

Abstract
A potent and selective inhibitor of KCNQ2, (S)-5 (ML252, IC50 = 69 nM), was discovered after a high-throughput screen of the MLPCN library was performed. SAR studies revealed a small structural change (ethyl group to hydrogen) caused a functional shift from antagonist to agonist activity (37, EC50 = 170 nM), suggesting an interaction at a critical site for controlling gating of KCNQ2 channels.
Keywords: KCNQ2, inhibitor, Kv7, ion channels, ML252, MLPCN probe, mode switch, CNS
Alzheimer’s disease (AD) is the most common neurodegenerative disorder, and the most common form of dementia, affecting a worldwide population of >20 million people. One of the major conditions of the disease is the decline of memory and loss of cognitive function leading to significant burden on the families of patients and the medical community with an estimated cost of more than $300 billion.1 Although there is no known cure, nor are there drugs that slow the progression of AD, there have been FDA approved drugs to help lessen the symptoms – though these drugs are only effective for a limited time frame. One class of drugs that has been approved are the cholinesterase inhibitors: compounds that prevent the break-down of acetylcholine (ACh).2 There has been significant research describing the loss of cholinergic tone in AD patients, which is correlated well with the progression of dementia2 and provides a rationale for therapeutic modulation of the cholinergic system.
In contrast to targeting cholinesterase inhibition, one area of research that has gained significant momentum over the past several years is use of voltage-gated ion channel modulators, specifically, potassium (K+) channel inhibitors, to enhance ACh release.2-3 KCNQ (also known as Kv7) channels belong to the voltage gated K+ channel superfamily, and contain five members (Kv7.1 – K v7.5) with each member containing six transmembrane segments. KCNQs are expressed in both the peripheral and central nervous systems (PNS and CNS), with the exception of Kv7.1 (KCNQ1) which is predominantly restricted to cardiac tissue, peripheral epithelial cells and smooth muscle cells. A number of compounds that modulate KCNQ2 channels have shown promise in pre-clinical models of memory enhancement (1, 1a, 24), with both 1a and 2 being advanced into clinical trials.5 A more recent example has been reported as a subtype selective blocker of KCNQ channels, 3.6 Unfortunately, the reported selectivity of these compounds is less than ideal and thus we embarked on a campaign to search for potent and selective KCNQ2 inhibitors.
The project initiated with a screen of the >300,000 NIH Molecular Library Small Molecule Repository (MLSMR) compound collection using a thallium influx assay (PubChem AID: 2156) to identify inhibitors of the KCNQ2 channel.7 Thallium can be used as a surrogate ion for potassium flux as it can permeate many potassium selective ion channels. Thallium influx was detected with a fluorescent dye (FluxOR) in a 384-well format.8 From the initial library screen, ~3,400 compounds were deemed “hits” and after a round of triage, ~1,000 compounds were counter screened against parental cells (PubChem AID: 493029) and cells expressing KCNQ1. From this round of results, 553 compounds were reconfirmed against KCNQ2 channels utilizing an automated patch clamp assay (PubChem AID: 588531) to yield 58 confirmed KCNQ2 inhibitors. From these experiments, compounds 4 and 5 were selected as starting “leads” compounds for medicinal chemistry (Table 1). These initial compounds displayed a significant SAR improvement moving from the 2-phenyl (4, 4.7 μM) to 2-pyrrolidine (5, 0.16 μM). As KCNQ2 inhibitors have been shown to be effective in enhancing cognition in some CNS behavioral models, these compounds represent excellent starting points for a CNS indication (MW ~ 300, cLogP <4.5, tPSA <40).
Table 1.
HTS Validated hits.
| 4 SID: 17415222 CID: 319496 |
5 SID: 49820623 CID: 24248347 |
|
|---|---|---|

|

|
|
| IC50 (μM) | 4.66 ± 1.06 | 0.161 ± 0.015 |
The initial SAR assessment started with evaluation of the right-hand portion of the molecule, keeping the 2-(pyrrolidin-1-yl)aniline portion constant (Table 2). The initial hit, 5, was resynthesized and reconfirmed from powder (IC50 = 163 nM).9 Modification of the ethyl side-chain led to unexpected results. Truncating the ethyl to a methyl group retained activity (6, 260 nM); however, deletion of the ethyl group or replacement with a single fluorine led to a mode switch resulting in potent KCNQ2 activators (7, EC50 = 743 nM; 8, EC50 = 990 nM). To the best of our knowledge, this is the first report of a “mode switch” in KCNQ2 channel modulators and the SAR will be explored further (vide infra). Extending the chain into the isopropyl group was not tolerated (9); however, activity could be returned by cyclizing into the cyclopropyl group (10, 3500 nM) with reduced activity compared to 5. Further chain extension into the sec-butyl group also was deleterious compared to 5 (11, 2220 nM). Interestingly, deletion of a methyl group, leaving the straight-chain propyl group was well tolerated (12, 320 nM). Cyclization of the pendant side-chain to the phenyl group resulting in the indane structure was also tolerated (13, 330 nM). Disubstitution on the methylene group with either a gem-dimethyl or cyclopropyl was not tolerated (14 and 15), nor was the 6-membered chromane structure (16). Lastly, reversing the amide moiety or alkylation of the amide resulted in complete loss of activity (17 and 18).
Table 2.
Initial SAR analysis of right-hand portion.

|
||
|---|---|---|
| Entry | R | KCNQ2 IC50 (μM)10 |
| 5 |

|
0.163 ± 0.042 |
| 6 |

|
0.26 ± 0.064 |
| 7 |

|
0.743 ± 0.225* (activator) |
| 8 |

|
0.99 ± 0.77* (activator) |
| 9 |

|
(−25.41%) |
| 10 |

|
3.50 ± 1.66 |
| 11 |

|
2.22 ± 1.35 |
| 12 |

|
0.32 ± 0.06 |
| 13 |

|
0.33 ± 0.05 |
| 14 |

|
>30 |
| 15 |

|
>30 |
| 16 |

|
>30 |
| 17 |

|
(−38%) |
| 18 |

|
>30 |
Next, we investigated the right-hand phenyl portion of the molecule utilizing the ethyl substituted inhibitor scaffold as well as the unbranched methylene activator scaffold in order to evaluate both the SAR surrounding both modes of pharmacology (Table 3). For the inhibitor scaffold, most of the substituents evaluated were well tolerated leading to nanomolar compounds. There were, however, a couple of exceptions. Namely, 3,4-dimethoxy substituents were not tolerated (19, 20.2 μM), a 125-fold loss of potency; and the 2-chloro substituent led to a 20-fold loss of potency (30, 3.3 μM). All of the other substitutions (halogen, trifluoromethyl, methoxy) led to compounds that retained activity similarly (2 – 3-fold loss of activity) to the unsubstituted phenyl group. Two compounds were of equal potency to the phenyl group, 4-fluoro (26, 130 nM) and 3-methoxy (34, 120 nM). It is of note that the 3,4-dimethoxy compound had a 125-fold loss of potency as both the 4-methoxy (38, 380 nM) and 3-methoxy (34, 120 nM) were individually very potent compounds. The activator SAR did not track exactly with the inhibitor SAR; however, a number of submicromolar compounds were discovered. The most active compound was the 3-chloro substituent (37, 170 nM). Unlike the inhibitors, the 3-methoxy (35, 2350 nM) and 4-methoxy (39, 1750 nM) activators were much less potent than the best compound. Although there was a diminution in potency for some compounds, for the most part, small substituents were tolerated on the right-hand phenyl group.
Table 3.
SAR evaluation of the phenyl substituent on the inhibitor and activator scaffolds.

|
||||
|---|---|---|---|---|
|
| ||||
| KCNQ2 (μM)10 | ||||
| Entry | R | Ri | IC50 | EC50 |
| 19 | 3,4-dimethoxy | Et | 20.23 ± 4.89 | |
| 20 | 3,4-difluoro | Et | 0.17 ± 0.02 | |
| 21 | H | 1.73 ± 0.34 | ||
| 22 | 3-fluoro | Et | 0.36 ± 0.03 | |
| 23 | H | 0.49 ± 0.12 | ||
| 24 | 3-methyl | Et | 0.33 ± 0.05 | |
| 25 | H | 1.32 ± 0.29 | ||
| 26 | 4-fluoro | Et | 0.13 ± 0.03 | |
| 27 | H | 0.70 ± 0.82 | ||
| 28 | 3,4-dichloro | Et | 0.47 ± 0.08 | |
| 29 | H | 1.07 ± 0.34 | ||
| 30 | 2-chloro | Et | 3.31 ± 0.87 | |
| 31 | H | 0.95 ± 0.27 | ||
| 32 | 3-trifluoromethyl | Et | 0.39 ± 0.14 | |
| 33 | H | 0.35 ± 0.21 | ||
| 34 | 3-methoxy | Et | 0.12 ± 0.03 | |
| 35 | H | 2.35 ± 2.25 | ||
| 36 | 3-chloro | Et | 0.24 ± 0.04 | |
| 37 | H | 0.17 ± 0.12 | ||
| 38 | 4-methoxy | Et | 0.38 ± 0.06 | |
| 39 | H | 1.75 ± 0.59 | ||
| 40 | 4-chloro | Et | 0.28 ± 0.03 | |
| 41 | H | 0.80 ± 0.31 | ||
| 42 | 2-fluoro | Et | 0.44 ± 0.09 | |
| 43 | H | (26.6%) | ||
After establishing the optimal right-hand substituent for the inhibitor (5) and activators (37), we next turned our attention to the left-hand portion of the molecule (Table 4). The initial HTS hits displayed a preference for the 2-pyrrolidine versus the 2-phenyl which was confirmed upon resynthesis (4, 1320 nM; 5, 163 nM). Moving the 2-pyrrolidine group around the ring produced orthogonal results when comparing the inhibitor and activator scaffolds. The 3-pyrrolidine substituent was completely devoid of activity in the inhibitor series (44, >30,000 nM) while the activator series retained some activity (although with a ~5-fold loss of activity, 45, 1020 nM). Further movement to the 4-position resulted in reversal of activity, where the inhibitor retained all activity (46, 210 nM) and the activator lost significant activity (47, 8660 nM). Enlarging the ring system to the 2-piperidine resulted in loss of activity in both series (48 and 49); although the activator series did retain weak activity (7090 nM). Further expansion of the ring to the seven-membered ring led to total loss of activity. Any other modification of the 2-(pyrrolidin-1-yl) ring system led to much reduced activity or complete loss of activity in both the inhibitor and activator series.
Table 4. SAR evaluation of the left-hand phenyl substituent on the inhibitor and activator scaffolds.
|
|
||||
|---|---|---|---|---|
|
| ||||
| KCNQ2 (μM)10 | ||||
| Entry | R | R1 | R2 = Et, IC50 | R2 = H, EC50 |
| 5 | 2-(pyrrolidin-1-yl) | H | 0.163 ± 0.042 | |
| 37 | Cl | 0.17 ± 0.12 | ||
| 44 | 3-(pyrrolidin-1-yl) | H | >30 | |
| 45 | Cl | 1.02 ± 0.05 | ||
| 46 | 4-(pyrrolidin-1-yl) | H | 0.21 ± 0.032 | |
| 47 | Cl | 8.66 ± 1.73 | ||
| 48 | 2-(piperidin-1-yl) | H | (−32%) | |
| 49 | Cl | 7.09 ± 2.50 | ||
| 50 | 2-(azepan-1-yl) | H | >30 | |
| 51 | Cl | >30 | ||
| 52 | 2-morpholino | H | 12.67 ± 0.76 | |
| 53 | Cl | >30 | ||
| 54 | 2-(NI-ethyl-NI-methylamine) | H | >30 | |
| 55 | Cl | >30 | ||
| 56 | 2-(dimethylamine) | H | (−59.77%) | |
| 57 | Cl | 27.42 ± 1.80 | ||
| 58 | 2-(pyrrolidine-2,5-dione) | H | >30 | |
| 59 | 2-(1H-imidazol-1-yl) | H | >30 | |
| 60 | 2-(1H-pyrrol-1-yl) | H | 5.27 ± 0.62 | |
| 61 | Cl | 12.27 ± 1.37 | ||
| 62 | 2-(2,5-dimethyl-1H-pyrrol-1-yl) | H | >30 | |
| 63 | 2-methoxy | H | (−28.05%) | |
| 64 | 2-(NI-benzyl-NI-methylamine) | H | (−27.09%) | |
| 65 | Cl | >30 | ||
| 4 | 2-phenyl | H | 1.32 ± 0.46 | |
| 66 | 2-chloro | H | 14.99 ± 2.31 | |
| 67 |

|
>30 | ||
As the inhibitor series represents mixtures of enantiomers, we next turned our attention to the synthesis of the discrete enantiomers in order to determine if there is an enantiospecific bias within this series. In the three examples analyzed, the (S)-enantiomer proved to be more potent than the (R)-enantiomer, with (S)-5 being the most active compound (69 nM) (Supplemental Material, Table 1). This represents the most potent KCNQ2 inhibitor reported to date, and was declared an MLPCN probe (ML252).11
Next, we evaluated the most potent inhibitor ((S)-5), several close analogs, and known compounds for selectivity against KCNQ1 (Kv7.1) (Supplemental Material, table 2). Each of the known compounds was either mid-nanomolar or micromolar against KCNQ2 in the reported assay. However, in order to compare (S)-5 with these compounds, we chose to re-evaluate all compounds in a common assay system (PubChem AID: 588426). In this assay system, compounds 1 and 2 were more potent (~8 – 10-fold) compared to previous values; however, 3 was ~7-fold less potent. (S)-5 and all analogs were all >40-fold selective versus KCNQ1; whereas the previously reported compounds were either not selective, or in some instances, more potent versus KCNQ1. Furthermore, (S)-5 and the previously reported compounds were further evaluated against a panel of other Kv7.x channels and (S)-5 was more selective versus KCNQ1/E1 and all compounds were not selective versus KCNQ2/3 or KNCQ4, with the exception of 3. In addition to the selectivity performed in our laboratories, 5 has been tested in >300 assays performed within the MLPCN network; and was only active in four assays not dependent on KCNQ2 and was weakly active (>15 μM) in each of these assays (See Supplemental Material). Lastly, to more fully assess the selectivity of this probe molecule, (S)-5 was evaluated on Ricerca Bioscience’s Lead Profiling Screen, which is a radioligand binding assay profiling >68 GPCRs, ion channels and transporters. (S)-5 was found to bind with only one of the 68 assays conducted (melantonin, MT1, 61% inhibition at 10 μM). Several ion channels are included in this profiling assay (calcium channel, L- and N-type; potassium channel (KATP and hERG)) which were not inhibited by (S)-5, highlighting the selectivity profile of (S)-5.
Lastly, we evaluated (S)-5 and other close analogs in our Tier 1 in vitro PK assays including an assessment of intrinsic clearance (CLINT/CLHEP) in hepatic microsomes.12 Intrinsic clearance allows for the rank ordering of compounds with respect to their tendency to undergo oxidative metabolism, which provides a prediction for subsequent clearance in in vivo PK evaluation. All of the compounds evaluated in this assay showed high clearance in both human and rat microsomes (Supplemental Material, Table 3). As the unsubstituted right-hand phenyl group provides an attractive site for oxidation, compounds that contain substituents meant to block this oxidation were evaluated and all of these compounds also showed high clearance. For a better understanding of the nature of the instability of these compounds, we performed a metabolic soft-spot analysis using rat hepatocytes. This analysis showed that the major site of metabolism was on the pyrrolidine ring, α-methylene to the nitrogen. Unfortunately, our initial SAR evaluation showed that modification of this ring system was not well tolerated; thus potentially limiting the in vivo dosing regimen used for this probe molecule.
Although the metabolic stability of (S)-5 suggests it would not be a candidate for oral administration, due to the therapeutic importance of KCNQ2 inhibitors as CNS agents, we further profiled (S)-5 in a snapshot in vivo PK study to assess the plasma and brain levels after a single time point and single dose following IP administration (Table 5). (S)-5 was observed to be highly brain penetrant with a B:P ratio of 1.9 and absolute brain levels of 672 nM, nearly 10-fold greater than the IC50 value.
Table 5. In vitro and in vivo characterization of (S)-5.

| |
|---|---|
|
| |
| (S)-5 | |
| Parameter | |
|
| |
| MW | 308.4 |
| TPSA | 32.3 |
| cLogP | 4.27 |
|
| |
| In Vitro Pharmacology | IC50 (μM) |
|
| |
| KCNQ2 | 0.07 ± 0.01 |
| KCNQ1 | 2.92 ± 3.90 |
| KCNQ1/E1 | 8.12 ± 1.47 |
| KCNQ2/Q3 | 0.12 ± 0.02 |
| KCNQ4 | 0.20 ± 0.06 |
| CYP (1A2, 2C9, 3A4, 2D6) | 6.1, 18.9, 3.9, 19.9 |
|
| |
| In Vitro PK | |
|
| |
| Rat CLHEP (mL/min/kg) | 67.3 |
| rPPB (%fu) | 0.6 |
| rBHB (%fu) | 0.3 |
|
| |
| In Vivo PK, IP PBL (10 mg/kg, 1 hr) | |
|
| |
| Plasma (ng/mL) | 115.8 |
| Brain (ng/g) | 207.5 |
| B/P | 1.9 |
CONCLUSION
In summary, we have discovered a potent, selective and brain penetrant KCNQ2 inhibitor, (S)-5, from a high-throughput screening campaign of the MLSMR library collection. After IP dosing, (S)-5 shows brain levels significantly higher than the in vitro IC50 for the inhibition of KCNQ2 and thus can be used as an in vivo tool molecule. In addition, during the course of the SAR campaign, we discovered a site on (S)-5 where small structural changes caused a functional shift from antagonist to agonist. Further SAR of this series identified a potent KCNQ2 activator, 37, which is equipotent to many previously reported KCNQ2 activators, including those from our laboratories (ML213).13 Further studies are planned to evaluate the effects of (S)-5 on acetylcholine release and will be reported in due course.
EXPERIMENTAL SECTION
Chemistry
The synthesis of (S)-5 is described below. The general chemistry, experimental information, and syntheses of all other compounds are supplied in the Supporting Information. Purity of all final compounds was determined by HPLC analysis is >95%.
(S)-2-Phenyl-N-(2-(pyrrolidin-1-yl)phenyl)butanamide ((S)-5)
To a stirred solution of (S)-(−)-2-phenylbutyric acid (50 mg, 0.304 mmol), HATU (116 mg, 0.304 mmol) in DMF (1.5 mL) was added ethyldiisopropylamine (79 mL, 0.456 mmol) followed by 2-(1-pyrrolidinyl)aniline (49 mg, 0.304 mmol) and the reaction mixture was stirred at room temperature overnight. The reaction was diluted with water (1.5 mL) and extracted with EtOAc (2× 2mL). The organic extracts were combined, concentrated and the residue was purified by RP prepHPLC eluting with 10 to 90% CH3CN/H2O (0.1% TFA) to give the product as the TFA salt which was dissolved in methylene chloride (5 mL), washed with aqueous saturated sodium bicarbonate solution, dried (MgSO4), filtered and concentrated. The free base was dissolved in methylene chloride (2 mL) and treated with 4M HCl in dioxane (0.2 mL). After 20 min, the reaction mixture was concentrated to give the product (60 mg, 57%). 1H NMR (400 MHz, DMSO-d6) δ 10.10 (br s, 1 H), 7.52 – 7.41 (m, 3 H), 7.41 – 7.30 (m, 3 H), 7.31 – 7.10 (m, 3 H), 3.76 (t, J = 7.5 Hz, 1 H), 3.72 – 3.42 (m, 1 H), 3.84 (br s, 4 H), 2.20 – 2.02 (m, 1 H), 1.88 (br s, 4 H), 1.81 – 1.66 (m, 1 H), 0.89 (t, J = 7.3 Hz, 3 H). LCMS: RT = 0.66 min., >98% @ 215 nm and 254 nm, MS (ESI+) m/z = 309.3 [M + H]+. HRMS (TOF, ES+), calc’d for C20H25N2O [M + H]+: 309.1967, found 309.1969. Chiral HPLC: 94.2% (S)-isomer; RT = 3.97 min. 5.75% (R)-isomer; RT = 4.29 min. 88.4% ee.
Supplementary Material
Figure 1.
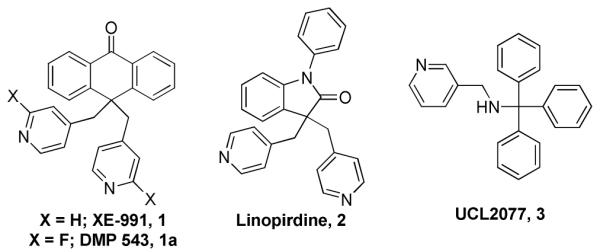
Structures of XE-991, DMP 543, Linopirdine and UCL2077.
ACKNOWLEDGMENT
The authors would like to thank Tammy Santomango, Frank Byers, and Ryan Morrison for technical assistance with the PK experiments and Nathan Kett and Sichen Chang for the purification of compounds. Vanderbilt is a member of the MLPCN and houses the Vanderbilt Specialized Chemistry Center for Accelerated Probe Development. Johns Hopkins is a member of the MLPCN and houses the Johns Hopkins Ion Channel Center.
Funding Sources
This work was generously supported by the NIH/MLPCN Grants R03 DA027716-01, U54 MH084691 (M.L.), and U54 MH084659 (C.W.L.).
ABBREVIATIONS
- HATU
2-(1H-7-azabenzotriazol-yl)-1,1,3,3-tetramethyl uranium hexafluorophosphate methanaminium
- SID
Pub-Chem structure ID
- CID
PubChem compound ID
- AID
PubChem assay ID
- CNS
central nervous system.
Footnotes
Author Contributions
Professors Lindsley and Hopkins directed and designed the chemistry and pharmacokinetic studies. Dr. Cheung performed the synthetic chemistry. Drs. Yu, Wu and Zou performed experiments. Drs. Lindsley, Hopkins, Li, McManus, Yu and Wu participated in writing or editing the manuscript
Mailing address: Department of Pharmacology, Vanderbilt University Medical Center, 2213 Garland Ave., Nashville, TN 37232, USA. (M.L.) Telephone: 410-614-5131. Fax: 410-614-1001. minli@jhmi.edu. Mailing address: Johns Hopkins University School of Medicine, 733 N. Broadway, BRB 319, Baltimore, MD 21205, USA.Present Addresses
REFERENCES
- 1. http://www.alz.org/alzheimers_disease_facts_and_figures.asp.
- 2.Camps P, Muñoz-Torrero D. Cholinergic drugs in pharmacotherapy of Alzheimer’s disease. Mini Rev. Med. Chem. 2002;2:11–25. doi: 10.2174/1389557023406638. [DOI] [PubMed] [Google Scholar]
- 3.Wickenden AD, Roeloffs R, McNaughton-Smith G, Rigdon GC. KCNQ potassium channels: drug targets for the treatment of epilepsy and pain. Expert Opin. Ther. Patents. 2004;14(4):1–13. [Google Scholar]
- 4.(a) Zaczek R, Chorvat RJ, Saye JA, Pierdomenico ME, Maciag CM, Logue AR, Rominger DH, Earl RA. Two new potent neurotransmitter release enhancers, 10,10-bis(4-pyridinylmethyl)-9(10H)-anthrancenone and 10,10-bis(2-fluoro-4-pyridinylmethyl)-9(10H)-anthracenone: comparison to linopirdine. J. Pharmacol. Exp. Ther. 1998;285(2):724–730. [PubMed] [Google Scholar]; (b) Earl RA, Zaczek R, Teleha CA, Fisher BN, Maciag CM, Marynowski ME, Logue AR, Tam SW, Tinker WJ, Huang S-M, Chorvat RJ. 2-Fluoro-4-pyridinylmethyl analogues of linopirdine as orally active acetylcholine release-enhancing agents with good efficacy and duration of action. J. Med. Chem. 1998;41(23):4615–4622. doi: 10.1021/jm9803424. [DOI] [PubMed] [Google Scholar]
- 5.Pieniaszek HJ, Jr., Fiske WD, Saxton TD, Kim YS, Garner DM, Xilinas M, Martz R. Single-dose pharmacokinetics, safety, and tolerance of linopirdine (DuP 996) in healthy young adults and elderly volunteers. J. Clin. Pharmacol. 1995;35(1):22–30. doi: 10.1002/j.1552-4604.1995.tb04741.x. [DOI] [PubMed] [Google Scholar]
- 6.Soh H, Tzingounis AV. The specific slow afterhyperpolarization inhibitor UCL2077 is a subtype-selective blocker of the epilepsy associated KCNQ channels. Mol. Pharm. 2010;78(6):1088–1095. doi: 10.1124/mol.110.066100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) For information on the Molecular Libraries Probe Production Centers Network (MLPCN), see: http://mli.nih.gov/mli/;; (b) For information on the Vanderbilt Specialized Chemistry Center, see: http://www.mc.vanderbilt.edu/centers/mlpcn/index.html.
- 8. For assay details, see Supporting Information. Assay ID’s (AIDs) can be viewed via the PubChem website.
- 9. For synthetic procedures and compound characterization, please see Supplemental Material.
- 10. IC50/EC50’s were generated from 8-point concentration response curves with 3-fold dilutions starting from the maximal concentration (30 μM) with quadruplicate determinations in the automated patch clamp assay. In cases in which a saturating response was not obtained at the highest tested concentration, (% at 30 μM) is listed. IC50/EC50 values are expressed as IC50/EC50 ± SD, using estimated standard deviations provided by the fitting software (Origin 6.0)
- 11. ML252 has been declared a probe via the Molecular Libraries Probe Production Centers Network (MLPCN) and is available through the network, see: http://mli.nih.gov/mli/
- 12.(a) Varma MVS, Obach RS, Rotter C, Miller HR, Chang G, Steyn SJ, El-Kattan A, Troutman MD. Physicochemical space for optimum oral bioavailability: contribution of human intestinal absorption and first-pass elimination. J. Med. Chem. 2010;53(3):1098–1108. doi: 10.1021/jm901371v. [DOI] [PubMed] [Google Scholar]; (b) Chang G, Steyn SJ, Umland JP, Scott DO. Strategic Use of Plasma and Microsome Binding To Exploit in Vitro Clearance in Early Drug Discovery. ACS Med. Chem. Lett. 2010;1(1):50–63. doi: 10.1021/ml900012h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu H, Wu M, Townsend SD, Zou B, Long S, Daniels JS, McManus OB, Li M, Lindsley CW, Hopkins CR. Discovery, synthesis, and structure-activity relationship of a series of N-aryl-bicyclo[2.2.1]heptane-2-carboxamides: characterization of ML213 as a novel KCNQ2 and KCNQ4 potassium channel opener. ACS Chem. Neurosci. 2011;2:572–577. doi: 10.1021/cn200065b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.