

Abstract
Prostate cancer is responsible for major deaths globally after lung cancer. However, etiology of prostate cancer is still unknown. Individual risk and incidence of prostate cancer may result from the interaction of genetic susceptibility with exposure to environmental factors such as infectious agents, tobacco, occupational exposure, dietary carcinogens, and/or hormonal imbalances leading to injury of the prostate and to the development of chronic inflammation. About 30% of all human cancers are caused by tobacco smoking and inhaled pollutants. Inflammation is now regarded as an important hallmark of cancer. The present study has been aimed to explore the pro-inflammatory levels in prostate carcinoma patients by examining the serum levels of novel cytokine interleukin-18 (IL-18) expression in tobacco exposed population. A total of 578 (n = 284 biopsy proven prostate cancer patients, n = 294 controls with and without tobacco exposed population) were recruited. Serum IL-18 (Interleukin-18) level was done by ELISA. The IL-18 levels between cancer patients and controls within same mode tobacco exposure as tobacco smoking (overall) showed significant difference (P < 0.0001) and further we compared within stratified group, it significantly differ (P < 0.0001) in bidi and cigarette smoking than control non users. Furthermore, IL-18 levels in tobacco chewers (overall) with gutkha and khaini chewers showed significant difference (P < 0.01) than controls non users. Moreover, the IL-18 levels between cancer patients and controls with in of combined mode chewers smokers and alcohol (CSA), smokers with alcohol showed significant difference (P < 0.01) than controls. The IL-18 levels also differed significantly (P < 0.05) with smokers and chewers in higher stages of III and IV, and showed non significant with in lower stages. Tobacco exposure enhance the inflammation in prostate carcinoma patients in stratified group as it have been represented as a risk factors in various cancers, but this study provide further its role that seems to influence inflammation especially in prostate carcinoma.
Keywords: Interleukin-18, NF-KB, prostate cancer, tobacco chewers, tobacco smokers
INTRODUCTION
Prostate cancer is the second most commonly diagnosed cancer, as well as the sixth leading cause of death in males with cancer worldwide.[1] Cancer of the prostate (PCa) is now recognized as one of the most important medical problems facing the male population. The six hallmarks of carcinogenesis have been proposed as sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, and activating invasion and metastasis.[2] Recently, seventh hallmark as cancer-related inflammation (CRI) is added, which involved in the induction of genetic instability by inflammatory mediators, leading to accumulation of random genetic alterations in cancer cells.[3] Inflammation has been found to be linked with various carcinomas such as lung,[4] gastric,[5] cervical,[6] hepato-cellular,[7] gall bladder,[8] urinary bladder,[9] pancreatic,[10] esophageal,[11] melanoma[12] and prostate.[13,14] Inflammation has also been linked with tumor since 1863, when Rudolf Virchow discovered leucocytes in neoplastic tissue.[15] Since then, chronic inflammation has been identified as a risk factor for cancer and even as means to prognosticate and diagnose cancer. The causes which stimulate this inflammatory response are multiple and include exposure to asbestos and cigarette smoke among others.[15] Several inflammatory interleukins including IL-1, IL-6, IL-8, and IL-18 have been linked with tumorigenesis, which suggests that inflammation is associated with cancer development.[16] The inflammation may cause genomic alterations affecting intrinsic cellular programs, e.g., cell cycle check-point control, programmed cell death, differentiation, metabolism and cell adhesion, in combination with epigenetic alterations affecting extrinsic programs, such as immune response, matrix metabolism, tissue oxygenation and vascular status and thus underlie human cancer development.[17,18] Only a minority of cancers are caused by germline mutations, while the vast majorities (90%) are linked to somatic mutations and environmental factors. Chronic inflammation has been implicated as an important environmental influence that can cause cancer. A recent study showed that the cause of chronic inflammation in cancer patients was chronic infection in 20%, tobacco smoking and inhaled pollutants in 30% and 35% to dietary factors.[16] Inflammation has also been associated in various steps including tumorigenesis, cellular transformation, promotion, survival, proliferation, invasion, angiogenesis and metastasis.[17,19]
Tobacco consumption has been linked to various human cancers including lung, oral cavity, breast, esophagus, pharynx, larynx, urinary bladder, pancreas and liver cancers[20–24] but its association with prostate carcinoma is controversial. The mortality burden attributed to tobacco use may be influenced by inflammation, which may promote cancer development and progression. There are more than 5,000 identified chemicals present in cigarette smoke[25,26] and 55 of these have been evaluated by the International Agency for Research on Cancer (IARC) as showing ‘sufficient evidence for carcinogenicity’ in either laboratory animals or humans.[24] Tobacco contain carcinogens like polycyclic aromatic hydrocarbons, aldehydes, benzo[alpha]pyrene, ethylene oxide, 4-aminobiphenyl and nitrosamines which are metabolically activated by hydrolysis, reduction, or oxidation by xenobiotic metabolism through phases I and II enzymes.
Among the identified environmental risk factors for cancers, tobacco exposure is the leading preventable risk factor.[27] The habit of smoking and betel quid chewing is common in many Asian countries including India.[28] Several studies have showed elevated levels of pro-inflammatory cytokines IL (IL-6, IL-12, IL-15, IL-17) in various malignancies.[29–31] Interleukin-18 (IL-18), a novel pro-inflammatory cytokine (a recently described member of the IL-1 cytokine super-family), is now recognized as an important regulator of innate and acquired immune responses. IL-18, (previously known as interferon-gamma (IFN-γ)-inducing factor), is expressed at sites of chronic inflammation in a variety of cancers and in the context of numerous infectious diseases.[32] The role of IL-18 is well-documented in various human carcinomas but studies evaluating its role in prostate carcinoma are very rare.[14,33] Limited data have shown elevated levels of IL-18 in prostate carcinoma. Data linking chronic inflammation with tobacco intake in prostate cancer is lacking. This study was done on men with prostate cancer in densely-populated north Indian region who enrolled for treatment. The present study has been carried out to evaluate the association of various modes of tobacco exposure and inflammation status with disease, the comparative association of tobacco consumption by various modes with the expression of IL-18.
MATERIALS AND METHODS
Patient and control selection
All newly diagnosed and previously untreated 284 men with cancer prostate attending urologic clinics of Sanjay Gandhi Post Graduate Institute of Medical Sciences, Lucknow and Chhatrapati Shahuji Maharaj Medical University, Lucknow, India between April 2007 and January 2012 were included in the study. During the same period, age-matched 294 independent (of patients) controls in which 239 were using tobacco in any mode but non cancerous and 55 healthy subjects with in controls as a non users (free from tobacco and alcohol exposures) but all controls were free from diseases, were enrolled by organizing various camps. The patients and controls suffering with diabetes, arthritis, cardiovascular disease, hepatitis, AIDS and other inflammatory diseases including prostatitis were excluded. The ethical clearance was taken from the Institutional Ethics committee. Following an informed consent, a standardized clinical proforma was filled by interviewing the patients for information concerning age, gender, tobacco chewing, smoking and alcohol intake (recall basis) and medical and family history for any cancer. All study subjects completed a questionnaire covering medical, residential and occupational history. Information concerning dietary habits, family history of disease, smoking, tobacco chewing and alcohol drinking was also obtained from the questionnaire filled by the patients.
Exposure factors
The exposure factors were recorded in cases and controls, which included tobacco use (smoking and chewing form tobacco), alcohol intake. Tobacco habit was categorized into smokers and chewers (use of non-smoking tobacco as powder or in beetle leaf or areca nut, catechu) and non users as those who were not smoking, chewing and drinking. Smokers were defined as those who have been smoking last ten years or more in the form of cigarettes, bidis (hand-manufactured cigarettes consisting of tobacco wrapped in a tendu or temburini leaf) or any other smoked form as hookah (Indian water pipe), chillum, or any other smoked form not less than 20 times weekly for the last 10 years or mores. Similarly, tobacco chewers were defined as those who have been using more than 20 packets of chewable tobacco products: Khaini (tobacco-lime mixtures), gutkha (tobacco with betel nut, catechu, lime and flavorings), or betel quid (zarda paan) with tobacco for last 10 years or mores. Alcohol drinkers were defined as those who consumed any alcoholic beverages (e.g., beer, wine and spirits) not less than 750 ml/week for 10 years or more. Moreover we also sub grouped the various combination of exposure as smokers with alcohol, chewers with alcohol and combination of the above two exposures with alcohol and alcohol alone. Information was gathered on the age of initiation of smoking and the self-reported quantity of specific tobacco products consumed by the users. Special emphasis was laid on the form of tobacco, which was used by the subjects and the duration of consumption was noted. The numbers of cigarettes/bidis being consumed daily were also noted so as to quantify tobacco consumption in terms of pack years but in this paper we are not giving such details as it will be more elaborative besides limited space. All exposures as tobacco chewing or smoking and drinking alcohol were included who have the exposure history not less than 10 years.
Sample collection
Blood (5 ml in EDTA) was collected by venipuncture and immediately after blood sampling; serum was obtained by centrifugation at 2000 rpm for 15 min at 4°C and stored at -80°C until later analysis. Serum PSA, IL-18 (Bender Med Systems, ELISA kits Vienna, Austria) levels were determined using ELISA kits as per standard protocol of manufacturers. All newly diagnosed men with carcinoma prostate received treatment according to the stage of disease from the OPD. They were then followed 3 monthly for 4 years and 9 months and other tests to document recurrence or progression.
Statistical analysis
Data were summarized as Mean±SE and in percentages. Initially the analysis of variance ANOVA was applied among tobacco users, non users control and cancerous patients group, if found significant, then pairwise comparison was done by using independent unpaired t-test. All the analysis was carried out by using SPSS 15.0 and Graph Pad Prism (version 5.0). The P value < 0.05 was considered as statistically significant.
RESULTS
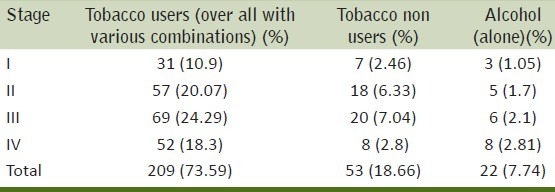
Men (284) with prostate cancer fulfilled the inclusion criteria were included in this study. Of these, 26.8% men were tobacco chewers (betel quid, gutkha and Khaini), 33.8% men were tobacco smokers (cigarette, bidi etc), 5.6% men were smokers and also consumed alcohol, 3.2% men were chewers and also consumed alcohol, 7.7% men consumed alcohol only, 4.2% men were smokers, chewers and alcoholics and 18.7% were not using any tobacco product and non-alcoholic [Table 1]. IL-18 levels of pure tobacco chewers,smokers and nonusers with stages were summarized in [Table 2]. IL-18 levels of all tobacco exposure groups (smokers, chewers alone and in combinations,) of control and cancer groups were summarized in Tables 3–5. About one-third (37%) patients were in grade III followed by grade-II (27%), grade IV (22.5%) and grade I (13%) [Table 6].
Table 1.
Distribution of cancer patients by use of tobacco and alcohol
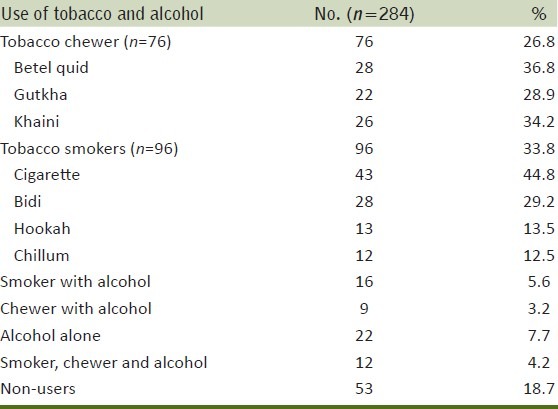
Table 2.
Relation of prostate cancer stage between non-users, smokers, and tobacco chewers with Interleukin-18#
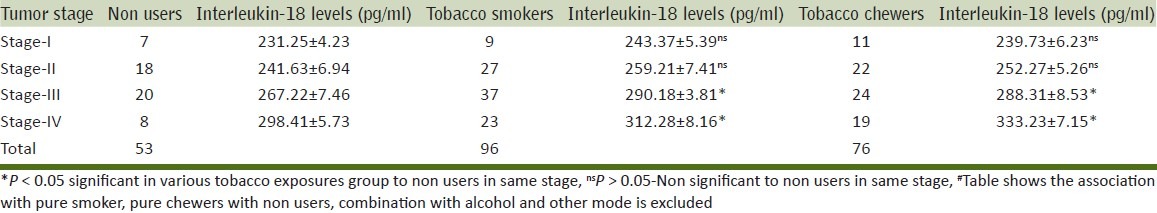
Table 3.
Tobacco chewers with various modes and their association with pro-inflammatory (IL-18) expression in prostate carcinoma patients and control
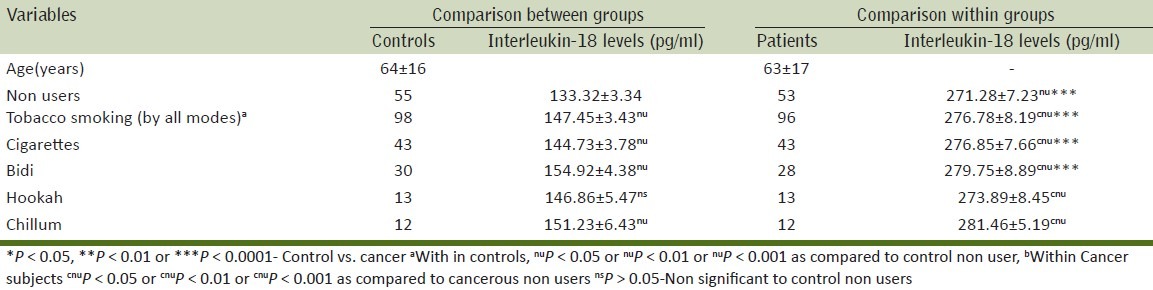
Table 5.
Relationship of IL-18 in controls and cancer patients according to alcohol use either alone or in various combinations
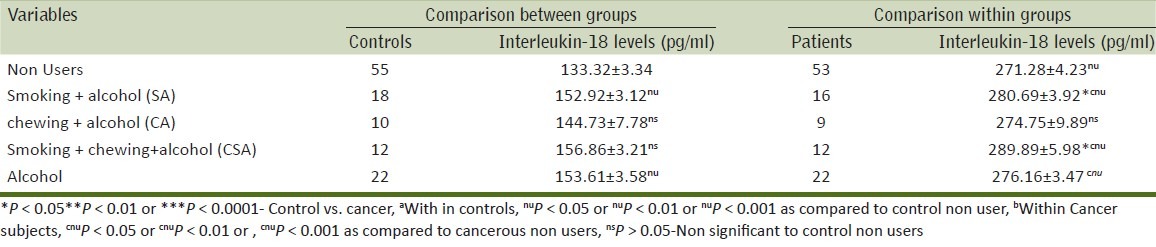
Table 6.
Distribution of cancer patients by various stages, tobacco users (combinations), non users and alcohol users
The Interleukin-18 (Pro-inflammatory marker) trends and tobacco exposure
Tobacco smokers
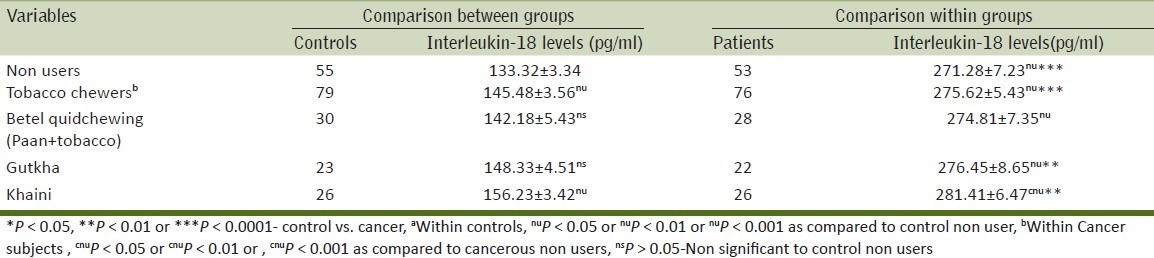
Table 3 shows the relation between various modes of smoking on IL-18 levels as compared to the control population. We found that the IL-18 levels in tobacco smokers were high as compared to controls. The IL-18 levels differed according to the mode of tobacco exposure and the highest level was seen in bidi smokers, followed by cigarette smokers, chillum, hookah (bidis > cigarette > chillum > Hookah).
Within control group, IL-18 levels with statistically high in tobacco smokers as compared to non-users. Similarly, significantly high levels of IL-18 were found in cigarette and bidi smokers as compared to non-users while it was not significant with chillum and hookah. In men with cancer, the IL-18 levels differed significantly (P < 0.05) among cigarette, bidi, hookah, chillum smokers as compared to non-users.
On comparing the IL-18 levels between cancer patients and controls with the same mode of exposure statistically significantly (P < 0.0001) difference in levels were found in cancer patients who were bidi and cigarette smokers than control bidi and cigarette users.
Tobacco chewers
Table 4 shows the relation between IL-18 levels with various forms of chewed tobacco. Among tobacco chewers groups the mean IL-18 levels showed a specific trend with the levels being highest in khaini chewers, followed by gutkha and betel quid chewers (khaini > gutkha > betel quid chewers). The levels of IL-18 were statistically significantly raised in cancer patients (who chewed tobacco in any form) as compared to controls. However, on comparing non-users controls with betel quid and gutkha users controls then the difference was not statistically significant.
Table 4.
Tobacco chewers with various modes and their association with pro.inflammatory (IL-18) expression in prostate carcinoma patients and control
Tobacco exposure in combination with alcohol intake
The relation between IL-18 levels with alcohol use either alone or in various combinations of tobacco use has been presented in Table 5. The mean level of IL-18 was highest in men who consumed all three [chewing, smoking and alcohol intake (CSA)]. This was followed by men who were smokers and alcohol consumers (SA) followed by men was consumed alcohol alone and then followed by chewers and alcohol consumers (CA) [CSA > SA > alcohol (alone) > CA]. The IL-18 levels in cancer patients were significantly higher in men who were CSA users, SA users and men who were drinkers as compared to cancer patients who were non-users. On comparing the IL-18 levels between cancer patients and controls with the same exposure, men in CSA and SA groups showed significant difference (P < 0.01) than controls. The tobacco exposures in patients and controls and its corresponding level of pro-inflammatory levels were summarized in various Tables 3 and 5.
Tobacco exposure and various stages of cancer
The IL-18 levels were significantly higher (P < 0.05) in men who were chewers and smokers as compared to non-users for stages III and IV cancer. The IL-18 levels were higher in men who were chewers and smokers as compared to non-users in stages I and II although, this difference was not significant, the results were presented in Table 2 and its percentage distribution in Table 6.
DISCUSSION
Exposure of different exogenous agents as chemicals in working environment for a long time may influence the physiological and biochemical metabolism. It may influence the prostate gland, the principal that such chemicals can alter the enzymatic activity has been established.[34,35] Moreover, animal studies demonstrated that prostate tumors can be induced by administration of chemicals.[36] Furthermore, many studies suggest various exogenous chemicals may affect hormone levels which may in turn, affect estrogen levels and androgenic stimulation of the prostate.[37–40] This study was conducted in densely populated north-Indian region to evaluate the association of various modes of tobacco consumption with inflammation in prostate cancer patients by measuring the serum IL-18 pro-inflammatory levels. Our study provides evidence that tobacco chewing and smoking may be important contributors for inflammation. The patients who were involved in tobacco smoking alone and smoking in combination with other mode as chewing and alcohol drinking showed significant increase in inflammation in carcinoma patients than non-users. The subjects who were involved in tobacco chewing alone and in combination with smoking with alcohol also showed significantly increased in inflammatory levels but slightly lower than smokers.
IL-18 and IL-6 are important in the recruitment and activation of inflammatory cells. The reason for the aggravation and induction of these pro-inflammatory mediators is probably due to the activation of redox-sensitive transcription factor NF-kB.[41,42] This transcription factor has been shown to be activated by a wide variety of agents including stress, cigarette smoke, viruses, bacteria, inflammatory stimuli, cytokines and free radicals.[43,44] Tobacco smoke is a heterogeneous mixture that contains approximately 4,000 chemical compounds, including 40 different substances classified as carcinogenic to humans or animals.[45]
Indices of increased local and systemic oxidative stress have been shown in cigarette smokers.[46–48] Several studies showed that both the gas and particulate fractions of cigarette smoke are rich sources of radicals, but the former are short lived.[47] Exposure to toxic agents in chewers can be estimated from biological markers. Some perceive hookah is not harmful[49–51] because of the belief that the smoke gets filtered in the water.[52]
One study suggested that regular smoking of marijuana and/or tobacco by young adults is associated with a high frequency of central airway inflammation. This injury is visually evident by bronchoscopy and is sometimes quite striking. At the microscopic level, there is evidence of airway inflammation in almost all smokers. These changes occur even in the absence of any symptoms or physiologic evidence of injury. The evidence for small airways inflammation was less striking in smokers of marijuana or tobacco alone, but quite prevalent in combined marijuana tobacco smoke. Collectively, these findings strongly suggest that smoking marijuana and/or tobacco has significant injurious effects on the central and peripheral airways, even in young and otherwise asymptomatic adults.[53] Many studies have found that there is a synergic effect of cigarette smoking, alcoholic consumption and betel quid chewing in carcinogenesis of oral cavity mucosa.[54–56] Bidis contain tobacco and also contain other chemicals like hydrogen cyanide and ammonia. Bidis deliver more nicotine and contains more N-nitrosonornicotine (NNN) and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) in comparison to cigarettes. Furthermore, compared to cigarettes, the mainstream smoke of bidi contains a higher concentration of several toxic and mutagenic substances, including hydrogen cyanide, carbon monoxide, volatile phenols and carcinogenic hydrocarbons such as benz[a]anthracene and benzo[a]pyrene in greater amounts than found in regular cigarettes.[57] Bidis could actually be worse than cigarettes due to a lot many reasons. They contain less tobacco but more nicotine than regular cigarettes. They pose the same risks for cancer, emphysema, heart diseases, etc. as cigarettes. Also, to keep bidis lit, smokers have to take more frequent and deeper puffs as compared to cigarettes. So, smokers may end up inhaling more smoke and taking it deeper into lungs. Also, due to the misconception that bidis are herbal and, therefore less harmful. Thus, it may be the reason of increased pro-inflammatory levels than cigarette smokers. A recent study by the Canadian government found that cannabis smoke contained more toxic substances than tobacco smoke. The study determined that marijuana smoke contained 20 times more ammonia and five times more hydrogen cyanide and nitrogen oxides than tobacco smoke.[58]
Smokers have increased rates of many cancers, especially those arising in the lung, head and neck, bladder and cervix and rare research on prostate cancer. It contains 1,014-1,016 free radicals/puff, which include reactive aldehydes, quinines and benzo(a)pyrene.[59] Many of these are relatively long lived, such as tar-semiquinone, ROS has been implicated in initiating inflammatory responses in the lungs through the activation of transcription factors, such as nuclear factor NF-kB and activator protein (AP)-1 and other signal transduction pathways, such as mitogen-activated protein (MAP) kinases and phosphoinositide-3-kinase (PI-3K), leading to enhanced gene expression of pro-inflammatory mediators.[60–62] Recently, it has been shown that oxidative stress and the redox status of the cells can also regulate nuclear histone modifications, such as acetylation, methylation and phosphorylation, leading to chromatin remodelling and recruitment of basal transcription factors and RNA polymerase II leading to the induction of proinflammatory mediators.[60,61] Among these, NF-kB has been reported to play an important role in mediating cell survival and the up-regulation of many cytokines and pro-inflammatory mediators essential to the host and ERK1/2 has been reported to mediate transcription of proteases and cytokines in response to a variety of stimuli, including cigarette smokes.[62]
In experimental systems, exposure to chewable tobacco products was associated with the generation of reactive oxygen species, modulation of inflammatory mediators and inhibition of collagen synthesis and impairment of DNA repair capacity.[63] This study also showed an unique trends of IL-18 expression in subjects with prostate cancer, betel chewers alone and betel chewers with alcohol drink showed only slight increase in inflammatory levels as compared to non-users. This could be because of the anti-oxidant properties of betel as shown previously by researchers.[64,65] Similarly, areca nut or seed is consumed simultaneously with betel and gutkha chewing which also has a strong antioxidant activity[66] but slight increase may be due to tobacco use with it.
Mechanistic studies on murine model showed a concentration dependent decrease in the extracellular production of nitric oxide in peritoneal macrophages. This decrease in the generation of reactive nitrogen species was mediated by the down-regulating transcription of inducible nitric oxide synthase in macrophages with concomitant decrease in the expression of interleukin-12. This study indicates the ability of betel leaf to down-regulate T-helper 1 pro-inflammatory responses.[67] Another reason of difference in level of IL-18 during chewing and smoking may be due to its way of exposure as it may comes quick contact in blood through smoking via lungs and chewing take quite more time and it acted by several enzymes of gastro-intestinal tract.
In this study, the difference in Interleukin-18 levels in various modes may be due to processed and unprocessed tobacco or its products in its chewing form and smoking form. Our study does not prove that tobacco is an etiological factor for cancer prostate. Our study however, shows that the levels of inflammation (as measured by IL-18) are higher in man with prostate cancer who are smokers, gutkha users and combined tobacco and alcohol users. In our previous study, we demonstrated that the levels of serum IL-18 were well correlated with disease progression (TNM staging) of various groups and elevated in patients of carcinoma prostate as compared to controls.[14]
The current novel pro-inflammatory markers of IL-18 is also documented with its higher expression in various other carcinoma as gastric,[29] breast[30] and oesophageal carcinoma.[31] The pathways for IL-18 production are well documented but its clear mode of action in patients with prostate carcinoma is not well documented. The IL-18 performs its various biological activities via its capacity of stimulating innate immunity and both Th1 and Th2-mediated responses.[68] It can also be speculated that IL-18 production by the normal adjacent prostate cells may reflect the degree of defense mechanism against tumor growth and dissemination of prostate carcinoma.[69] This study may become more imperative if larger sample size included and some parameters like various occupational exposures may be included, but we try to reduce biases and confounding as patients from all occupational sites included, who come for treatment during study time. Strongest point of this study is this novel pro-inflammatory marker (IL-18) is measured first time in tobacco exposed group and with various modes and stratified manners. This type of study will definitely solve the puzzle of tobacco exposure in development of various untreated disease like cancers.
CONCLUSIONS
Tobacco exposure has been represented as important risk factors in the development of various cancers, but this study provides further its role that seems to influence inflammation, especially in prostate carcinoma. Inflammation is now established as an important hallmark in development and progression of all cancers. Because these exposures factors are preventable, in light of the results found in this study and others like it, it is more imperative to stress to people the need to quit smoking or chewing tobacco for better life span.
ACKNOWLEDGEMENT
The authors are thankful to the department of Pharmacology and Therapeutics for providing the research facility for the study. Mr. Shailendra Dwivedi is thankful to Indian Council of Medical Research, New Delhi for providing the grant of Senior Research Fellowship. The present research work was also supported by the grant from NCD-III. Authors are also thankful to Dr. R. P. Mishra, statistician for helping in data analysis and interpretation of results.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
- 1.Verma M, Patel P, Verma M. Biomarkers in Prostate Cancer. Epidemiol Cancers. 2011;3:3773–98. doi: 10.3390/cancers3043773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 3.Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis. 2009;30:1073–81. doi: 10.1093/carcin/bgp127. [DOI] [PubMed] [Google Scholar]
- 4.Martey CA, Pollock SJ, Turner CK, Oreilly KM, Baglole CJ, Phipps RP, et al. Cigarette smoke induces cyclooxygenase-2 and microsomal prostaglandin E2 synthase in human lung fibroblasts: Implications for lung inflammation and cancer. Am J Physiol Lung Cell Mol Physiol. 2004;287:981–91. doi: 10.1152/ajplung.00239.2003. [DOI] [PubMed] [Google Scholar]
- 5.Peek RM, Jr, Crabtree JE. Helicobacter infection and gastric neoplasia. J Pathol. 2006;208:233–48. doi: 10.1002/path.1868. [DOI] [PubMed] [Google Scholar]
- 6.Castle PE, Hillier SL, Rabe LK, Hildesheim A, Herrero R, Bratti MC, et al. An association of cervical inflammation with high-grade cervical neoplasia in women infected with oncogenic human papillomavirus (HPV) Cancer Epidemiol Biomarkers Prev. 2001;10:1021–7. [PubMed] [Google Scholar]
- 7.Di Bisceglie AM. Hepatitis C and hepatocellular carcinoma. Hepatology. 1997;26(Suppl 1):34S–8. doi: 10.1002/hep.510260706. [DOI] [PubMed] [Google Scholar]
- 8.Kanoh K, Shimura T, Tsutsumi S, Suzuki H, Kashiwabara K, Nakajima T, et al. Significance of contracted cholecystitis lesions as high risk for gallbladder carcinogenesis. Cancer Lett. 2001;169:7–14. doi: 10.1016/s0304-3835(01)00523-7. [DOI] [PubMed] [Google Scholar]
- 9.Offersen BV, Knap MM, Marcussen N, Horsman MR, Hamilton-Dutoit S, Overgaard J. Intense inflammation in bladder carcinoma is associated with angiogenesis and indicates good prognosis. Br J Cancer. 2002;87:1422–30. doi: 10.1038/sj.bjc.6600615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcea G, Dennison AR, Steward WP, Berry DP. Role of inflammation in pancreatic carcinogenesis and the implications for future therapy. Pancreatology. 2005;5:514–29. doi: 10.1159/000087493. [DOI] [PubMed] [Google Scholar]
- 11.Murphy SJ, Anderson LA, Johnston BT, Fitzpatrick DA, Watson PR, Monaghan P, et al. Have patients with esophagitis got an increased risk of adenocarcinoma? Results from a population-based study. World J Gastroenterol. 2005;11:7290–5. doi: 10.3748/wjg.v11.i46.7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berwick M, Armstrong BK, Ben-Porat L, Fine J, Kricker A, Eberle C, et al. Sun exposure and mortality from melanoma. J Natl Cancer Inst. 2005;97:195–9. doi: 10.1093/jnci/dji019. [DOI] [PubMed] [Google Scholar]
- 13.Nelson WG, De Marzo AM, De Weese TL, Isaacs WB. The role of inflammation in the pathogenesis of prostate cancer. J Urol. 2004;172(5 Pt 2):6–1. doi: 10.1097/01.ju.0000142058.99614.ff. [DOI] [PubMed] [Google Scholar]
- 14.Dwivedi S, Goel A, Natu SM, Mandhani A, Khattri S, Pant KK. Diagnostic and Prognostic Significance of Prostate Specific Antigen and Serum Interleukin 18 and 10 in Patients with Locally Advanced Prostate Cancer: A Prospective Study. Asian Pacific J Cancer Prev. 2011;12:1843–8. [PubMed] [Google Scholar]
- 15.Balkwill F, Mantovani A. Inflammation and cancer: Back to Virchow? Lancet. 2001;357:539–45. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 16.Aggarwal BB, Vijayalekshmi RV, Sung B. Targeting inflammatory pathways for prevention and therapy of cancer: Short-term friend, longterm foe. Clin Cancer Res. 2009;15:425–30. doi: 10.1158/1078-0432.CCR-08-0149. [DOI] [PubMed] [Google Scholar]
- 17.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–7. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 19.Mantovani A. Cancer: Inflammation by remote control. Nature. 2005;435:752–3. doi: 10.1038/435752a. [DOI] [PubMed] [Google Scholar]
- 20.WHO report on the global tobacco epidemic. 2008. [Last accessed on 2010 Aug 9]. www.http://who.int/tobacco .
- 21.Magnusson C, Wedren S, Rosenberg LU. Cigarette smoking and breast cancer risk: A population-based study in Sweden. Br J Cancer. 2007;97:1287–90. doi: 10.1038/sj.bjc.6604007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boffetta P, Aagnes B, Weiderpass E, Andersen A. Smokeless tobacco use and risk of cancer of the pancreas and other organs. Int J Cancer. 2005;114:992–5. doi: 10.1002/ijc.20811. [DOI] [PubMed] [Google Scholar]
- 23.Brennan P, Bogillot O, Greiser E, Chang-Claude J, Wahrendorf J, Cordier S. The contribution of cigarette smoking to bladder cancer in women (pooled European data) Cancer Causes Control. 2001;12:411–7. doi: 10.1023/a:1011214222810. [DOI] [PubMed] [Google Scholar]
- 24.Tobacco: A Major International Health Hazard. Vol. 74. Lyon: IARC; 1986. International Agency for Research on Cancer; pp. 145–65. [Google Scholar]
- 25.83rd ed. Lyon, France: IARC; 2004. International Agency for Research on Cancer, Tobacco Smoke and Involuntary Smoking. [Google Scholar]
- 26.Woga GN, Hecht SS, Felton JS, Conney AH, Loeb LA. “Environmental and chemical carcinogenesis,”. Semin Cancer Biol. 2004;14:473–86. doi: 10.1016/j.semcancer.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 27.Terry PD, Rohan TE. Cigarette smoking and the risk of breast cancer in women: A review of the literature. Cancer Epidemiol Biomarkers Prev. 2002;11:953–71. [PubMed] [Google Scholar]
- 28.Gupta PC, Ray CS. Epidemiology of betel quid usage. Ann Acad Med Singapore. 2004;33:31–6. [PubMed] [Google Scholar]
- 29.Kawabata T, Ichikura T, Majima T, Seki S, Chochi K, Takayama E, et al. Prospective serum interleukin-18 level as a postoperative prognostic marker in patients with gastric carcinoma. Cancer. 2001;92:2050–5. doi: 10.1002/1097-0142(20011015)92:8<2050::aid-cncr1544>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 30.Gunel N, Cokun U, Sancak B, Gunel U, Hasdemir O, Bozkurt S. Clinical importance of serum interleukin-18 and nitric oxide activities in breast carcinoma patients. Cancer. 2002;95:663–7. doi: 10.1002/cncr.10705. [DOI] [PubMed] [Google Scholar]
- 31.Tsuboi K, Miyazaki T, Nakajima M, Fukai Y, Masuda N, Manda R, et al. Serum IL- 12 and IL-18 levels as tumor marker in patients with esophageal carcinoma. Cancer Letter. 2004;205:207–14. doi: 10.1016/j.canlet.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 32.Gracie JA, Robertson SE, McInnes IB. Interleukin-18. J Leukoc Biol. 2003;73:213–24. doi: 10.1189/jlb.0602313. [DOI] [PubMed] [Google Scholar]
- 33.Shoujun N, Duangai W, Caibin F, Ouyang J. Clinical value of serum interleukin- 18 in patients with prostate cancer. Chinese-German J Clin Oncol. 2007;6:574–8. [Google Scholar]
- 34.Lee IP, Suzuki K. Induction of aryl hydrocarbon hydroxylase activity in the rat prostate glands by 2, 3, 7, 8-tetrachlorodibenzop-dioxin. J Pharmacol Exp Ther. 1980;215:601–5. [PubMed] [Google Scholar]
- 35.Lee IP, Suzuki K, Lee SD, Dixon RL. Aryl hydrocarbon hydroxylase induction in rat lung, liver, and male reproductive organs following inhalation exposure to diesel emission. Toxicol Appl Pharmacol. 1980;52:181–4. doi: 10.1016/0041-008x(80)90258-6. [DOI] [PubMed] [Google Scholar]
- 36.Waalkes MP, Rehm S, Perantoni AO, Coogan TP. Cadmium exposure in rats and tumours of the prostate. In: Nordberg GF, Herber RF, Alessio L, editors. Cadmium in the human environment: Toxicity and carcinogenicity. Vol. 118. Lyon, France: International Agency for Research on Cancer; 1992. pp. 391–404. [Google Scholar]
- 37.Brawley OW, Knopf K, Thompson I. The epidemiology of prostate cancer part II: The risk factors. Semin Urol Oncol. 1998;16:193–201. [PubMed] [Google Scholar]
- 38.Dich J, Wiklund K. Prostate cancer in pesticide applicators in Swedish agriculture. Prostate. 1998;34:100–12. doi: 10.1002/(sici)1097-0045(19980201)34:2<100::aid-pros4>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 39.Ross R, Schottenfeld D. Prostate cancer. In: Schottenfeld D, Fraumeni JF Jr, editors. Cancer epidemiology and prevention. 2nd ed. New York, NY: Oxford University Press; 1996. pp. 1180–206. [Google Scholar]
- 40.Golden RJ, Noller KL, Titus-Ernstoff L, Kaufman RH, Mittendorf R, Stillman R, et al. Environmental endocrine modulators and human health: An assessment of the biological evidence. Crit Rev Toxicol. 1998;28:109–227. doi: 10.1080/10408449891344191. [DOI] [PubMed] [Google Scholar]
- 41.Aggarwal BB, Shishodia S, Sandur SK, Pandey MK, Sethi G. Inflammation and cancer: How hot is the link? Biochem Pharmacol. 2006;72:1605–21. doi: 10.1016/j.bcp.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 42.Rahman I, Gilmour PS, Jimenez LA, MacNee W. Oxidative stress and TNF-alpha induce histone acetylation and NF-kappa B/ AP-1 activation in alveolar epithelial cells: Potential mechanism in gene transcription in lung inflammation. Mol Cell Biochem. 2002;234:239–28. [PubMed] [Google Scholar]
- 43.Ahn KS, Aggarwal BB. Transcription Factor NF.kB: A Sensor for Smoke and Stress Signals. Ann N Y Acad Sci. 2005;1056:218–33. doi: 10.1196/annals.1352.026. [DOI] [PubMed] [Google Scholar]
- 44.Anto RJ, Mukhopadhyay A, Shishodia S, Gairola CG, Aggarwal BB. Cigarette smoke condensate activates nuclear transcription factor-kB through phosphorylation and degradation of IkB: Correlation with induction of cyclooxygenase-2. Carcinogenesis. 2002;23:1511–8. doi: 10.1093/carcin/23.9.1511. [DOI] [PubMed] [Google Scholar]
- 45.Rockville, MD: DHHS Publication; 1990. US Department of Health and Human Services. The Health Benefits of Smoking Cessation: A Report of the Surgeon General. US Department of Health and Human Services, Public Health Service, Centers for Disease Control, Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health. [Google Scholar]
- 46.Rahman I, MacNee W. Role of oxidants/antioxidants in smoking induced lung diseases. Free Radic Biol Med. 1996;21:669–81. doi: 10.1016/0891-5849(96)00155-4. [DOI] [PubMed] [Google Scholar]
- 47.Pryor W. Cigarette smoke radicals and the role of free radicals in chemical carcinogenicity. Environ Health Perspect. 1997;105:875–82. doi: 10.1289/ehp.97105s4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mac Nee W. Pulmonary and systemic oxidant/antioxidant imbalance in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2005;2:50–60. doi: 10.1513/pats.200411-056SF. [DOI] [PubMed] [Google Scholar]
- 49.Ward KD, Vander Weg MW, Relyea G, Debon M, Klesges RC. Water pipe smoking among American military recruits. Prevent Med. 2006;43:92–7. doi: 10.1016/j.ypmed.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 50.Primack BA, Sidani J, Agarwal AA, Shadel WG, Donny EC, Eissenberg TE. Prevalence of and associations with water pipe tobacco smoking among U.S. University students. Ann Behav Med. 2008;36:81–6. doi: 10.1007/s12160-008-9047-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jackson D, Aveyard P. Waterpipe smoking in students: Prevalence, risk factors, symptoms of addiction, and smoke intake.Evidence from one British university. BMC Public Health. 2008;8:174. doi: 10.1186/1471-2458-8-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kandela P. Nargile smoking keeps Arabs in Wonderland. Lancet. 2000;356:1175. doi: 10.1016/s0140-6736(05)72871-3. [DOI] [PubMed] [Google Scholar]
- 53.Roth MD, Arora A, Barsky SH, Kleerup EC, Simmons M, Tashkin DP. Airway Inflammation in Young Marijuana and Tobacco Smokers. Am J Respir Crit Care Med. 1998;157:928–37. doi: 10.1164/ajrccm.157.3.9701026. [DOI] [PubMed] [Google Scholar]
- 54.Castellsagué X, Quintana MJ, Martinez MC, Nieto A, Sanchez MJ, Juan A, et al. The role of type of tobacco and type of alcoholic beverage in oral carcinogenesis. Int J Cancer. 2004;108:741–9. doi: 10.1002/ijc.11627. [DOI] [PubMed] [Google Scholar]
- 55.Wen CP, Tsai MK, Chung WS, Hsu HL, Chang YC, Chan HT, et al. Cancer risks from betel quid chewing beyond oral cancer: A multiple site carcinogen when acting with smoking. Cancer Causes Control. 2010;21:1427–35. doi: 10.1007/s10552-010-9570-1. [DOI] [PubMed] [Google Scholar]
- 56.Yen TT, Lin WD, Wang CP, Wang CC, Liu SA. The association of smoking, alcoholic consumption, betel quid chewing and oral cavity cancer: A cohort study. Eur Arch Otorhinolaryngol. 2008;265:1403–1407. doi: 10.1007/s00405-008-0659-z. [DOI] [PubMed] [Google Scholar]
- 57.Gupta PC, Murti PR, Bhonsle RB. Epidemiology of cancer by tobacco products and the significance of TSNA. Crit Rev Toxicol. 1996;26:183–98. doi: 10.3109/10408449609017930. [DOI] [PubMed] [Google Scholar]
- 58.David M, William SR, Genevieve L, Yolande L, Rebecca M, Paul W, et al. A Comparison of Mainstream and Sidestream Marijuana and Tobacco Cigarette Smoke Produced under Two Machine Smoking Conditions. Chem Res Toxicol. 2008;21:494–502. doi: 10.1021/tx700275p. [DOI] [PubMed] [Google Scholar]
- 59.Church DF, Pryor WA. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect. 1985;64:111–26. doi: 10.1289/ehp.8564111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rahman I, MacNee W. Role of transcription factors in inflammatory lung diseases. Thorax. 1998;53:601–12. doi: 10.1136/thx.53.7.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNF alpha induced death and sustained JNK activiation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–61. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 62.Mortaz E, Redegeld FA, Sarir H, Karimi K, Raats D, Nijkamp FP, et al. Cigarette smoke stimulates the production of chemokines in mast cells. J Leukoc Biol. 2008;83:575–80. doi: 10.1189/jlb.0907625. [DOI] [PubMed] [Google Scholar]
- 63.Lyon: IARC monographs volume; 1989. Smokeless Tobacco: Summary of Data Reported and Evaluation; pp. 363–70. [Google Scholar]
- 64.Choudhary D, Kale RK. Antioxidant and non-toxic properties of Piper betel leaf extract: In vitro and in vivo studies. Phytother Res. 2002;16:461–6. doi: 10.1002/ptr.1015. [DOI] [PubMed] [Google Scholar]
- 65.Jeng JH, Chen SY, Liao CH, Tung YY, Lin BR, Hahn LJ, et al. Modulation of platelet aggregation by areca nut and betel leaf ingredients: Roles of reactive oxygen species and cyclooxygenase. Free Radic Biol Med. 2002;32:860–71. doi: 10.1016/s0891-5849(02)00749-9. [DOI] [PubMed] [Google Scholar]
- 66.Zhang X, Wu J, Han Z, Mei WL, Dai HF. Antioxidant and cytotoxic phenolic compounds of areca nut (Areca catechu) Chem Res Chinese Univ. 2010;26:161–4. [Google Scholar]
- 67.Ganguly S, Mula S, Chattopadhyay S, Chatterjee M. An ethanol extract of Piper betle Linn. mediates its anti-inflammatory activity via down regulation of nitric oxide. J Pharma Pharmacol. 2007;59:711–8. doi: 10.1211/jpp.59.5.0012. [DOI] [PubMed] [Google Scholar]
- 68.Tanaka F, Hashimoto W, Okamura H, Robbins PD, Lotze MT, Tahara H. Rapid regeneration of potent and tumor- specific cytotoxic T lymphocytes by interleukin 18 using dendrite cells and natural killer cells. Cancer Res. 2000;60:4838–44. [PubMed] [Google Scholar]
- 69.Pages F, Berger A, Henglein B, Piqueras B, Danel C, Zinzindohoue F, et al. Modulation of interleukin -18 expression in human colon carcinoma: Consequences for tumor immune surveillance. Int J Cancer. 1999;84:326–30. doi: 10.1002/(sici)1097-0215(19990621)84:3<326::aid-ijc22>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]