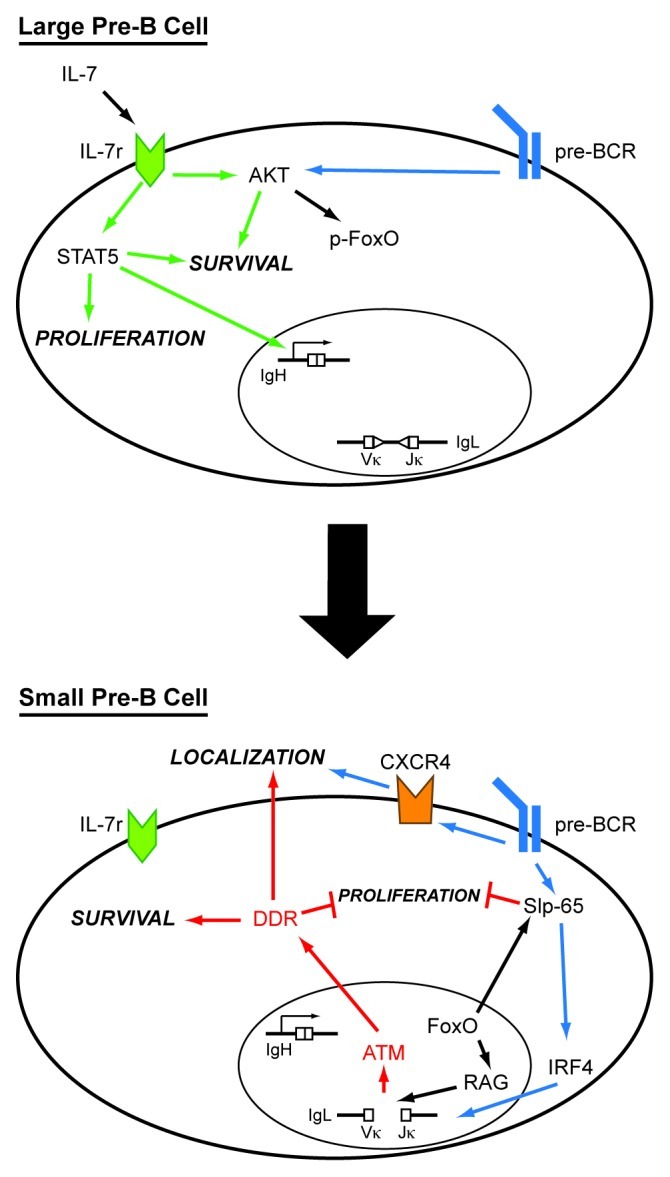
Figure 1. Integration of surface receptor signals and DNA damage responses in pre-B cells. In developing pre-B cells, signals from the IL-7r, pre-BCR and RAG DSBs coordinate to direct continued maturation. In large pre-B cells, IL-7r and pre-BCR signals support proliferation and survival to optimize clonal expansion of IgH-expressing cells. This proliferation phase occurs in bone marrow niches rich in IL-7-producing cells. IL-7r signals through STAT5 and Akt to maintain proliferation and survival. Additionally, STAT5 supports IgH chain gene transcription and suppresses IgL chain gene accessibility while Akt inhibits RAG expression. Pre-BCR signals support Akt signaling during this expansion phase. Attenuation of IL-7r signaling, which may be mediated by migration of pre-B cells to bone marrow niches devoid of IL-7-producing cells, results in transition to the small pre-B cell developmental stage. Loss of IL-7 results in termination of STAT5 signals and associated cell cycle arrest. The cessation of Akt activity results in stabilization of FoxO transcription factors and expression of RAG and SLP-65. SLP-65 redirects pre-BCR signaling to expression of IRF4 and transcription of IgL chain genes, which permits IgL locus accessibility to RAG. RAG DSBs trigger ATM-dependent DNA damage responses (DDR), which intersect with pre-BCR signals to promote pre-B cell survival, suppress IL-7-driven proliferation and trigger expression of proteins that support cellular re-localization.
Acknowledgments
This work was supported by the National Institutes of Health grants CA136470, AI074953 and AI47829 to B.P.S. J.J.B. was supported by a Hyundai Hope on Wheels Scholar Award.
Glossary
Abbreviations:
- RAG
recombinase-activating gene complex
- DSB
double-stranded DNA break
- NHEJ
non-homologous end joining
- BCR
B cell receptor
- IL-7r
interleukin-7 receptor
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/22021
References
- 1.Tonegawa S. Somatic generation of antibody diversity. Nature. 1983;302:575–81. doi: 10.1038/302575a0. [DOI] [PubMed] [Google Scholar]
- 2.Fugmann SD, Lee AI, Shockett PE, Villey IJ, Schatz DG. The RAG proteins and V(D)J recombination: complexes, ends, and transposition. Annu Rev Immunol. 2000;18:495–527. doi: 10.1146/annurev.immunol.18.1.495. [DOI] [PubMed] [Google Scholar]
- 3.Gellert MV. V(D)J recombination: RAG proteins, repair factors, and regulation. Annu Rev Biochem. 2002;71:101–32. doi: 10.1146/annurev.biochem.71.090501.150203. [DOI] [PubMed] [Google Scholar]
- 4.Oettinger MA. V(D)J recombination: on the cutting edge. Curr Opin Cell Biol. 1999;11:325–9. doi: 10.1016/S0955-0674(99)80044-1. [DOI] [PubMed] [Google Scholar]
- 5.Helmink BA, Sleckman BP. The response to and repair of RAG-mediated DNA double-strand breaks. Annu Rev Immunol. 2012;30:175–202. doi: 10.1146/annurev-immunol-030409-101320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rooney S, Chaudhuri J, Alt FW. The role of the non-homologous end-joining pathway in lymphocyte development. Immunol Rev. 2004;200:115–31. doi: 10.1111/j.0105-2896.2004.00165.x. [DOI] [PubMed] [Google Scholar]
- 8.Herzog S, Reth M, Jumaa H. Regulation of B-cell proliferation and differentiation by pre-B-cell receptor signalling. Nat Rev Immunol. 2009;9:195–205. doi: 10.1038/nri2491. [DOI] [PubMed] [Google Scholar]
- 9.Nemazee D, Kouskoff V, Hertz M, Lang J, Melamed D, Pape K, et al. B-cell-receptor-dependent positive and negative selection in immature B cells. Curr Top Microbiol Immunol. 2000;245:57–71. doi: 10.1007/978-3-642-59641-4_3. [DOI] [PubMed] [Google Scholar]
- 10.Corfe SA, Paige CJ. The many roles of IL-7 in B cell development; mediator of survival, proliferation and differentiation. Semin Immunol. 2012;24:198–208. doi: 10.1016/j.smim.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 11.Erlandsson L, Licence S, Gaspal F, Lane P, Corcoran AE, Mårtensson IL. Both the pre-BCR and the IL-7Ralpha are essential for expansion at the pre-BII cell stage in vivo. Eur J Immunol. 2005;35:1969–76. doi: 10.1002/eji.200425821. [DOI] [PubMed] [Google Scholar]
- 12.Desiderio S, Lin WC, Li Z. The cell cycle and V(D)J recombination. Curr Top Microbiol Immunol. 1996;217:45–59. doi: 10.1007/978-3-642-50140-1_4. [DOI] [PubMed] [Google Scholar]
- 13.Durocher D, Jackson SP. DNA-PK, ATM and ATR as sensors of DNA damage: variations on a theme? Curr Opin Cell Biol. 2001;13:225–31. doi: 10.1016/S0955-0674(00)00201-5. [DOI] [PubMed] [Google Scholar]
- 14.Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3:155–68. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 15.Smith GC, Jackson SP. The DNA-dependent protein kinase. Genes Dev. 1999;13:916–34. doi: 10.1101/gad.13.8.916. [DOI] [PubMed] [Google Scholar]
- 16.Hill R, Lee PW. The DNA-dependent protein kinase (DNA-PK): More than just a case of making ends meet? Cell Cycle. 2010;9:3460–9. doi: 10.4161/cc.9.17.13043. [DOI] [PubMed] [Google Scholar]
- 17.Gapud EJ, Sleckman BP. Unique and redundant functions of ATM and DNA-PKcs during V(D)J recombination. Cell Cycle. 2011;10:1928–35. doi: 10.4161/cc.10.12.16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gapud EJ, Dorsett Y, Yin B, Callen E, Bredemeyer A, Mahowald GK, et al. Ataxia telangiectasia mutated (Atm) and DNA-PKcs kinases have overlapping activities during chromosomal signal joint formation. Proc Natl Acad Sci USA. 2011;108:2022–7. doi: 10.1073/pnas.1013295108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, et al. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1996;86:159–71. doi: 10.1016/S0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 20.Elson A, Wang Y, Daugherty CJ, Morton CC, Zhou F, Campos-Torres J, et al. Pleiotropic defects in ataxia-telangiectasia protein-deficient mice. Proc Natl Acad Sci USA. 1996;93:13084–9. doi: 10.1073/pnas.93.23.13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lavin MF, Shiloh Y. The genetic defect in ataxia-telangiectasia. Annu Rev Immunol. 1997;15:177–202. doi: 10.1146/annurev.immunol.15.1.177. [DOI] [PubMed] [Google Scholar]
- 22.van der Burg M, Ijspeert H, Verkaik NS, Turul T, Wiegant WW, Morotomi-Yano K, et al. A DNA-PKcs mutation in a radiosensitive T-B- SCID patient inhibits Artemis activation and nonhomologous end-joining. J Clin Invest. 2009;119:91–8. doi: 10.1172/JCI37141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, Baltimore D. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev. 1996;10:2411–22. doi: 10.1101/gad.10.19.2411. [DOI] [PubMed] [Google Scholar]
- 24.Bredemeyer AL, Sharma GG, Huang CY, Helmink BA, Walker LM, Khor KC, et al. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature. 2006;442:466–70. doi: 10.1038/nature04866. [DOI] [PubMed] [Google Scholar]
- 25.Goodarzi AA, Yu Y, Riballo E, Douglas P, Walker SA, Ye R, et al. DNA-PK autophosphorylation facilitates Artemis endonuclease activity. EMBO J. 2006;25:3880–9. doi: 10.1038/sj.emboj.7601255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma Y, Pannicke U, Schwarz K, Lieber MR. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell. 2002;108:781–94. doi: 10.1016/S0092-8674(02)00671-2. [DOI] [PubMed] [Google Scholar]
- 27.Savic V, Yin B, Maas NL, Bredemeyer AL, Carpenter AC, Helmink BA, et al. Formation of dynamic gamma-H2AX domains along broken DNA strands is distinctly regulated by ATM and MDC1 and dependent upon H2AX densities in chromatin. Mol Cell. 2009;34:298–310. doi: 10.1016/j.molcel.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A. H2AX: the histone guardian of the genome. DNA Repair (Amst) 2004;3:959–67. doi: 10.1016/j.dnarep.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 29.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 30.Helmink BA, Tubbs AT, Dorsett Y, Bednarski JJ, Walker LM, Feng Z, et al. H2AX prevents CtIP-mediated DNA end resection and aberrant repair in G1-phase lymphocytes. Nature. 2011;469:245–9. doi: 10.1038/nature09585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bednarski JJ, Nickless A, Bhattacharya D, Amin RH, Schlissel MS, Sleckman BP. RAG-induced DNA double-strand breaks signal through Pim2 to promote pre-B cell survival and limit proliferation. J Exp Med. 2012;209:11–7. doi: 10.1084/jem.20112078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guidos CJ, Williams CJ, Grandal I, Knowles G, Huang MT, Danska JS. V(D)J recombination activates a p53-dependent DNA damage checkpoint in scid lymphocyte precursors. Genes Dev. 1996;10:2038–54. doi: 10.1101/gad.10.16.2038. [DOI] [PubMed] [Google Scholar]
- 33.Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–31. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 34.Perkins EJ, Nair A, Cowley DO, Van Dyke T, Chang Y, Ramsden DA. Sensing of intermediates in V(D)J recombination by ATM. Genes Dev. 2002;16:159–64. doi: 10.1101/gad.956902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–9. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 36.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 37.Rowh MA, DeMicco A, Horowitz JE, Yin B, Yang-Iott KS, Fusello AM, et al. Tp53 deletion in B lineage cells predisposes mice to lymphomas with oncogenic translocations. Oncogene. 2011;30:4757–64. doi: 10.1038/onc.2011.191. [DOI] [PubMed] [Google Scholar]
- 38.Difilippantonio MJ, Petersen S, Chen HT, Johnson R, Jasin M, Kanaar R, et al. Evidence for replicative repair of DNA double-strand breaks leading to oncogenic translocation and gene amplification. J Exp Med. 2002;196:469–80. doi: 10.1084/jem.20020851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gladdy RA, Taylor MD, Williams CJ, Grandal I, Karaskova J, Squire JA, et al. The RAG-1/2 endonuclease causes genomic instability and controls CNS complications of lymphoblastic leukemia in p53/Prkdc-deficient mice. Cancer Cell. 2003;3:37–50. doi: 10.1016/S1535-6108(02)00236-2. [DOI] [PubMed] [Google Scholar]
- 40.Zhu C, Mills KD, Ferguson DO, Lee C, Manis J, Fleming J, et al. Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell. 2002;109:811–21. doi: 10.1016/S0092-8674(02)00770-5. [DOI] [PubMed] [Google Scholar]
- 41.Rooney S, Sekiguchi J, Whitlow S, Eckersdorff M, Manis JP, Lee C, et al. Artemis and p53 cooperate to suppress oncogenic N-myc amplification in progenitor B cells. Proc Natl Acad Sci USA. 2004;101:2410–5. doi: 10.1073/pnas.0308757101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bredemeyer AL, Helmink BA, Innes CL, Calderon B, McGinnis LM, Mahowald GK, et al. DNA double-strand breaks activate a multi-functional genetic program in developing lymphocytes. Nature. 2008;456:819–23. doi: 10.1038/nature07392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peschon JJ, Morrissey PJ, Grabstein KH, Ramsdell FJ, Maraskovsky E, Gliniak BC, et al. Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. J Exp Med. 1994;180:1955–60. doi: 10.1084/jem.180.5.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.von Freeden-Jeffry U, Vieira P, Lucian LA, McNeil T, Burdach SE, Murray R. Lymphopenia in interleukin (IL)-7 gene-deleted mice identifies IL-7 as a nonredundant cytokine. J Exp Med. 1995;181:1519–26. doi: 10.1084/jem.181.4.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goetz CA, Harmon IR, O’Neil JJ, Burchill MA, Farrar MA. STAT5 activation underlies IL7 receptor-dependent B cell development. J Immunol. 2004;172:4770–8. doi: 10.4049/jimmunol.172.8.4770. [DOI] [PubMed] [Google Scholar]
- 46.Malin S, McManus S, Cobaleda C, Novatchkova M, Delogu A, Bouillet P, et al. Role of STAT5 in controlling cell survival and immunoglobulin gene recombination during pro-B cell development. Nat Immunol. 2010;11:171–9. doi: 10.1038/ni.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bertolino E, Reddy K, Medina KL, Parganas E, Ihle J, Singh H. Regulation of interleukin 7-dependent immunoglobulin heavy-chain variable gene rearrangements by transcription factor STAT5. Nat Immunol. 2005;6:836–43. doi: 10.1038/ni1226. [DOI] [PubMed] [Google Scholar]
- 48.Mandal M, Powers SE, Ochiai K, Georgopoulos K, Kee BL, Singh H, et al. Ras orchestrates exit from the cell cycle and light-chain recombination during early B cell development. Nat Immunol. 2009;10:1110–7. doi: 10.1038/ni.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li WQ, Jiang Q, Khaled AR, Keller JR, Durum SK. Interleukin-7 inactivates the pro-apoptotic protein Bad promoting T cell survival. J Biol Chem. 2004;279:29160–6. doi: 10.1074/jbc.M401656200. [DOI] [PubMed] [Google Scholar]
- 50.Amin RH, Schlissel MS. Foxo1 directly regulates the transcription of recombination-activating genes during B cell development. Nat Immunol. 2008;9:613–22. doi: 10.1038/ni.1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Herzog S, Hug E, Meixlsperger S, Paik JH, DePinho RA, Reth M, et al. SLP-65 regulates immunoglobulin light chain gene recombination through the PI(3)K-PKB-Foxo pathway. Nat Immunol. 2008;9:623–31. doi: 10.1038/ni.1616. [DOI] [PubMed] [Google Scholar]
- 52.Lazorchak AS, Liu D, Facchinetti V, Di Lorenzo A, Sessa WC, Schatz DG, et al. Sin1-mTORC2 suppresses rag and il7r gene expression through Akt2 in B cells. Mol Cell. 2010;39:433–43. doi: 10.1016/j.molcel.2010.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ochiai K, Maienschein-Cline M, Mandal M, Triggs JR, Bertolino E, Sciammas R, et al. A self-reinforcing regulatory network triggered by limiting IL-7 activates pre-BCR signaling and differentiation. Nat Immunol. 2012;13:300–7. doi: 10.1038/ni.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bankovich AJ, Raunser S, Juo ZS, Walz T, Davis MM, Garcia KC. Structural insight into pre-B cell receptor function. Science. 2007;316:291–4. doi: 10.1126/science.1139412. [DOI] [PubMed] [Google Scholar]
- 55.Bergman Y. Allelic exclusion in B and T lymphopoiesis. Semin Immunol. 1999;11:319–28. doi: 10.1006/smim.1999.0188. [DOI] [PubMed] [Google Scholar]
- 56.Brady BL, Steinel NC, Bassing CH. Antigen receptor allelic exclusion: an update and reappraisal. J Immunol. 2010;185:3801–8. doi: 10.4049/jimmunol.1001158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thompson EC, Cobb BS, Sabbattini P, Meixlsperger S, Parelho V, Liberg D, et al. Ikaros DNA-binding proteins as integral components of B cell developmental-stage-specific regulatory circuits. Immunity. 2007;26:335–44. doi: 10.1016/j.immuni.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 58.Johnson K, Hashimshony T, Sawai CM, Pongubala JM, Skok JA, Aifantis I, et al. Regulation of immunoglobulin light-chain recombination by the transcription factor IRF-4 and the attenuation of interleukin-7 signaling. Immunity. 2008;28:335–45. doi: 10.1016/j.immuni.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 59.Marshall AJ, Fleming HE, Wu GE, Paige CJ. Modulation of the IL-7 dose-response threshold during pro-B cell differentiation is dependent on pre-B cell receptor expression. J Immunol. 1998;161:6038–45. [PubMed] [Google Scholar]
- 60.Schebesta M, Pfeffer PL, Busslinger M. Control of pre-BCR signaling by Pax5-dependent activation of the BLNK gene. Immunity. 2002;17:473–85. doi: 10.1016/S1074-7613(02)00418-1. [DOI] [PubMed] [Google Scholar]
- 61.Rolink A, Kudo A, Karasuyama H, Kikuchi Y, Melchers F. Long-term proliferating early pre B cell lines and clones with the potential to develop to surface Ig-positive, mitogen reactive B cells in vitro and in vivo. EMBO J. 1991;10:327–36. doi: 10.1002/j.1460-2075.1991.tb07953.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rolink A, Streb M, Melchers F. The kappa/lambda ratio in surface immunoglobulin molecules on B lymphocytes differentiating from DHJH-rearranged murine pre-B cell clones in vitro. Eur J Immunol. 1991;21:2895–8. doi: 10.1002/eji.1830211137. [DOI] [PubMed] [Google Scholar]
- 63.Amaravadi R, Thompson CB. The survival kinases Akt and Pim as potential pharmacological targets. J Clin Invest. 2005;115:2618–24. doi: 10.1172/JCI26273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Casellas R, Shih TA, Kleinewietfeld M, Rakonjac J, Nemazee D, Rajewsky K, et al. Contribution of receptor editing to the antibody repertoire. Science. 2001;291:1541–4. doi: 10.1126/science.1056600. [DOI] [PubMed] [Google Scholar]
- 65.Derudder E, Cadera EJ, Vahl JC, Wang J, Fox CJ, Zha S, et al. Development of immunoglobulin lambda-chain-positive B cells, but not editing of immunoglobulin kappa-chain, depends on NF-kappaB signals. Nat Immunol. 2009;10:647–54. doi: 10.1038/ni.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Domen J, van der Lugt NM, Acton D, Laird PW, Linders K, Berns A. Pim-1 levels determine the size of early B lymphoid compartments in bone marrow. J Exp Med. 1993;178:1665–73. doi: 10.1084/jem.178.5.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mikkers H, Nawijn M, Allen J, Brouwers C, Verhoeven E, Jonkers J, et al. Mice deficient for all PIM kinases display reduced body size and impaired responses to hematopoietic growth factors. Mol Cell Biol. 2004;24:6104–15. doi: 10.1128/MCB.24.13.6104-6115.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Feng C, Woodside KJ, Vance BA, El-Khoury D, Canelles M, Lee J, et al. A potential role for CD69 in thymocyte emigration. Int Immunol. 2002;14:535–44. doi: 10.1093/intimm/dxf020. [DOI] [PubMed] [Google Scholar]
- 69.Pearce G, Angeli V, Randolph GJ, Junt T, von Andrian U, Schnittler HJ, et al. Signaling protein SWAP-70 is required for efficient B cell homing to lymphoid organs. Nat Immunol. 2006;7:827–34. doi: 10.1038/ni1365. [DOI] [PubMed] [Google Scholar]
- 70.Rosen SD. Ligands for L-selectin: homing, inflammation, and beyond. Annu Rev Immunol. 2004;22:129–56. doi: 10.1146/annurev.immunol.21.090501.080131. [DOI] [PubMed] [Google Scholar]
- 71.Shiow LR, Rosen DB, Brdicková N, Xu Y, An J, Lanier LL, et al. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature. 2006;440:540–4. doi: 10.1038/nature04606. [DOI] [PubMed] [Google Scholar]
- 72.Schwab SR, Cyster JG. Finding a way out: lymphocyte egress from lymphoid organs. Nat Immunol. 2007;8:1295–301. doi: 10.1038/ni1545. [DOI] [PubMed] [Google Scholar]
- 73.Galkina E, Florey O, Zarbock A, Smith BR, Preece G, Lawrence MB, et al. T lymphocyte rolling and recruitment into peripheral lymph nodes is regulated by a saturable density of L-selectin (CD62L) Eur J Immunol. 2007;37:1243–53. doi: 10.1002/eji.200636481. [DOI] [PubMed] [Google Scholar]
- 74.Subramanian H, Grailer JJ, Ohlrich KC, Rymaszewski AL, Loppnow JJ, Kodera M, et al. Signaling through L-selectin mediates enhanced chemotaxis of lymphocyte subsets to secondary lymphoid tissue chemokine. J Immunol. 2012;188:3223–36. doi: 10.4049/jimmunol.1101032. [DOI] [PubMed] [Google Scholar]
- 75.Allende ML, Tuymetova G, Lee BG, Bonifacino E, Wu YP, Proia RL. S1P1 receptor directs the release of immature B cells from bone marrow into blood. J Exp Med. 2010;207:1113–24. doi: 10.1084/jem.20092210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pereira JP, Xu Y, Cyster JG. A role for S1P and S1P1 in immature-B cell egress from mouse bone marrow. PLoS One. 2010;5:e9277. doi: 10.1371/journal.pone.0009277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sherman MH, Kuraishy AI, Deshpande C, Hong JS, Cacalano NA, Gatti RA, et al. AID-induced genotoxic stress promotes B cell differentiation in the germinal center via ATM and LKB1 signaling. Mol Cell. 2010;39:873–85. doi: 10.1016/j.molcel.2010.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Walsh NC, Teitell M. B-cell differentiation stimulated by physiologic DNA double strand breaks. Cell Cycle. 2011;10:176–7. doi: 10.4161/cc.10.2.14474. [DOI] [PubMed] [Google Scholar]