

Abstract
This report provides a synopsis of the esterase processing of short chain fatty acid (SCFA)-derivatized hexosamine analogs used in metabolic glycoengineering by demonstrating that the extracellular hydrolysis of these compounds is comparatively slow (e.g., with a t1/2 of ~ 4 hours to several days) in normal cell culture as well as in high serum concentrations intended to mimic in vivo conditions. Structure activity relationship (SAR) analysis of common sugar analogs revealed that O-acetylated and N-azido ManNAc derivatives were more refractory against extracellular inactivation by FBS than their butanoylated counterparts consistent with in silico docking simulations of Ac4ManNAc and Bu4ManNAc to human carboxylesterase 1 (hCE1). By contrast, all analogs tested supported increased intracellular sialic acid production within two hours establishing that esterase processing once the analogs are taken up by cells is not rate limiting.
Keywords: ManNAc analogs, metabolic oligosaccharide engineering, esterase, prodrugs, drug metabolism
Ester-containing small molecules are common in nature, exemplified by acetylsalicylic acid (aspirin), and they also are common prodrugs and profluorophores used for medical, diagnostic, and research purposes.1 The biological activity or therapeutic effects of these compounds depend intricately on the stability or lability of the ester group as well as the organ, tissue, or subcellular site of hydrolysis. A group of these compounds with emerging significance, hexosamine analogs with ester-linked short chain fatty acid (SCFA) groups used in metabolic glycoengineering (illustrated by Ac4ManNAc and Bu4ManNAc, or their “R”-modified analogs), face a conflicting set of challenges to be used successfully in both in vitro and animal experiments as outlined in Figure 1. Metabolic glycoengineering originated with efforts to exploit N-acyl modified monosaccharides – epitomized by using N-acetylmannosamine (ManNAc analogs) to target the sialic acid pathway – to redecorate the cell surface with non-natural sugar epitopes.2,3 The versatility of metabolic glycoengineering was expanded considerably when the Bertozzi group used monosaccharide analogs to install bioorthogonal chemical functional groups into the glycocalyx of living cells in the form of ketone4 and azide groups.5 Finally, the chemical options available through this technique have continued to expand with the recent metabolic incorporation of thiols,6 aryl azides,7 alkynes,8 and diazarines9 into cell surface displayed glycans.
Figure 1. Overview of extra- and intracellular processing of SCFA-hexosamine analogs.
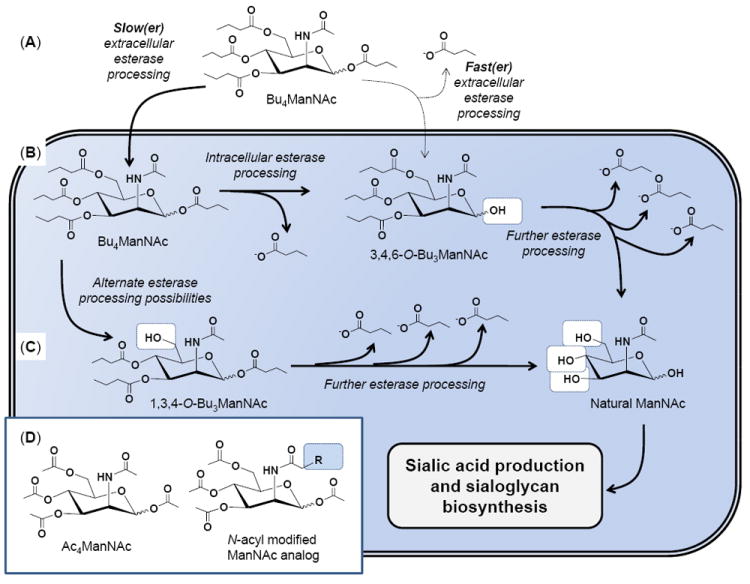
(A) The lead compound Bu4ManNAc is used for illustrative purposes; if extracellular esterase/lipase degradation is relatively slow (left) then the intact molecule will enter the cell; alternately if ester hydrolysis is relatively fast (right) a partially degraded metabolite such as 3,4,6-O-Bu3ManNAc will enter the cell (B). In either case, the butanoylated analog is subject to further esterase processing to ultimately produce fully deprotected ManNAc (B), which can also be derived from 1,3,4-O-Bu3ManNAc (or other partially deprotected intermediates, not shown) (C) and used by the sialic acid biosynthetic pathway (as described in detail elsewhere14,35) to produced sialic acid and sialylated glycans. (D) Similar esterase processing occurs for peracetylated ManNAc (illustrated) and its hydrolysis products as well as to N-acyl, R-modified analogs (such as the azido analogs shown in Fig. 2D).
There are many potential biomedical applications for metabolic glycoengineering ranging from imaging, diagnosis, to the treatment of diseases ranging from cancer10,11 to viral infections12 with additional uses reviewed elsewhere.13,14 Unfortunately the in vivo use of non-natural monosaccharide analogs has been slowed by low efficiency by which cells take up and metabolize these compounds; typically concentrations in the tens of millimolar are required in cell culture, which translates to unacceptably high doses for large animal studies. This pitfall has been rectified in part by the introduction of short chain fatty acid (SCFA) protecting groups, most commonly acetate, into analog design to increase cellular uptake by 600 to 900-fold.15,16 We have found that metabolic efficiency can be increased further, by ~1,800 and 2,100 fold for propionate- and butanoyl-modified hexosamines, respectively.17 This protecting group strategy, which renders the core sugar more lipophilic and thus increases plasma membrane permeability, is contingent on the hope that the SCFA moieties will be ignored by extracellular esterases and lipases and reach the targeted cell unscathed.
The extracellular stability of a SCFA-protected monosaccharide prodrug is important for two reasons. First, when the primary intention is for the compound to enter a cell and intercept a glycosylation pathway, the SCFA moieties that enhance membrane permeability must remain attached to the core sugar until the analog reaches the membrane of the target cell. Alternately, if the intention is to elicit “whole molecule” therapeutic effects exhibited by some analogs (e.g., by Bu4ManNAc or 3,4,6-O-Bu3ManNAc (Fig. 1B) as described in more detail below and in other publications18-20), the exact pattern of regioisomeric SCFA derivatization must remain intact until the analog has time to interact with a target cell. To illustrate this concept with a specific example, if the C6-substitutent of Bu4ManNAc were hydrolyzed extracellularly, certain of the resulting molecules (e.g., 1,3,4-O-Bu3ManNAc, Fig. 1C) would not be able to elicit the anti-oncogenic properties held by the parent molecule.21 Instead, this analog is a facile intermediate for further debutanoylation and incorporation into the sialic acid pathway and thus actually increases the oncogenic propensities of late stage cancer cells by increasing the sialylation of adhesion molecules implicated in metastasis.22
Once a sugar analog used in metabolic glycoengineering is taken into a target cell, the SCFA protecting groups must be fully removed from the core sugar to intercept the targeted glycosylation pathway (which can also, in addition to sialic acid pathway targeted by ManNAc analogs in the current study, include the biosynthetic routes for fucose, GalNAc, or O-GlcNAcylation14). Thus, unlike in the extracellular milieu where the analog will ideally be refractory to hydrolysis of the SCFA groups, rapid deprotection inside the cell is necessary for facile metabolic glycoengineering of cellular glycans. This paper addresses these issues by providing experimental evidence that esterase/lipase inactivation of SCFA-derivatized ManNAc analogs is relatively slow outside of a cell (Fig. 1A) allowing these compounds to be used for routine in vitro cell culture and, most likely in vivo applications, without undue concern for rapid degradation. By contrast, once taken up by a cell, esterase processing of analogs is relatively fast; or, to put it another way, cells have ample esterase activity to ensure that analog processing is not the rate limiting step for the incorporation of SCFA-derivatized ManNAc analogs into the sialic acid pathway.
In the first set of experiments a functional assay was used to monitor the effects of analog incubation in fetal bovine serum. This assay, which has been described in detail previously in our study of Ac4ManNAc uptake from tissue culture medium and incorporation into the sialic acid biosynthetic pathway16 monitors the removal of SCFA groups, which reduces cellular uptake of the core sugar and subsequently results in a measurable decrease in sialic acid biosynthesis compared to control cells incubated with non-esterase or serum-treated analog. Ac4ManNAc was refractory to serum inactivation over a 48 h time period (Fig. 2A), thereby providing evidence that peracetylated monosaccharides widely used in metabolic glycoengineering are sufficiently resistant to serum esterases for a wide range of in vivo applications. For example, in some applications such as treatment of hereditary inclusion body myopapthy (HIBM) with exogenously delivery ManNAc, sialic acid production peaked as soon as four hours after administration of Ac4ManNAc23 while in other cases the administration of SCFA-derivatized ManNAc analogs, e.g., Ac4ManNProp, required repeated daily administration over several weeks to achieve maximal in vivo replacement of natural sialic acids with the non-natural, metabolically engineered counterparts.24
Figure 2. “Extracellular” esterase processing time course comparing various SCFA-derivatized ManNAc analogs.
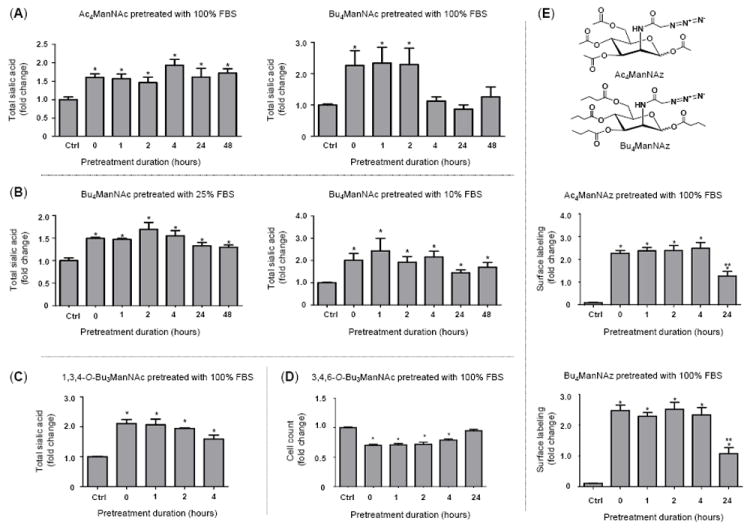
Several lines of evidence establish that SCFA-ManNAc analogs are relatively refractory against extracellular esterase/lipase hydrolysis: (A) Enhanced sialic acid production emanating from FBS-treated Ac4ManNAc continued for much longer (e.g., ≥48 h) compared to Bu4ManNAc (e.g., for ~ 2 to 4 h); (B) Under conditions encountered in typical cell culture, e.g., with 10% (or even 25%) FBS, butanoylated ManNAc was resistant to esterases/lipase degration. Tributanoylated ManNAc analogs showed moderate resistance to FBS degradation with (C) the “1,3,4” analog evaluated in the sialic acid flux assay used in panels A and B while (D) the “3.4,6” analog was tested by evaluating enhanced cytotoxity of analogs with this particular SAR, as measured by cell counts as previously described.18 (E) Azido-derivatived ManNAc analogs are shown (note that the structures of analogs used in panels A-C are given in Figure 1) along with surface labeling of Ac4ManNAz and Bu4ManNAz that had been pretreated with FBS. Detection of surface azido groups was performed by flow cytometry as previously described25 but by using single step labeling with Acetylene-Fluor 488 (Cat. No. TA106-25, Click Chemistry Tools, Scottsdale, AZ). Analogs were synthesized and characterized as previously described: Ac4ManNAc and Bu4ManNAc;17 1,3,4-O-Bu3ManNAc and 3,4,6-O-Bu3ManNAc;18 and Ac4ManNAz and Bu4ManNAc.25 All data shown represents a minimum of n = 3 replicates; the “*” symbol indicates p < 0.05 compared to the control and the double “**” symbols in panel E indicate a statistical difference (p < 0.05) at 24 h compared to both the control and earlier time points.
Despite the serum stability of acetylated ManNAc analogs, butanoylated analogs have the advantage of 3- to 4-fold more efficient metabolic incorporation17,25 and importantly, offer added bioactivity of potential therapeutic value. For example, hydrolyzed n-butyrate can provide therapeutic benefit by functioning as a histone deacetylase inhibitor.26,27 In addition, “whole molecule” activity – where biological responses emanate from the intact compound before esterase processing occurs – exemplify an emerging concept that sugar-based templates offer an attractive platform for drug development;28 the whole molecule effects of butanoylated hexosamines include suppression of NF-κB and metastatic oncogenes21 and are described in more detail elsewhere.18-20 In the current work we found that fully n-butanoylated ManNAc (Bu4ManNAc) was more rapidly inactivated by serum esterases than Ac4ManNAc, with the butanoylated analog showing a measurable reduction in sialic acid production in indicator cells after exposure to FBS for less than four hours while the acetylated sugar was stable for 48 h, the maximal time interval tested (Fig. 2A). The reduction in sialic acid production observed for Bu4ManNAc required the removal of at least 2 of the 4 butyrate groups during the pre-incubation period, which renders the dibutanoylated analog membrane impermeable.18 By contrast, lower concentrations of FBS (e.g., 10% or 25%) did not result in a statistically significant reversion of Bu4ManNAc-enhanced sialic acid production in the indicator cells (Fig. 2B). These results have two implications: one is that under typical in vitro cell culture conditions, extracellular degradation of butanoylated analogs is not a substantial concern. Conversely, extrapolating the 100% serum treatment condition to the in vivo milieu (because, disregarding the cells present, blood can be regarded as 100% serum), the degradation of butanoylated analog after only a few hours shows that the in vivo stability of these compounds is a potential concern when translating metabolic glycoengineering to animal studies. However, pending detailed pharmacokinetic evaluation (which is beyond the scope of the present report), the half life of butanoylated analogs is “in the ballpark” for at least some in vivo applications (e.g., the ~ four hour kinetics for maximal sialic acid production observed in the Ac4ManNAc-treated hereditary inclusion body myopathy mouse model23).
The finding that Bu4ManNAc likely has only marginally sufficient serum stability for effectively eliciting “whole molecule” effects in vivo raised the concern that tributanoylated analogs with otherwise favorable properties (for example, 3,4,6-O-Bu3ManNAc has stronger “whole molecule” responses than Bu4ManNAc19,20 and 1,3,4-O-Bu3ManNAc has “high flux” properties with negligible side effects such as cytotoxicity18,25) might be even more rapidly inactivated by extracellular esterases. To evaluate this possibility, we compared 1,3,4-O-Bu3ManNAc and 3,4,6-O-Bu3ManNAc in relevant assays. First the high flux analog 1,3,4-O-Bu3ManNAc did not lose its ability to supplement sialic acid production noticeably faster than Bu4ManNAc, showing only a relatively minor reduction in analog-enhanced sialic acid production after four hours of serum treatment (Fig. 2C), which was similar to the kinetics observed for the perbutanoylated analog (as shown in Fig. 2B). Similarly when growth inhibition, a “whole molecule” property of 3,4,6-O-Bu3ManNAc,18 was tested by monitoring the number of cells present after various time intervals of serum pretreatment, comparable kinetics were observed where loss of analog-enhanced cell growth inhibition began to be manifest between two and four hours, with a complete reversal after 24 hours (Fig. 2D).
As a final test of the extracellular esterase processing of ManNAc analogs, we evaluated azido-modified analogs pioneered by the Bertozzi group who first exploited them in modified Staudinger bio-orthogonal reactions;5 these compounds are now increasingly being used for metabolic glycoengineering because of their ability to be exploited in click reactions after installation into cellular glycoconjugates. By testing surface expression of azido-modified sialic acids in indicator cells, both Ac4ManNAz and Bu4ManNAz were refractory to a loss of signal over the first four hours of FBS treatment but did experience a reduction, but not complete reversal, of surface expression after 24 hours of pre-treatment (Fig. 2E). This assay suggested that the butanoylated analog Bu4ManNAz was more refractory to serum deactivation than Bu4ManNAc, possibly because the presence of the N-azido group interfered with binding of the butanoylated analog to the esterase(s). Despite this ambiguity, this result established that azido-modified analogs are not rapidly (e.g., within minutes) deactivated by serum enzymes but instead should again have sufficient longevity for in vivo applications using once a day (or less frequent) dosing regimens.
The requirement that intracellular esterase activity – in contrast to extracellular esterase activity as outlined above – occurs rapidly and thus is not a rate limiting step for sialic acid production was confirmed experimentally by monitoring production of this monosaccharide in cells treated with butanoylated ManNAc analogs over early time points. This issue is relevant because in organisms such as Escherichia coli strains that lack robust non-specific esterase activity, the strategy of providing esterified precursors for metabolic engineering is not viable.29 In our experiments in human cells, the perbutanoylated analog Bu4ManNAc, the “high flux” analog 1,3,4-O-Bu3ManNAc, and the 3,4,6-O-Bu3ManNAc analog with enhanced “whole molecule” effects all gave rise to statistically identical levels of increased sialic acid production over the first 12 hours (Fig. 3A). These data diverge from previous results that showed that each compound supported different levels of sialic acid production when measured after 2 to 3 days of exposure to analog; for example, 1,3,4-O-Bu3ManNAc was dubbed the “high flux” analog because of the very high sialic acid levels found in cells after 48 to 72 hours or exposure.18,25 The current results indicate that this analog does not necessarily support a higher rate of metabolic flux per se, but instead the high levels of sialic acid observed in analog-treated cells after extended exposure are likely a consequence of the lack of cytotoxicity of this analog. For context, analogs that inhibit the robust growth of cells reduce ManNAc analog-mediated sialic acid production16,17,30 and the lower amounts of sialic acid observed after 2 or 3 days of exposure to Bu4ManNAc or 3,4,6-O-Bu3ManNAc likely result from the cytotoxicity of these analogs and not because esterase processing is a rate limiting step for incorporation into the sialic acid pathway. Instead, in mammalian cells downstream enzymes such as sialic acid synthase likely comprise a bottleneck for ManNAc-supported flux through the sialic acid biosynthetic pathway.31,32
Figure 3. Intracellular esterase processing and an explanation for slow extracellular and fast intracellular analog hydrolysis.
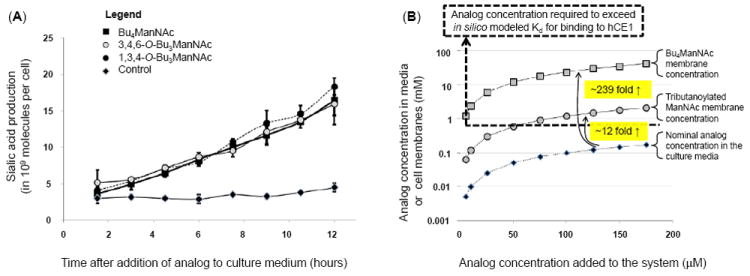
(A) Sialic acid production in LS174T “indicator cells” incubated with different forms of butanoylated ManNAc analogs shows statistical no difference over the first several hours. (B) Theoretical explanation for the slow extracellular and rapid intracellular processing of SCFA-ManNAc analogs: tributanoylated and Bu4ManNAc concentrations are estimated in solution and in (or near) membranes based on logP values calculated with ChemDraw. The Kd for association with hCE1 (a plausible model esterase) indicated with the dashed line, showing that the nominal concentration for analog in solution in media is typically below this threshold resulting in slow extracellular hydrolysis of SCFA groups while membrane-associated analog is typically at concentrations higher than this threshold, thus explaining the rapid intracellular hydrolysis of SCFA groups.
To address a final loose end, we were curious about the apparent disparity (albeit a fortunate one) where esterase cleavage of SCFA groups was much slower extracellularly (e.g., at least four hours was generally needed to remove one or two SCFA groups, Fig. 2) than intracellularly (where, as shown in Figure 3A, sialic acid production was observed within two hours for butanoylated ManNAc analogs. We have previously reported similar kinetics for intracellular sialic acid production from Ac4ManNAc33, which is only possible upon complete removal of SCFA groups from the core sugar. One explanation for these results is that the enzymes implicated in detoxification and most likely analog processing, such as human carboxylesterase 1 (hCE1), are intracellular while extracellular enzymes (e.g., butylcholinesterases) are not capable of facile hydrolysis of SCFA from monosaccharides. A second non-exclusive explanation is based on in silico docking simulations performed with AutoDock (following previously described procedures19) to evaluate the interaction of Ac4ManNAc (-2.9 kcal/mol and Kd = 7.2 mM) and Bu4ManNAc (-4.1 kcal/mol and Kd = 0.9 mM) with hCE1. While relatively weak, these binding values fall within the range of other carbohydrate ligands docked to proteins,34 making these results plausible and thereby providing an attractive mechanism to explain why SCFA-ManNAc analogs are hydrolyzed slowly outside of a cell when in solution but rapidly inside a cell.
To explain in more detail, analog concentrations are sketched in Figure 3B, showing that the nominal concentration in solution in culture medium is below the Kd for binding to hCE1 (e.g., 0.9 mM for Bu4ManNAc) at concentrations typically used in metabolic glycoengineering experiments (e.g., <200 μM). As a result, the extracellular processing of the analog is slow and inefficient. Considering, however, that cellular membranes act as sinks for peracetylated ManNAc analogs16 and this effect is exacerbated for the more lipophilic butanoylated analogs, we were able to extrapolate membrane concentrations for these compounds based on log P values calculated by ChemDraw. Specifically, tributanoylated analog concentration is expected to be enriched at least 12-fold in membranes compared to aqueous solution with an additional ~20 fold gain (to ~239 fold in total) for perbutanoylated analogs. Because hCE1 is a membrane associated enzyme, it is reasonable that the local concentration of analog near membranes thereby becomes sufficiently high to meet or exceed the in silico predicted Kd for this enzyme for concentrations of butanoylated analogs added to culture media in the 25 to 200 μM range typically used. Overall, although speculative at present, this mechanism satisfactorily explains the slow extracellular processing of analog (because, even if appropriate esterases are present in serum, analog concentrations in solution are considerably below the Kd, resulting in slow processing) as well as the facile intracellular processing (as just explained by the membrane sink effect).
In summary, this report provides experimental evidence based on an in vitro model system that two potential pitfalls for the in vivo use of metabolic glycoengineering substrates – the potential rapid extracellular or the too slow intracellular esterase-mediated removal of the SCFA groups – are not a major concern. Consequently, this work provides a foundation for the continued development of metabolic glycoengineering by establishing a rationale for expanded translation from the “chemical glycobiology” origins of this methodology into more disease oriented animal model systems and biomedical applications.
Acknowledgments
Funding for this paper was provided by the National Institutes of Health, grants R01CA112314 and R01AR054005.
Abbreviations
- FBS
fetal bovine serum
- GalNAc
N-acetyl-d-galactosamine
- GlcNAc
N-acetyl-d-glucoosamine
- hCE1
human carboxylesterase 1
- ManNAc
N-acetyl-d-mannosamine
- SCFA
short chain fatty acids
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lavis LD. ACS Chem Biol. 2008;3:203. doi: 10.1021/cb800065s. [DOI] [PubMed] [Google Scholar]
- 2.Kayser H, Zeitler R, Kannicht C, Grunow D, Nuck R, Reutter W. J Biol Chem. 1992;267:16934. [PubMed] [Google Scholar]
- 3.Yarema KJ, Mahal LK, Bruehl RE, Rodriguez EC, Bertozzi CR. Metabolic delivery of ketone groups to sialic acid residues. J Biol Chem. 1998;273:31168. doi: 10.1074/jbc.273.47.31168. [DOI] [PubMed] [Google Scholar]
- 4.Mahal LK, Yarema KJ, Bertozzi CR. Science. 1997;276:1125. doi: 10.1126/science.276.5315.1125. [DOI] [PubMed] [Google Scholar]
- 5.Saxon E, Bertozzi CR. Science. 2000;287:2007. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- 6.Sampathkumar S-G, Li AV, Jones MB, Sun Z, Yarema KJ. Nat Chem Biol. 2006;2:149. doi: 10.1038/nchembio770. [DOI] [PubMed] [Google Scholar]
- 7.Han S, Collins BE, Bengtson P, Paulson JC. Nat Chem Biol. 2005;1:93. doi: 10.1038/nchembio713. [DOI] [PubMed] [Google Scholar]
- 8.Sawa M, Hsu T-L, Itoh T, Sugiyama M, Hanson SR, Vogt PK, Wong C-H. Proc Natl Acad Sci U S A. 2006;103:12371. doi: 10.1073/pnas.0605418103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanaka Y, Kohler JJ. J Am Chem Soc. 2008;130:3278. doi: 10.1021/ja7109772. [DOI] [PubMed] [Google Scholar]
- 10.Mahal LK, Yarema KJ, Lemieux GA, Bertozzi CR. In: Sialobiology and Other Novel Forms of Glycosylation. Inoue Y, Lee YC, Troy FA III, editors. Gakushin Publishing Company; Osaka, Japan: 1999. p. 273. [Google Scholar]
- 11.Chefalo P, Pan Y, Nagy N, Guo Z, Harding CV. Biochemistry. 2006;45:3733. doi: 10.1021/bi052161r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keppler OT, Horstkorte R, Pawlita M, Schmidt C, Reutter W. Glycobiology. 2001;11:11R. doi: 10.1093/glycob/11.2.11r. [DOI] [PubMed] [Google Scholar]
- 13.Campbell CT, Sampathkumar S-G, Weier C, Yarema KJ. Mol Biosyst. 2007;3:187. doi: 10.1039/b614939c. [DOI] [PubMed] [Google Scholar]
- 14.Du J, Meledeo MA, Wang Z, Khanna HS, Paruchuri VDP, Yarema KJ. Glycobiology. 2009;19:1382. doi: 10.1093/glycob/cwp115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarkar AK, Fritz TA, Taylor WH, Esko JD. Proc Natl Acad Sci U S A. 1995;92:3323. doi: 10.1073/pnas.92.8.3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones MB, Teng H, Rhee JK, Baskaran G, Lahar N, Yarema KJ. Biotechnol Bioeng. 2004;85:394. doi: 10.1002/bit.10901. [DOI] [PubMed] [Google Scholar]
- 17.Kim EJ, Sampathkumar S-G, Jones MB, Rhee JK, Baskaran G, Yarema KJ. J Biol Chem. 2004;279:18342. doi: 10.1074/jbc.M400205200. [DOI] [PubMed] [Google Scholar]
- 18.Aich U, Campbell CT, Elmouelhi N, Weier CA, Sampathkumar SG, Choi SS, Yarema KJ. ACS Chem Biol. 2008;3:230. doi: 10.1021/cb7002708. [DOI] [PubMed] [Google Scholar]
- 19.Elmouelhi N, Aich U, Paruchuri VDP, Meledeo MA, Campbell CT, Wang JJ, Srinivas R, Khanna HS, Yarema KJ. J Med Chem. 2009;52:2515. doi: 10.1021/jm801661m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Z, Du J, Che P-L, Meledeo MA, Yarema KJ. Curr Opin Chem Biol. 2009;13:565. doi: 10.1016/j.cbpa.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campbell CT, Aich U, Weier CA, Wang JJ, Choi SS, Wen MM, Maisel K, Sampathkumar S-G, Yarema KJ. J Med Chem. 2008;51:8135. doi: 10.1021/jm800873k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Almaraz RT, Tian Y, Bhattarcharya R, Tan E, Chen S-H, Dallas MR, Chen L, Zhang Z, Zhang H, Konstantopoulos K, Yarema KJ. Mol Cell Proteomics. 2012 doi: 10.1074/mcp.M1112.017558. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malicdan MCV, Noguchi S, Tokutomi T, Goto Y-i, Nonaka I, Hayashi YK, Nishino I. J Biol Chem. 2012;287:2689. doi: 10.1074/jbc.M111.297051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gagiannis D, Gossrau R, Reutter W, Zimmermann-Kordmann M, Horstkorte R. Biochim Biophys Acta. 2007;1770:297. doi: 10.1016/j.bbagen.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 25.Almaraz RT, Aich U, Khanna HS, Tan E, Bhattacharya R, Shah S, Yarema KJ. Biotechnol Bioeng. 2012;109:992. doi: 10.1002/bit.24363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sampathkumar S-G, Campbell CT, Weier C, Yarema KJ. Drug Future. 2006;31:1099. [Google Scholar]
- 27.Sampathkumar S-G, Jones MB, Meledeo MA, Campbell CT, Choi SS, Hida K, Gomutputra P, Sheh A, Gilmartin T, Head SR, Yarema KJ. Chem Biol. 2006;13:1265. doi: 10.1016/j.chembiol.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 28.Meutermans W, Le GT, Becker B. ChemMedChem. 2006;1:1164. doi: 10.1002/cmdc.200600150. [DOI] [PubMed] [Google Scholar]
- 29.Antonczak AK, Simova Z, Tippmann EM. J Biol Chem. 2009;284:28795. doi: 10.1074/jbc.M109.027409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim EJ, Jones MB, Rhee JK, Sampathkumar S-G, Yarema KJ. Biotechnol Prog. 2004;20:1674. doi: 10.1021/bp049841q. [DOI] [PubMed] [Google Scholar]
- 31.Jacobs CL, Goon S, Yarema KJ, Hinderlich S, Hang HC, Chai DH, Bertozzi CR. Biochemistry. 2001;40:12864. doi: 10.1021/bi010862s. [DOI] [PubMed] [Google Scholar]
- 32.Viswanathan K, Lawrence S, Hinderlich S, Yarema KJ, Lee YC, Betenbaugh M. Biochemistry. 2003;42:15215. doi: 10.1021/bi034994s. [DOI] [PubMed] [Google Scholar]
- 33.Yarema KJ. In: Cell Engineering 3. Glycosylation. Al-Rubeai M, editor. Vol. 3. Kluwer Academic Publishers; Dordrecht, The Netherlands: 2002. p. 171. [Google Scholar]
- 34.Laederach A, Reilly PJ. J Comput Chem. 2003;24:1748. doi: 10.1002/jcc.10288. [DOI] [PubMed] [Google Scholar]
- 35.Tanner ME. Bioorg Chem. 2005;33:216. doi: 10.1016/j.bioorg.2005.01.005. [DOI] [PubMed] [Google Scholar]