

Abstract
The evolutionary adaptations of thermophilic water-soluble proteins required for maintaining stability at high temperature have been extensively investigated. Little is known about the adaptations in membrane proteins, however. Here, we compare many properties of mesophilic and thermophilic membrane protein structures, including side-chain burial, packing, hydrogen bonding, transmembrane kinks, loop lengths, hydrophobicity, and other sequence features. Most of these properties are quite similar between mesophiles and thermophiles although we observe a slight increase in side-chain burial and possibly a slight decrease in the frequency of transmembrane kinks in thermophilic membrane protein structures. The most striking difference is the increased hydrophobicity of thermophilic transmembrane helices, possibly reflecting more stringent hydrophobicity requirements for membrane partitioning at high temperature. In agreement with prior work examining transmembrane sequences, we find that thermophiles have an increase in small residues (Gly, Ala, Ser, and Val) and a strong suppression of Cys. We also find a relative dearth of most strongly polar residues (Asp, Asn, Glu, Gln, and Arg). These results suggest that in thermophiles, there is significant evolutionary pressure to offload destabilizing polar amino acids, to decrease the entropy cost of side chain burial, and to eliminate thermally sensitive amino acids.
Keywords: stability, folding, structure, transmembrane helix, hydrogen bonds, polar amino acids, burial, kink, packing
Introduction
The proteins of thermophilic organisms are able to maintain stable folds at high temperatures. The adaptations required to maintain stability at high temperature have been of interest not only from the perspective of evolutionary mechanisms, but also because it may teach us how to stabilize proteins. As a result, the stabilizing mechanisms have been extensively explored for water-soluble proteins. The mechanisms observed include an increase in secondary structure propensity;1,2 changes that decrease unfolded state entropy such as the introduction of Pro residues, reduction of Gly residues,3 smaller loops,4 and the addition of disulfide bonds;5–7 an increase of hydrogen bonds and salt bridges;8–12 and better optimized hydrophobicity.13 Szilágyi and Závodszky3 made an extensive study of many protein families and found that few of these mechanisms were general, except an increase in salt bridges. Rather, many of these mechanisms may be used for stabilization of a given protein, but not necessarily all of them. Berezovsky and Shakhnovich11 make the interesting observation that the preferred stabilization mechanisms can change depending on the evolutionary path. For example, proteins from organisms that evolved at high temperature show increased compactness, whereas proteins from organisms that evolved at lower temperatures and then adapted to higher temperatures are less compact and have increased salt bridges.
Because of the dearth of membrane protein structures, the evaluation of differences between mesophiles and thermophiles has been largely restricted to sequence analysis. Schneider et al.14 studied sequence differences between predicted transmembrane helices in the genomes of thermophilic and mesophilic membrane proteins. They observed a striking depletion of Cys residues in thermophiles and an increase in Gly, Ser, and Ala pair motifs, suggesting a preference for the packing of small residues. By comparing mesophilic and thermophilic reaction center sequences and structures, Shlyk-Kerner et al.15 showed that subtle changes in cavity volumes in photosynthetic reaction centers were a key factor in switching between thermophilic and mesophilic properties, implying an important role for packing in thermal adaptation.
Recent advances in structure determination of membrane proteins prompted us to investigate whether the database had been enlarged enough to examine the generality of prior results and look for additional structural features that change between thermophilic and mesophilic proteins. We were able to develop a database of 25 independent thermophilic/hyperthermophilic α-helical membrane protein structures and 101 independent mesophilic structures. Although a far cry from what is available for soluble proteins, this database enables us to examine whether there are prominent structural features that distinguish mesophiles and thermophiles.
Results and Discussion
Thermophile–mesophile database construction
To explore the structural differences between thermophilic and mesophilic membrane proteins that may contribute to thermostability, we constructed a database of high-resolution thermophilic and mesophilic membrane protein structures. We were able to find independent structures from 25 thermophiles (growth temperature >50°C) and 101 mesophiles (see Materials and Methods section). The database allows us to compare the features of thermophilic and mesophilic structures globally. Different folds may have different global properties, however. For example, Hildebrand et al.16 showed that proteins that undergo large domain movement are less well-packed than other membrane proteins. Ideally, we would like to compare proteins from the same family,3 but there is currently not enough structural data. We therefore built a database of similar structural segments to compare the similar local structures between thermophiles and mesophiles. We refer to global comparisons of the properties of all thermophiles to all mesophiles as unpaired data and the comparisons between similar structured regions only as paired data.
Burial and packing
Van der Waals packing is thought to play an important role in stabilizing the transmembrane domains of membrane proteins.17 We have found that membrane proteins increase van der Waals packing contributions relative to soluble proteins by increasing the overall level of side-chain burial, rather than by improved packing efficiency (volume occupied by atoms).18,19 Nevertheless, either increased packing efficiency or increased burial could be an important factor in stabilizing membrane proteins in hot environments.15 We therefore investigated whether packing density and burial are different in mesophiles and thermophiles.
When comparing the water-soluble domains, we found essentially no difference between mesophiles and thermophiles in burial or packing for both the paired and the unpaired data (Table I). We also found no significant difference in packing density in the transmembrane domains.
Table I.
Differences Between Thermophilic and Mesophilic Membrane Protein Structures
| Transmembrane | Soluble | P-value of Mann-Whitney U-Test | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Average and standard errora | Average and standard errora | Transmembrane | Soluble | |||||||
| Property | Thermophile | Mesophile | Paired differenceb | Thermophile | Mesophile | Paired differenceb | Unpaired | Paired | Unpaired | Paired |
| Burial (fractional side-chain buried) | 0.780 ± 0.027 | 0.733 ± 0.012 | 0.054 ± 0.045 | 0.672 ± 0.021 | 0.661 ± 0.008 | 0.004 ± 0.025 | 0.026* | 0.133 | 0.124 | 0.496 |
| Packing | 0.719 ± 0.006 | 0.727 ± 0.002 | −0.002 ± 0.012 | 0.718 ± 0.004 | 0.717 ± 0.002 | 0.003 ± 0.004 | 0.063 | 0.433 | 0.296 | 0.234 |
| Surface loop length (number of residues) | N/A | 19.126 ± 0.735 | 19.189 ± 0.747 | −0.063 ± 0.203 | 0.491 | |||||
| Helix length (number of residues) | 25.627 ± 0.723 | 26.969 ± 0.789 | 0.404 ± 1.074 | N/A | 0.269 | 0.304 | ||||
| Kinks (number per TM-helix) | 0.070 ± 0.008 | 0.073 ± 0.004 | −0.011 ± 0.009 | 0.455 | 0.190 | |||||
| Interhelical H-bonds (number per residue) | 0.102 ± 0.020 | 0.115 ± 0.008 | −0.028±0.025 | 0.205 | 0.160 | |||||
| Hydrophobicity | 0.126 ± 0.024 | 0.220 ± 0.015 | 0.039 ± 0.034 | 0.005* | 0.336 | |||||
For unpaired structure or direct homolog data sets.
Mean difference (thermophiles–mesophiles) with standard error (A paired difference of 0 indicates no difference between paired thermophiles and mesophiles).
Statistically significant (P < 0.05).
We did observe a roughly 5% increase in the average fraction of the side-chain surface area buried in the transmembrane domains of thermophiles (P = 0.026, Mann–Whitney U-test). The increase was also seen in the paired data, albeit without statistical significance. Although the increase is small in magnitude, even mesophilic transmembrane domains bury a high fraction of their potential surface area and hence any increase is likely to be difficult to achieve and therefore hard to detect. Thus, we suggest that thermophiles may bury slightly more surface area on average in the transmembrane domains.
Surface loop length in thermophiles and mesophiles
Prior work on water-soluble proteins showed that thermophiles can have shorter loops between secondary structure elements, which is thought to be a mechanism for reducing unfolded state entropy.4 In a similar fashion, we might expect to see shorter loop lengths between transmembrane helices of thermophiles compared to mesophiles. On the other hand, it is possible that longer surface loop lengths may have evolved to help structure the transmembrane domains.
To investigate changes in loop length, we would ideally compare proteins within the same family, but as noted above there were few homologous thermophile/mesophile pairs. Consequently, we augmented our structural database with homologous sequences. For every structure, we identified homologous mesophile and thermophile sequences. We then utilized the sequence alignment with the protein of known structure to define the transmembrane segment endpoints. In this manner, we could use structural information to estimate loop lengths in all the homologous proteins. We found an average loop length of 19.189 ± 0.747 residues for mesophiles and 19.126 ± 0.735 for thermophiles (Table I). Thus, there is apparently no major difference in loop length between the two classes of proteins (P = 0.49, Mann–Whitney U-test).
Transmembrane helix length in thermophiles and mesophiles
Another way to reduce the entropy cost of folding would be to decrease order in the folded state. This could be accomplished by increased fraying at the ends of transmembrane helices, reducing their length on average. We found a very small (5%) decrease in the average length of thermophilic transmembrane helices for the unpaired databases (P = 0.27, Mann–Whitney U-test). This difference completely disappears in the paired data, however. Thus, if transmembrane helices are shorter, it is not a large difference.
Transmembrane kinks in thermophiles and mesophiles
Kinks and other helix distortions should be higher energy features relative to regular helices.20 Consequently, it is possible that these distortions would be minimized in the transmembrane helices of membrane proteins. In the overall analysis of all thermophiles and mesophiles, we found an average of 0.073 ± 0.004 kinks per transmembrane residue in mesophiles and 0.070 ± 0.008 kinks in thermophiles (Table I). Although within margin of error, we also found a consistent decrease in the number of kinks in thermophiles when comparing the paired structures. Thus, there may be modest evolutionary pressure to reduce the number of kinks in transmembrane helices. Helix distortions are likely to be important for function, however, and hence there are likely to be limits on how much kinking can be reduced.
Interhelical hydrogen bonding in thermophiles and mesophiles
An increase in hydrogen bonds and salt bridges is a commonly observed trend when comparing water-soluble proteins from mesophiles and thermophiles.8–12 Although most hydrogen-bonded side-chain interactions that have been experimentally measured in membrane proteins appear to make relatively modest contributions,21–27 some have been measured as high as 1.8 kcal/mol and in theory it is possible that they could be made even stronger.26,28 Thus, it seems reasonable to expect that the number of transmembrane interhelical hydrogen bonds might increase in thermophiles. However, we find a slight decrease in the number of interhelical hydrogen bonds in thermophiles relative to mesophiles in both the unpaired and the paired comparisons. This result is consistent with the view that strongly polar residues are generally present in the transmembrane domains for functional reasons, not to stabilize the structure.26,27 As discussed below, the imperative to maintain hydrophobicity may trump any benefit accrued by additional hydrogen bonds.
Thermophilic transmembrane helices are more hydrophobic
Prior work comparing the amino acid composition of thermophilic and mesophilic transmembrane helices relied on predictions from sequence information.14 Here, we employed structures to define the transmembrane segments. To bolster our database, we added aligned sequences of close homologs of the known structures, using the structures to identify transmembane helices.
The observed differences in amino acid composition are shown in Figure 1. In agreement with the earlier results from predicted transmembrane helices, we find a dramatic depletion of Cys in thermophiles, likely reflecting its higher reactivity at high temperature. We also find an enrichment of the small residues Ala and Gly and to a lesser extent Ser and Val. Schneider et al.14 found a significant increase in Asp and Glu among thermophiles and essentially no difference for the other strongly polar amino acids except Gln which was found to be depleted in thermophiles. We, however, find a large reduction in all the strongly polar residues (Asp, Asn, Glu, Gln, and Arg) with the exception of Lys and His. We also see a compensatory increase in the large apolar residues Phe, Leu, and Ile.
Figure 1.
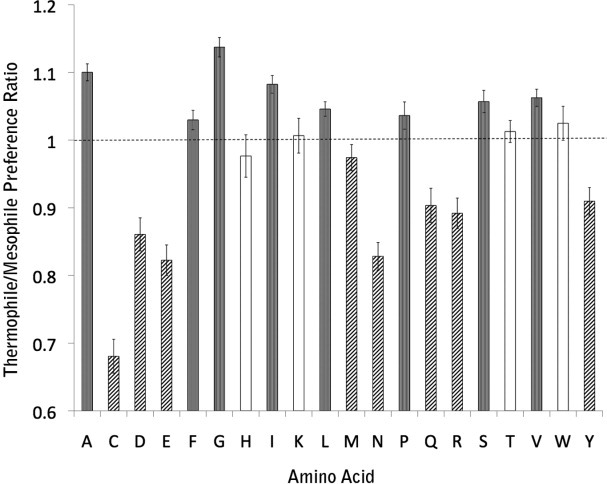
Transmembrane residues composition differences between thermophiles and mesophiles. The “Thermophile/Mesophile Preference Ratio” axis describes the ratio of fraction of each amino acid type in thermophile relative to mesophiles. Gray bars indicate significant differences >1, hatched bars indicate significant differences <1, and the white bars indicate no significant difference.
Why do we find a clear depletion of polar residues in thermophilic transmembrane helices that were not observed using transmembrane helices predicted from sequence data? It appears that the transmembrane helices of thermophiles are simply more hydrophobic on average than those of mesophiles (Supporting Information Table S1). Indeed, using the biological hydrophobicity scale,29 the average hydrophobicity of structurally observed transmembrane helices is +0.220 ± 0.015 kcal/mol per residue in mesopohiles and +0.126 ± 0.024 kcal/mol per residue in thermophiles (P = 0.005, Mann–Whitney U-test). Thus, a uniform prediction criteria for both mesophiles and thermophiles would tend to reject more of the less hydrophobic transmembrane helices in mesophiles.
Conclusions
The most striking difference we observe between the membrane proteins in mesophiles and thermophiles is a general increase in the hydrophobicity of the thermophile transmembrane helices. This result implies more stringent requirements for transmembrane helix insertion at high temperature. To the extent that the translocon measures the thermodynamic preference for a sequence segment to insert in the bilayer or remain in an aqueous environment,29 it makes sense that a lower free energy well would be required at high temperature to ensure the insertion decision is made. Theoretical calculations and experimental measurements indicate that the free energy of partitioning peptides into bilayers is relatively constant with temperature,43,44 and hence higher temperatures should decrease the probability of insertion. This imperative may explain the general depletion of strongly polar residues that is observed. It may also preclude the deployment of more stabilizing hydrogen bonding interactions. If so, membrane proteins need to find other ways to stabilize their structures at higher temperature. How might they do this? One observation made by Scheider et al.14 and confirmed here is the increase in small amino acids in thermophiles. This could have two stabilizing effects: (1) packing small residues rather than large residues lowers the entropy cost of packing,45,46 which could be particularly important at higher temperature; and (2) small residues can allow for more intimate interactions between helices. Consistent with this latter view, we also find a modest increase in the average fraction of side-chain surface area buried in thermophiles. Finally, we observe a possible small reduction in the number of high energy kinks.
A significant caveat with a global comparison of mesophiles and thermophiles is that we interpret all changes as reflecting the effects of growth temperature, the only common parameter that changes between the two groups of proteins analyzed here. But clearly there are other parameters that change when comparing proteins from different organisms. Moreover, some proteins in mesophiles are not any less stable than a thermophilic protein, but we do not have stability data for most membrane proteins at this time. Also, as pointed out by Szilágyi and Závodszky,3 any given protein may evolve a different predominant method of stabilization. As a result of all these variables, many actual stabilizing mechanisms may disappear in the noise. A careful parsing of the different effects will be more feasible when we have many examples of mesophile and thermophile structures from the same family, along with stability measurements, so that direct comparisons can be made.
Materials and Methods
Structure database
A nonredundant database of mesophiles and thermophiles was created using α-helical membrane proteins from Stephen White's database (http://blanco.biomol.uci.edu/mpstruc/listAll/list).30 Growth temperatures were obtained from the American Type Culture Collection and web searches (Supporting Information Table S2). Structures were divided into mesophiles and thermophiles based on a 50°C optimal growth temperature cutoff.
Each group of structures (mesophiles and thermophiles) was made nonredundant separately. Within each group, none of the protein sequences was similar to each other at the level of an expectation value of 1 × 10−10 according to BLAST.31
For each structure, the author-recommended oligomer was chosen as described by REMARK 350 in each Protein Data Bank (PDB) file. Only unique chains (i.e., nonidentical) were used for analysis. Monotopic membrane proteins, nuclear magnetic resonance, and electron crystallography structures were eliminated. We also eliminated structures that had fewer than three transmembrane helices per oligomer. Prosthetic groups for each structure were retained, but lipids and other nonintrinsic components were removed from each structure. The PDB codes for the two groups are as follows.
Mesophiles (101): 1AIJ, 1BCC, 1BGY, 1COW, 1EFR, 1FFT, 1KPL, 1KQF, 1L0V, 1LGH, 1NEK, 1NKZ, 1PW4, 1Q16, 1Q90, 1QO1, 1U7G, 1V54, 1YEW, 1ZCD, 1ZOY, 1ZRT, 2A79, 2ACZ, 2BHW, 2BL2, 2BS2, 2C3E, 2CK3, 2E74, 2EI4, 2EXW, 2GMH, 2GSM, 2H8A, 2I37, 2J58, 2JLN, 2NUU, 2O01, 2O7L, 2OAR, 2OAU, 2Q7M, 2QCU, 2QI9, 2QKS, 2QTS, 2RCR, 2RDD, 2V8N, 2VQG, 2W5J, 2WCD, 2WSS, 2WSW, 2X2V, 2XUT, 2YFY, 2ZUQ, 2ZW3, 2ZXE, 3B8E, 3B9W, 3B9Y, 3BF0, 3BKD, 3CHX, 3CK6, 3CX5, 3D31, 3DQB, 3EAR, 3EHZ, 3FH6, 3G5U, 3H90, 3HD6, 3JQO, 3K07, 3KCU, 3KG2, 3KP9, 3LIM, 3LRB, 3LUT, 3M71, 3MK7, 3MKT, 3NAF, 3NVO, 3O7Q, 3OAA, 3P0G, 3PJZ, 3QAK, 3QE7, 3QNQ, 3RLB, 3RVY, and 3S0X.
Thermophiles (25): 1EHK, 1JB0, 1ORQ, 2A0L, 2B2F, 2F2B, 2HN2, 2ONK, 2QJU, 2VPZ, 2ZY9, 3A5C, 3AQP, 3ARC, 3B4R, 3BVD, 3DIN, 3GIA, 3HYW, 3K5B, 3KBC, 3KDS, 3M9S, 3MP7, and 3ORG.
Thermophile–mesophile structural pair assignment
Structurally homologous pairs were identified using the structural alignment tool TM-align.32 One set of pairs was constructed with no resolution cutoff that could be used for sequence-based analysis, and a second set was constructed using a 3.2 Å or better resolution cutoff that was used for analyzing more detailed features. Only unique chains were used for structural pairing. Each thermophile structure was structurally aligned to every mesophile structure. A pair was considered similar if the alignment involved more than 100 residues and the root mean square deviation was less than 5 Å. The longest alignment was chosen as the representative pairing for each thermophile structure. Each thermophile was matched to one mesophile sequentially and in a nonredundant fashion so that no thermophile was matched to the same mesophile. Analysis (e.g., burial, packing, helix length, etc.) of structural pairs was restricted to aligned regions only.
Transmembrane region assignment
Transmembrane regions were obtained from a consensus database constructed from the Protein Data Bank of Transmembrane Proteins (PDBTM) and Orientations of Proteins in Membranes (OPM) database, or using TMDET to identify transmembrane regions for those proteins not present in these databases.33–35 The assigned transmembrane regions are summarized in Supporting Information Table S3.
Burial calculation
Burial data were obtained by calculating relative solvent accessibilities per residue using a fast version of the Shrake and Rupley algorithm36 implemented in the Ezprot library (http://www.doe-mbi.ucla.edu/local/software/ezprot).37 Only unpaired and paired structures of 3.2Å or better resolution were analyzed. Burial values were calculated separately for soluble and transmembrane regions of unpaired thermophiles and mesophiles, as well as for the paired thermophile–mesophile structures. For the unpaired structures, values were averaged over all mesophiles and thermophiles separately. In computing the average, burial values were weighted by the number of residues in a given protein to obtain a weighted burial average over all proteins; this ensured that burial values from smaller proteins did not unnecessarily bias the results calculated over all proteins. For the paired structures, difference values between each thermophile–mesophile pair were averaged over all thermophile–mesophile pairs.
Packing calculation
Packing values were calculated using Rother et al.'s Voronoia program for computing packing densities.38 Surface residues were excluded from the calculation. A 0.20-Å grid distance and the ProtOr radii were chosen as parameters based on similar ones chosen by Hildebrand et al.16 Only unpaired and paired structures of 3.2 Å or better resolution were analyzed. Packing values were calculated separately for soluble and transmembrane regions of unpaired thermophiles and mesophiles, as well as for the thermophile–mesophile paired structures (as was done above for burial). For the unpaired structures, the values were averaged over all thermophiles and mesophiles separately. In computing the average, packing values were weighted by the number of residues in a given protein to obtain a weighted packing average over all proteins. For the paired structures, difference values between each thermophile–mesophile pair were averaged over all thermophile–mesophile pairs.
Interhelical hydrogen bonding calculation
HBPLUS was used to identify hydrogen bonds.39–41 Only structures of 3.2 Å or better resolution were analyzed and we only counted hydrogen bonds between transmembrane helices (interhelical hydrogen bonds). Interhelical hydrogen bonds were counted for each unpaired thermophile or mesophile structure and normalized by the number of transmembrane residues in each structure. The average number of interhelical hydrogen bonds per transmembrane residue was then calculated for all thermophile or mesophile proteins separately. Similarly, for paired structures, interhelical hydrogen bonds were counted for each paired thermophile or mesophile structure and normalized by the number of transmembrane residues in each paired structure. The difference in this value between each thermophile–mesophile paired structure was then calculated. These differences were then averaged over all paired structures.
Transmembrane helix length calculation
Transmembrane helix length was examined in unpaired and paired structures with no restriction on structure resolution. To define the extent of transmembrane helices, we first started with the helical transmembrane segment in the hydrocarbon core region of the bilayer, and then continued the helices out to the helix endpoints as defined by DSSP.40,41 No structures with fewer than three transmembrane helices were considered in analyzing helix length. For unpaired structures, helix lengths were averaged over each structure and then over all thermophiles (or mesophiles). For paired thermophile–mesophile structures, helix lengths were averaged over the chain of each paired structure for aligned residues. The difference in average helix length between the two paired structures was then calculated. These differences were then averaged over all paired structures.
Transmembrane amino acid hydrophobicity calculation
The biological hydrophobicity scale was used for this calculation.29 For each unpaired structure, hydrophobicity was averaged over each transmembrane segment. The average for each unpaired structure was then averaged over all thermophile or mesophile structures. Similarly, for each paired structure, hydrophobicity was averaged over each transmembrane segment. The difference in average hydrophobicity between paired structures was then calculated. Finally, the average of differences in hydrophobicity between paired structures was calculated.
Transmembrane helical kink calculation
Transmembrane helical kinks were identified as described previously.42 Only structures of 3.2 Å or better resolution were analyzed. Kinks were counted for each unpaired thermophile or mesophile structure and normalized by the number of transmembrane residues in each structure. The average number of kinks per transmembrane residue was then calculated for all thermophile or mesophile proteins separately. For the paired structures, kinks were normalized by the number of transmembrane residues in each paired structure. The difference in this value between each thermophile–mesophile paired structure was then calculated. These differences were then averaged over all paired structures.
Direct homolog identification for surface loop length and transmembrane amino acid preference studies
Neither the unpaired nor the paired structures provided sufficient statistical power to study surface loop length and transmembrane amino acid preferences. As a result, we identified direct sequence homologs of the unpaired mesophile structures using BLAST (identifying both mesophiles and thermophiles).31 We used an expectation cutoff value of 1 × 10−10 and limited the number of homologs identified to 10,000 for each unpaired mesophile structure's sequence. These BLAST results were screened for thermophile and mesophile homologs according to species name. We have provided the species names used in the Supporting Information. Only homolog alignments whose length was 70% or greater than the length of the query mesophile sequence were retained. The average number of mesophile sequences obtained in this manner was 955 and the average number of thermophile sequences was 34.
Surface loop length calculation
Using the direct homologs of mesophiles to identify additional thermophiles and mesophiles, we analyzed loop length between thermophiles and mesophiles. Loops were identified as the intervening regions between transmembrane segments. We ignored loops longer than 50 residues to eliminate folded water-soluble domains. Only loops that were entirely contained within direct homolog alignments were considered. We analyzed one randomly chosen thermophile or mesophile homolog for every original, unique chain of each mesophile structure to avoid double counting homologs. Loop lengths were then averaged over all mesophiles or thermophiles.
Transmembrane amino acid preferences calculation
Using the direct homologs of mesophiles to identify additional thermophiles and mesophiles, we analyzed transmembrane amino acid preferences between thermophiles and mesophiles. Only transmembrane amino acids that were contained within direct homolog alignments were considered. Sequence gaps were not counted. We employed a weighting scheme to prevent over counting of highly similar sequences. For example, if two aligned sequences are 100% identical, each amino acid at each position would be counted twice if unweighted although the second sequence provides no new information. Counts were therefore weighted according to the average sequence identity between the original structure and the homolog sequence. The first homolog's sequence was given full weight for each amino acid, whereas additional homolog amino acids were given a weight of (1-% identity). In other words, if the homolog sequence was 90% identical to the sequence of the structure, the count for every amino acid in the sequence was incremented only by 0.1 rather than 1. The number of amino acids of each type was counted for mesophiles or thermophiles. These counts were then normalized by the total number of amino acids for mesophiles or thermophiles. Finally, we calculated the ratio between thermophiles and mesophiles of each amino acid type.
Acknowledgments
The authors thank members of the Bowie lab for comments on the manuscript.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Warren GL, Petsko GA. Composition analysis of alpha-helices in thermophilic organisms. Protein Eng. 1995;8:905–913. doi: 10.1093/protein/8.9.905. [DOI] [PubMed] [Google Scholar]
- 2.Menéndez-Arias L, Argos P. Engineering protein thermal stability. Sequence statistics point to residue substitutions in alpha-helices. J Mol Biol. 1989;206:397–406. doi: 10.1016/0022-2836(89)90488-9. [DOI] [PubMed] [Google Scholar]
- 3.Szilágyi A, Závodszky P. Structural differences between mesophilic, moderately thermophilic and extremely thermophilic protein subunits: results of a comprehensive survey. Structure. 2000;8:493–504. doi: 10.1016/s0969-2126(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 4.Thompson MJ, Eisenberg D. Transproteomic evidence of a loop-deletion mechanism for enhancing protein thermostability. J Mol Biol. 1999;290:595–604. doi: 10.1006/jmbi.1999.2889. [DOI] [PubMed] [Google Scholar]
- 5.Beeby M, O'Connor BD, Ryttersgaard C, Boutz DR, Perry LJ, Yeates TO. The genomics of disulfide bonding and protein stabilization in thermophiles. PLoS Biol. 2005;3:e309. doi: 10.1371/journal.pbio.0030309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mallick P, Boutz DR, Eisenberg D, Yeates TO. Genomic evidence that the intracellular proteins of archaeal microbes contain disulfide bonds. Proc Natl Acad Sci USA. 2002;99:9679–9684. doi: 10.1073/pnas.142310499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jorda J, Yeates TO. Widespread disulfide bonding in proteins from thermophilic archaea. Archaea. 2011;2011:409156. doi: 10.1155/2011/409156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vetriani C, Maeder DL, Tolliday N, Yip KS-P, Stillman TJ, Britton KL, Rice DW, Klump HH, Robb FT. Protein thermostability above 100°C: a key role for ionic interactions. Proc Natl Acad Sci USA. 1998;95:12300–12305. doi: 10.1073/pnas.95.21.12300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vogt G, Woell S, Argos P. Protein thermal stability, hydrogen bonds, and ion pairs. J Mol Biol. 1997;269:631–643. doi: 10.1006/jmbi.1997.1042. [DOI] [PubMed] [Google Scholar]
- 10.Vogt G, Argos P. Protein thermal stability: hydrogen bonds or internal packing? Fold Des. 1997;2:S40–S46. doi: 10.1016/s1359-0278(97)00062-x. [DOI] [PubMed] [Google Scholar]
- 11.Berezovsky IN, Shakhnovich EI. Physics and evolution of thermophilic adaptation. Proc Natl Acad Sci USA. 2005;102:12742–12747. doi: 10.1073/pnas.0503890102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cambillau C, Claverie JM. Structural and genomic correlates of hyperthermostability. J Biol Chem. 2000;275:32383–32386. doi: 10.1074/jbc.C000497200. [DOI] [PubMed] [Google Scholar]
- 13.Spassov VZ, Karshikoff AD, Ladenstein R. The optimization of protein-solvent interactions: thermostability and the role of hydrophobic and electrostatic interactions. Protein Sci. 1995;4:1516–1527. doi: 10.1002/pro.5560040809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneider D, Liu Y, Gerstein M, Engelman DM. Thermostability of membrane protein helix-helix interaction elucidated by statistical analysis. FEBS Lett. 2002;532:231–236. doi: 10.1016/s0014-5793(02)03687-6. [DOI] [PubMed] [Google Scholar]
- 15.Shlyk-Kerner O, Samish I, Kaftan D, Holland N, Sai PSM, Kless H, Scherz A. Protein flexibility acclimatizes photosynthetic energy conversion to the ambient temperature. Nature. 2006;442:827–830. doi: 10.1038/nature04947. [DOI] [PubMed] [Google Scholar]
- 16.Hildebrand PW, Rother K, Goede A, Preissner R, Frömmel C. Molecular packing and packing defects in helical membrane proteins. Biophys J. 2005;88:1970–1977. doi: 10.1529/biophysj.104.049585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bowie JU. Solving the membrane protein folding problem. Nature. 2005;438:581–589. doi: 10.1038/nature04395. [DOI] [PubMed] [Google Scholar]
- 18.Joh NH, Oberai A, Yang D, Whitelegge JP, Bowie JU. Similar energetic contributions of packing in the core of membrane and water-soluble proteins. J Am Chem Soc. 2009;131:10846–10847. doi: 10.1021/ja904711k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oberai A, Joh NH, Pettit FK, Bowie JU. Structural imperatives impose diverse evolutionary constraints on helical membrane proteins. Proc Natl Acad Sci USA. 2009;106:17747–17750. doi: 10.1073/pnas.0906390106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao Z, Bowie JU. Shifting hydrogen bonds may produce flexible transmembrane helices. Proc Natl Acad Sci USA. 2012;109:8121–8126. doi: 10.1073/pnas.1201298109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hong H, Blois TM, Cao Z, Bowie JU. Method to measure strong protein-protein interactions in lipid bilayers using a steric trap. Proc Natl Acad Sci USA. 2010;107:19802–19807. doi: 10.1073/pnas.1010348107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen F, Degnin C, Laederich M, Horton WA, Hristova K. The A391E mutation enhances FGFR3 activation in the absence of ligand. Biochim Biophys Acta. 2011;1808:2045–2050. doi: 10.1016/j.bbamem.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fleming KG, Engelman DM. Specificity in transmembrane helix–helix interactions can define a hierarchy of stability for sequence variants. Proc Natl Acad Sci USA. 2001;98:14340–14344. doi: 10.1073/pnas.251367498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stanley AM, Fleming KG. The role of a hydrogen bonding network in the transmembrane β-barrel OMPLA. J Mol Biol. 2007;370:912–924. doi: 10.1016/j.jmb.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 25.Gratkowski H, Lear JD, DeGrado WF. Polar side chains drive the association of model transmembrane peptides. Proc Natl Acad Sci USA. 2001;98:880–885. doi: 10.1073/pnas.98.3.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bowie JU. Membrane protein folding: how important are hydrogen bonds? Curr Opin Struct Biol. 2011;21:42–49. doi: 10.1016/j.sbi.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joh NH, Min A, Faham S, Whitelegge JP, Yang D, Woods VL, Bowie JU. Modest stabilization by most hydrogen-bonded side-chain interactions in membrane proteins. Nature. 2008;453:1266–1270. doi: 10.1038/nature06977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ben-Tal N, Sitkoff D, Topol IA, Yang A-S, Burt SK, Honig B. Free energy of amide hydrogen bond formation in vacuum, in water, and in liquid alkane solution. J Phys Chem B. 1997;101:450–457. [Google Scholar]
- 29.Hessa T, Kim H, Bihlmaier K, Lundin C, Boekel J, Andersson H, Nilsson I, White SH, Heijne G von. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature. 2005;433:377–381. doi: 10.1038/nature03216. [DOI] [PubMed] [Google Scholar]
- 30.White SH. The progress of membrane protein structure determination. Protein Sci. 2004;13:1948–1949. doi: 10.1110/ps.04712004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008;36:W5–W9. doi: 10.1093/nar/gkn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. OPM: orientations of proteins in membranes database. Bioinformatics. 2006;22:623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- 34.Tusnády GE, Dosztányi Z, Simon I. PDB_TM: selection and membrane localization of transmembrane proteins in the protein data bank. Nucleic Acids Res. 2005;33:D275–D278. doi: 10.1093/nar/gki002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tusnády GE, Dosztányi Z, Simon I. TMDET: web server for detecting transmembrane regions of proteins by using their 3D coordinates. Bioinformatics. 2005;21:1276–1277. doi: 10.1093/bioinformatics/bti121. [DOI] [PubMed] [Google Scholar]
- 36.Shrake A, Rupley JA. Environment and exposure to solvent of protein atoms. Lysozyme and insulin. J Mol Biol. 1973;79:351–371. doi: 10.1016/0022-2836(73)90011-9. [DOI] [PubMed] [Google Scholar]
- 37.Pettit FK, Bare E, Tsai A, Bowie JU. HotPatch: a statistical approach to finding biologically relevant features on protein surfaces. J Mol Biol. 2007;369:863–879. doi: 10.1016/j.jmb.2007.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rother K, Hildebrand PW, Goede A, Gruening B, Preissner R. Voronoia: analyzing packing in protein structures. Nucleic Acids Res. 2009;37:D393–D395. doi: 10.1093/nar/gkn769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McDonald IK, Thornton JM. Satisfying hydrogen bonding potential in proteins. J Mol Biol. 1994;238:777–793. doi: 10.1006/jmbi.1994.1334. [DOI] [PubMed] [Google Scholar]
- 40.Joosten RP, Beek T, A h T, Krieger E, Hekkelman ML, Hooft RWW, Schneider R, Sander C, Vriend G. A series of PDB related databases for everyday needs. Nucl Acids Res. 2010 doi: 10.1093/nar/gkq1105. [Internet]. Available from: http://nar.oxfordjournals.org/content/early/2010/11/11/nar.gkq1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 42.Meruelo AD, Samish I, Bowie JU. TMKink: a method to predict transmembrane helix kinks. Protein Sci. 2011;20:1256–1264. doi: 10.1002/pro.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ulmschneider JP, Smith JC, White SH, Ulmschneider MB. In silico partitioning and transmembrane insertion of hydrophobic peptides under equilibrium conditions. J Am Chem Soc. 2011;133:15487–15495. doi: 10.1021/ja204042f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fernández-Vidal M, White S, Ladokhin A. Membrane partitioning: “Classical” and “nonclassical” hydrophobic effects. J Membr Biol. 2011;239:5–14. doi: 10.1007/s00232-010-9321-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.MacKenzie KR, Engelman DM. Structure-based prediction of the stability of transmembrane helix-helix interactions: the sequence dependence of glycophorin A dimerization. Proc Natl Acad Sci USA. 1998;95:3583–3590. doi: 10.1073/pnas.95.7.3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bowie JU. Solving the membrane protein folding problem. Nature. 2005;438:581–589. doi: 10.1038/nature04395. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.