

Abstract
Trichomonas vaginalis is a parasite of the urogenital tract in men and women, with a worldwide presence and significant implications for global public health. T. vaginalis research entered the age of genomics with the publication of the first genome sequence in 2007, yet subsequent utilization of other ‘omics’ technologies and methods has been slow. Here, we review some of the tools and approaches available to interrogate T. vaginalis biology, with an emphasis on recent advances and current limitations, and draw attention to areas where further efforts are needed to effectively examine the complex and intriguing biology of the parasite.
Keywords: Trichomonas vaginalis, population genetics, -omics, molecular tools, evolution
A trichy parasite
T. vaginalis, a protistan parasite of the human urogenital tract, causes trichomoniasis in about half of infected women, producing malodorous vaginal discharge, vulval irritation and inflammation, and punctate cervical microhaemorrhages (‘strawberry cervix’). Males typically remain asymptomatic, but can suffer urethral inflammation, urethral discharge and dysuria. Despite trichomoniasis having a prevalence nearly as high as chlamydia, gonorrhea and syphilis combined [1], T. vaginalis is neglected compared to other STD-causing organisms, and was long regarded as a self-clearing ‘nuisance’ [2]. In fact infections are associated with pelvic inflammatory disease, adverse pregnancy outcomes, infertility, an increased incidence of aggressive prostate cancers, and an up to two-fold increase in HIV-1 transmission [3, 4], potentially translating into a significant number of global HIV infections [5].
The apparently simple life-cycle of T. vaginalis consists of a free-swimming trophozoite transmitted between sexual partners and reproducing by mitosis, which adopts an amoeboid form to attach to the vaginal epithelium. Around 50% of isolates harbor a linear double-stranded RNA virus, possibly influencing virulence [6]. Parasites can also harbor Mycoplasma hominis, a bacterium of the lower genital tract which is implicated in pelvic inflammatory disease and pregnancy complications [7]. Currently only the 5-nitroimidazole drugs metronidazole and tinidazole are available to treat trichomoniasis, and resistance to the drugs has been noted since their first deployment [8].
Since the draft T. vaginalis genome was published in 2007 [9], the scientific community has begun to address essential topics such as parasite genetic diversity, population structure, mechanisms of pathogenesis, and parasite-microbiota interactions. Here we review recent scientific advances in such areas of T. vaginalis research, and note gaps in our knowledge that can now be addressed via advances in technology and our understanding of the parasite from genome sequencing.
What ‘-omics’ resources are available for T. vaginalis?
A first step in utilizing genomics to understand an organism’s biology involves establishing reference genomes of the species. Figure 1 summarizes sequencing projects and publications for several human parasites. Compared to the Plasmodium genus, which has the most complete genomic, proteomic and transcriptomic toolkit, Trichomonasis impoverished in terms of both genome projects and publications. ‘Next generation sequencing’ and its decreased costs should enable sequencing of multiple isolates and analysis of genome-wide variations in the coming years. Below we summarize the limited ‘-omic’ studies carried out so far to characterize T. vaginalis.
Figure 1.
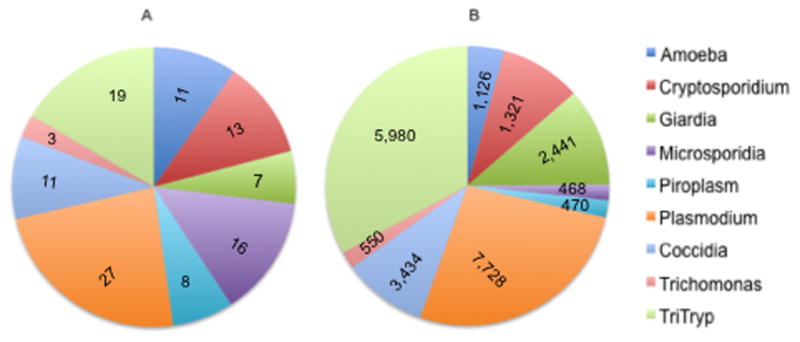
T. vaginalis is a ‘neglected’ parasite . Data are shown for human parasites that belong to nine different genera (Entamoeba, Cryptosporidium, Giardia, Plasmodium, Trichomonas) or groups (Microsporidia, piroplasms, coccidia, TriTryp) of closely-related prasites as far as (a) number of genome projects, and (b) number of research publications in PubMed. The number of PubMed articles was calculated from the past 10 years.
The T. vaginalis genome and its challenges
The ~160 Mb draft genome sequence of the T. vaginalis G3 lab isolate was published in 2007 as a highly fragmented assembly [9]. The genome contains ~ 60 000 predicted protein-coding genes, ~1100 ribosomal RNA genes, and at least 14 390 viral or transposable element ORFs (Table 1). While the current genome sequence is far from being assembled as six haploid T. vaginalis chromosomes [10], it sheds light on many interesting features of the parasite. For example, thegenome was found to be much larger than previously described, and a comparison with other genomes of Entamoeba (~20 Mb), Plasmodium (~25 Mb) and Toxoplasma (~63 Mb) shows that it is many times larger than several of these other parasite genomes. Theunusual size of the genome primarily reflects its complement of transposable elements, which comprise at least a quarter to perhaps over a third of the genome [11]. These highly duplicated sequences pose the major technical challenge to genome assembly, since sequence reads from first- and second-generation sequencing technology are too short in length to span multi-kilobase, high copy number transposable elements such as Mavericks [11] and the non-repetitive flanking sequences that could be used to differentiate one element from another. ‘Mate-pair’ sequencing–where read pairs are separated by a predetermined genomic distance of up to a few tens of kilobases–is a means to link a repetitive contig to another contig in the correct order, but is technically laborious and expensive [12]. Optical mapping, where a physical restriction map of the entire genome is produced and aligned against an in silico virtual restriction map of contigs produced by other sequencing methods [13], potentially could circumvent the limitations of these technologies. It has already improved the assembly of the complex maize genome, which consists of ~ 85% repetitive sequences [14]. Newly developed software that is able to combine optical mapping with next-generation sequencing (http://www.opgen.com/products-and-services/softwares) may provide a new cost effective solution for the complex assembly that will be required to produce useful genome sequences of other T. vaginalis isolates. Technologies that can read through vast genomic distances (e.g., PacBio, Oxford Nanopore) constitute another potential solution to the problem of assembling such repetitive genomes.
Table 1.
Omics data sets for T. vaginalisa
| Category | Number | Ref |
|---|---|---|
| Genomic Data Sets | ||
| No. published whole genome sequences | 1 | [9] |
| Total gene count | 99 009 | TrichDB |
| Predicted protein coding genes | 59 672 | TrichDB |
| No. homologues in other species | 54 715 | TrichDB |
| Repeat (transposable element and viral) ORF count | 14 390 | [9] |
| No. genes with Gene Ontology terms | 13 172 | TrichDB |
| No. genes with EC numbers | 880 | KEGG |
| Transcriptome Data Sets | ||
| No. redundant ESTs | >150,000 | [9, 15, 16] |
| No. microarrays | 1 | [15, 16] |
| No. Digital SAGE tags | 104 million | unpublished |
| No. RNA-Seq data sets | 0 | |
| Proteomic Data Sets | ||
| Trophozoite proteins | 164 | [26] |
| Hydrogenosomal proteins | 569 | [24] |
| No. metabolomic studies | 1 | unpublished |
Data summarized from the genome publication [9], TrichDB v1.3 [66], KEGG (Kyoto Encyclopedia of Genes and Genomes http://www.genome.jp/kegg/pathway.html), and other studies as referenced.
Gene expression in T. vaginalis
Gene expression studies as part of the T. vaginalis genome project included analysis of thousands of expressed sequence tags (ESTs) representing RNA from parasites cultured under different conditions (e.g., cold-stress, low iron, and low glucose) [9]. Recently, ~45 000 more ESTs were generated in order to study the expression of the largest gene family in T. vaginalis -- the BspA-like family -- and its role in pathogenicity [15]. Of the 911 genes, a third were shown to have an EST match, but different cDNA libraries derived from different culture conditions had little overlap in their TvBspA transcripts, suggesting differential expression of the genes under different conditions. Extending this analysis further, these authors used a small spotted array with ~5000 non-redundant cDNAs including 74 different TvBspA cDNAs. Almost 18% of these genes showed expression variation between the tested conditions. A second more extensive transcriptome project generated ~19 000 ESTs and used the same spotted array to study the role of iron in T. vaginalis gene expression [16]. Here ~4% of the genes studied were differentially expressed under different iron conditions. Additional experiments revealed that iron influences the expression of only some T. vaginalis paralogous genes, similar to the findings of Noel et al. [15], and indicating stringent regulation of gene expression in T. vaginalis.
While the ESTs and few microarray studies have provided useful data, more sophisticated and data-rich expression sets are required. Recent progress towards this has been the development of 104 million digital SAGE (serial analysis of gene expression) tags that map to genes expressed in T. vaginalis grown under aerobic and microaerobic conditions (Jeremy Mottram, unpublished).
T. vaginalis appears to express many of the ncRNAs (non-coding RNAs) involved in RNA processing such as snRNAs (small nuclear RNAs), snoRNAs (small nucleolar RNAs), ribonuclease P, and RNAse MRP (mitochondrial RNA processing) [17–20]. These are in addition to components of the microRNA (miRNA) pathway (Argonaute-like genes, a Dicer-like gene and multiple DEAD/DEAH-box helicases) identified in the genome sequence [9]. Indeed, next generation sequencing of small RNAs has identified multiple putative miRNAs in T. vaginalis, many of which share sequence similarity with known miRNAs from plants and animals [17, 21, 22]. Recently, malate dehydrogenase was identified as the first potential target of miRNA regulation in T. vaginalis [23], through comparison of mRNA and protein expression levels in trophozoite and amoeboid forms.The identification of ncRNA networks in T. vaginalis now provides opportunities for investigating the evolution of such systems in deep-branching eukaryotes.
T. vaginalis proteomics and metabolomics
High-throughput proteomic studies are starting to expand upon findings from the T. vaginalis genome. For example, in silico analysis of the genome predicted 138 proteins to be targeted to its hydrogenosome [9], but mass spectrometry of purified hydrogenosomes has now characterized 569 proteins–four times as many–indicating more complexity and functions for the organelle than was originally thought [24]. Another study used multidimensional protein identification (MuDPiT) technology to identify the surface proteome of six T. vaginalis strains with differing adherence capacities to vaginal epithelial cells, and from 411 proteins identified, 11 were found to be more abundant in highly adherent cells [25]. Another study used 2D gel electrophoresis combined with MALDI-TOF mass spectrometry analysis to characterize 164 proteins expressed in the T. vaginalis trophozoite stage [26]. Future proteomic studies could improve the annotation of hypothetical proteins (which constitute an extraordinary ~86% of proteins predicted to be encoded by the T. vaginalis genome) and, for example, elucidate the roles of proteins encoded within the genome’s enormous number of predicted transposable elements [27]. In addition, large-scale T. vaginalis proteomic studies should allow us to interrogate the differences in female versus male infections, and symptomatic versus asymptomatic infections.
Metabolomics involves comprehensive profiling of the final products of biological processes in cells [28], enabling analysis of metabolic pathway perturbations in, for example, parasites under drug treatment. Metabolomics is an emerging field, and we know of only one such study involving T. vaginalis to date (http://ebookbrowse.com/Trichomonas-metabolomics-pdf-d80829749) (Graham H. Coombs and Gareth D. Westrop, unpublished). This analysis compared metabolic pathways of T. vaginalis with the cattle parasite TriTrichomonas foetus, which causes a venereal disease in cattle (Box 1), using a hybrid mass spectrometer LTQ-Orbitrap with liquid chromatography. Significant differences were identified in multiple metabolic pathways, including arginine catabolism (upregulated in T. vaginalis) and sphingolipid metabolism (upregulated in T. foetus). Such lines of research hold promise for identifying genes involved in pathogenesis, as well as potential new drug targets.
Box 1. The importance of comparative analysis between different parabasalid species.
T. vaginalis belongs to phylum Parabasalia, a grouping of microareophilic, single-cell flagellates that harbor H2-producing organelles called hydrogenosomes [67]. Over 400 parabasalid species have been described, and a recent analysis of morphological and phylogenetic data classified these into six phylogenetic groups: Trichomonadea, Tritrichomonadea, Hypotrichomonadea, Spirotrichonymphea, Cristamonadea and Trichonymphea [68]. The majority of parabasalids are enteric commensals of metazoans, however, the Trichomonadea and Tritrichomonadea contain species of particular medical and veterinary importance, and include T. vaginalis(Figure I). For example, Trichomonas tenax, a sister taxon to T. vaginalis, infects the human oral cavity and is associated with pulmonary infections [69]; PentaTrichomonas hominis and Dientamoeba fragilis inhabit the intestinal tract of several animal species, and have been associated with human gastrointestinal disease [70, 71]; TriTrichomonas foetus is an important veterinary parasite, inhabiting the urogenital tract of cattle where it can cause infertility and spontaneous abortions, as well as infecting the guts of other vertebrate species such as cats, causing chronic large-bowel diarrhea [72]; finally, Trichomonas gallinae and TetraTrichomonas gallinarum are found in the oral cavity and gastrointestinal system of birds, the former causing avian trichomonosis responsible for a recent decline in common British garden birds [73], and the latter inhabiting the intestinal tract of several poultry species. In addition to parasitic and commensal parabasalids, several free-living species such as PseudoTrichomonas keilini have been described. Interestingly, these free-living species do not form one phylogenetic clade, and are instead dispersed throughout the parabasalid lineage [74]. It is therefore unclear whether symbiosis and parasitism evolved only once in the Parabasalia from a free-living common ancestor, or are the result of convergent evolution. This distribution of lifestyle and host-range diversity exhibited by the parabasalids provides a unique opportunity for investigations into the evolution of symbiosis and parasitism, and the adaptation of microorganisms to different environmental niches.
To date, most studies on phylum Parabasalia have focused on T. vaginalis and T. foetus as they have the largest impact on human and livestock health. Parabasalia show striking differences in genome size and chromosome number [10], with the genomes of T. vaginalis, T. tenax, and T. foetus (size range 133 – 177 Mb) nearly double the size of P. hominis (94 Mb). The single reference genome generated for T. vaginalis [15] revealed the large genome size (~160 Mb) to be the result of massively expanded gene families, including those whose function is important for interaction of the parasite with its immediate environment [9, 15]. Whether genomic expansion played a role in the adaptation of the organism to a urogenital environment, as has been hypothesized [9], remains an unanswered question. Thus, comparative examination of a range of parabasalids, including genomic, proteomic and metabolomic analyses, has the potential to be instrumental in the study of T. vaginalis host specificity and pathogenicity.
T. vaginalis population genetics comes of age
Until recently, molecular studies of T. vaginalis that investigated the genetic diversity, population structure and correlations of clinically relevant phenotypes to genetic relatedness, have been limited to analyses of fragment length polymorphism (RFLP), random amplified polymorphic DNA (RAPD), and sequence polymorphisms in rRNA genes and intergenic regions (see for example Refs. [29, 30]). Although these studies have provided insight into within-species diversity, they have also led to contradictory findings–most likely due to innate limitations with the genetic markers themselves. To address these shortcomings, three groups have recently taken advantage of the T. vaginalis genome sequence to develop panels of microsatellite (MS) markers [31, 32] and single nucleotide polymorphisms (SNPs) located within single-copy genes [31, 33]. Prokopi et al., developed four MS used to genotype 17 isolates [32]. Conrad et al. validated a panel of 21 MS markers in seven laboratory strains, tested for marker stability in long-term cultures, and localized 14 of the markers to four of the six T. vaginalis chromosomes using fluorescent in situ hybridization (FISH) [31]. Cornelius et al. published a multilocus sequence typing (MLST) scheme, targeting polymorphisms in seven single-copy housekeeping genes [33].
To what uses have these new genetic markers been put? The Conrad markers have been used in an extensive global population genetics study comprising ~235 isolates from Mexico, Chile, India, Australia, Papua New Guinea, Italy, Africa and the United States [34]. The study found high levels of T. vaginalis genetic diversity, and identified a two ‘type’ population structure, similar to previous studies [35, 36]. Type 1 and Type 2 appear to be present in near equal proportions worldwide, but were found to differ significantly in the rate at which they harbor the dsRNA virus and in their sensitivity to the anti-parasitic drug metronidazole. The authors postulate that this population structure, and the phenotypic differences associated with them, may explain some of the parasite’s variation in pathology, virulence and drug resistance. Cornelius et al. utilized their MLST scheme to genotype 68 isolates, including reference lab strains and isolates from patients in Mississippi, and similarly found high genetic diversity and a two-cluster population structure [33].
The availability of these new genetic markers radically expands the potential for T. vaginalis population genetics research. For example, MS markers have been used to differentiate between single and multiple strain infections, important because multi-strain infections have a broad range of clinically relevant effects in a number of human pathogens [37]. Moreover, genotyping will advance studies of recurrent T. vaginalis infections by providing an additional tool to differentiate between new infections and recrudescence of existing infections either due to drug resistance, latent infections or reinfections.
The human microbiome and T. vaginalis
Study of the ecological interactions between commensal bacteria and microbial pathogens that co-inhabit the human urogenital tract has recently gathered momentum due to next generation sequencing. Using this technology, researchers have expanded our understanding of what constitutes a ‘normal’ vagina or ‘healthy’ male urethra and have identified characteristics of bacterial composition that are associated with infection. For example, using data from 16S rRNA sequencing of vaginal samples collected from asymptomatic North American women from four ethnic groups (white, black, Hispanic, and Asian), Ravel et al. [38] identified five bacterial ‘community types’, four of which were dominated by one of four species of Lactobacillus (Lactobacillusiners , Lactobacillus crispatus, Lactobacillus gasseri, or Lactobacillus jensenii). The fifth community type had low proportions of lactic acid bacteria and high proportions of strictly anaerobic organisms. Interestingly, the proportions of each community group varied significantly by ethnic group, as did the vaginal pH. A follow-up study that used the same data set but restricted analysis to the eleven T. vaginalis-positive women in the cohort indicated that the presence of T. vaginalis in asymptomatic women could be associated with low Lactobacillus community types [39]. Unfortunately, no association of a particular T. vaginalis genotype with community type was found, due to the low number of parasite samples tested.
Less well studied is the microbiome of the male urogenital tract. A recent study cloned and sequenced 16S rRNA amplicons of the microbiota of first-catch urine [40], which has been shown to be indicative of the male urogenital tract microbiome [41]; a second study used 16S rRNA pyrosequencing to identify microbes isolated from the coronal sulci of circumcised and uncircumcised Ugandan men [42]. Despite differences in sequencing depth, both studies found that bacterial communities are complex and that a single, characteristic microbial community is not apparent. Furthermore, an overlap was found between the male urethral microbiome and the microbial communities of the superficial skin, colon, and vagina. The microbiomes of men infected with STIs tended to cluster together, distinct from those of non-infected men, and often included unidentified bacteria associated with female genital tract pathology [40]. Unfortunately, neither study specifically considered an association between T. vaginalis and the male urogenital tract microbiome.
The ability to characterize the diversity of microbial community types that inhabit the male and female urogenital tracts is enormously powerful, and will permit investigators to determine if disturbances in microbiomes allow sexually transmitted pathogens to infect sexual partners opportunistically, or if they cause specific types of disruption to urogenital microbial communities. Establishing causality will require following patient urogenital microbiomes longitudinally in the context of sexual activity, and hormonal changes, as reported recently [43]. In addition, these techniques can be used to understand the impact that metronidazole treatment (or other antibiotic usage) may have on the vaginal microbial community, and the short- and long-term implications for vaginal health [44, 45]. Ultimately, a robust in vivo model system is needed to facilitate investigation of ecological interactions between the microbial community and T. vaginalis. Although mouse [46] and Macaca models [47] have been described, they are less than ideal, and a rodent model with a humanized urogenital tract, or an in vitro system composed of differentiated vaginal cells, immune cells and commensal bacteria, will be needed to consider the role of microbial ecology in regulating T. vaginalis infection and virulence. By understanding the influence of particular bacteria on successful vaginal colonization, it may be possible to tailor preventative measures by identifying women most susceptible to T. vaginalis infection, and modify their indigenous microbial communities to protect them against infection.
T. vaginalis drug resistance: a neglected area of research
The 5-nitroimidazole, metronidazole, has been used to treat T. vaginalis infections since 1960, but several studies suggest emergence of resistant T. vaginalis strains with a prevalence of around 5% in the United States (for example, [48]), and as high as 17% elsewhere [49]. Tinidazole, introduced in the past decade, has a better molar efficacy against T. vaginalis isolates in vitro and fewer side effects [50]. However, because of the similarity in chemical structure between tinidazole and metronidazole, the former can fail to cure trichomoniasis [51] and recurrence rates already show a strong relationship between the MLC (maximum lethal concentration) for both drugs [50]. Despite the low prevalence of resistance, reliance on a single group of antimicrobial agents increases the possibility for resistance, thus drug resistance in T. vaginalis deserves more attention [52].
Genetic basis of drug resistance
The inherent problems and issues surrounding monitoring and assaying drug resistance in T. vaginalis are briefly discussed in Box 2. Recent studies exploring resistance mechanisms have focused on mining the genome sequence, through targeting candidate genes shown to have an important role in drug resistance in other organisms. For example, Pal and colleagues screened the T. vaginalis genome and identified homologs of bacterial nitroreductases (ntr) and nitroimidazole reductases (nims), absent from the majority of eukaryotes, but associated with reduced susceptibility to metronidazole in Helicobacter pylori (presence of premature stop codons in ntr genes) and in Bacteroides (overexpression of nim genes) [53]. Although neither of these genetic changes predicted metronidazole sensitivity in T. vaginalis, the enzymes were shown to most likely activate (NTR) or inactivate (NIM) metronidazole, and that metronidazole activation may possibly occur in both the cytosol and hydrogenosome, contrary to previous reports of activation occurring solely in the hydrogenosome. A more recent study showed down-regulation of flavin reductase and alcohol dehyrogenase activities in T. vaginalis strains with clinical metronidazole resistance [54]. The authors also found that thioredoxin reductase (previously hypothesized to play a role in resistance [55, 56]), had no role in clinical metronidazole resistance among the nine isolates examined, consistent with the idea that clinical resistance is not caused by a loss of drug activating pathways, as observed in anaerobic resistance.
Box 2. Challenges of monitoring and assaying drug resistance in T. vaginalis.
The antiparasitic effect of metronidazole is based upon its activation in the T. vaginalis mitochondrion- relict organelle, the hydrogenosome. The drug enters the organelle by passive diffusion and interferes with metabolic processes that occur on the hydrogenosome membrane. Several metabolic pathways such as those involving pyruvate: ferredoxin oxidoreductase[76, 77], thioredoxin reductase [55, 56], and nitroreductase [53] have been associated with metronidazole activation. These pathways can reduce the drug to its toxic form, although the reaction is reversible because oxygen can react with metronidazole and turn it back into an inactive form. Furthermore, a potential for drug resistance reversal was identified in laboratory-derived resistant parasites when grown for several generations without drug [77, 78]. Indeed, in anaerobically resistant lab strains, hydrogenosomal pathways and thioredoxin reductase are inactive [55, 76]. Thus, metronidazole resistance under aerobic and anaerobic conditions is likely two distinct processes (Table I), complicating assay methods and identification of molecular mechanisms of resistance.
Clinical isolates of T. vaginalis that are resistant to metronidazole only show resistance to the drug in the presence of oxygen [80] (with the exception of strain B7268 where anaerobic resistance developed in a patient [81]). The cause of aerobic resistance in clinical isolates is most likely due to a defect in oxygen scavenging, leading to elevated oxygen levels, re-oxidation of the metronidazole radical ion and detoxification of the drug [82]. It is also important to mention that metronidazole activating pathways are fully intact in clinical isolates [54, 83], while reduction of flavins is impaired [54].
Although an optimal assay with standardized cut-offs for determining metronidazole resistance has yet to be defined, two current methods used are: (i) an MLC test, which uses a gradient of drug concentration in order to determine the minimal lethal concentration (MLC) of drug at which no motile parasites are observed by microscopy; and (ii) a tritiated thymidine test, based upon scintillation counter measurement of radioactive nucleoside incorporation into DNA during DNA synthesis by viable parasites.
Table I.
Comparison of aerobic and anaerobic metronidazole resistance phenotypes in T. vaginalis.
| Characteristic | Aerobic ‘clinical’ resistance | Anaerobic ‘in vitro’ resistance | Refa |
|---|---|---|---|
| Population | Clinical isolates | Lab-induced isolates | |
| Level of resistance | Low | High | |
| Relationship with O2 | Manifests in presence of O2 | Manifests in absence of O2 | [80] |
| Isolates with both phenotypes? | Yes, can be induced in vitro | No (few exceptions) | |
| Hydrogenosome shape | Normal | Smaller | [84] |
| O2 scavenging | Lowered | No change | [82] |
| Hydrogenosomal pathways involved in drug activation | Fully active | Lost | [55, 56] |
| Glycolysis | Increased | No change | [83] |
| Lactate production | Higher | No change | [83] |
| Ethanol production | Reduced | No change | [83] |
| Example isolates | LA/03/CDC/1 [51]; CDC085 [85]; Fall River and C1:NIH [86] | BRIS/92/STDL/B7268 [87]; MR-5/30/50/100 [88]; IR78 [80] |
Where applicable.
However, comparative genomics of resistant versus parental strains is an even more powerful method to understand the genetic basis of drug resistance, as shown in recent bacterial studies [57]. A ‘functional genomics’ approach can also be used to screen for resistance in metagenomic samples or in individual parasites, since it does not require whole genome sequencing or prior knowledge about drug resistance candidates. In this method, whole genomes are sheared, DNA fragments cloned, and recombinant colonies selectively cultured under different drug concentrations (Figure 2) [58]. These studies also enable identification of drug resistance genes acquired by horizontal gene transfer from microbiota [59], although it must be noted that there is no evidence that lateral gene transfer directly contributes to metronidazole resistance in T. vaginalis [53]. A combination of novel genomic methods like these will improve our understanding concerning interactions between antimicrobial drugs, T. vaginalis and other microorganisms.
Figure 2. Genomic methods for the characterization of drug resistance.

Two approaches are shown: (1) Selection of drug resistant lines from culture-adapted sensitive strains grown in media containing increasing levels of drug (lab-induced anaerobic resistance); and (2) Isolation of drug resistant isolates from vaginal swabs (clinical aerobic resistance). In both (1) and (2), after quantifiable determination of the phenotype (minimal lethal concentration, or MLC) and DNA extraction, one of three steps or a combination of all can be applied: (a) Analysis of candidate genes known to be involved in drug resistance in other microbes; (b) Whole genome sequencing (WGS) and comparative genomics of sensitive and resistant strains; (c) Functional selection to characterize novel or confirm known resistance genes. In this latter process, genomic DNA is cloned into an expression system vector that can be cultured in the presence of drug. Recombinant clones containing putative resistance genes are able to grow in the presence of drug, and can then be sequenced for identification
The danger within: the T. vaginalis virus
T. vaginalis is parasitized by T. vaginalis virus (TVV), a double-stranded RNA (dsRNA) virus belonging to the Totiviridae family, members of which infect fungi and a variety of parasitic protists including Giardia lamblia and Leishmania braziliensis (reviewed in [6]). A recently identified TVV species brings the total number identified to four [60], each with a monosegmented dsRNA genome ~4.5 kb in length. This genome encodes two genes: a capsid protein and an RNA-dependent RNA polymerase, and since these ORFs are overlapping the polymerase is thought to be expressed as a fusion protein with the capsid following a ribosomal frameshift [60]. The extent to which the presence of these viruses influences parasite pathogenicity is unknown. Previous studies have correlated TVV infection with upregulation of parasite virulence factors, including cysteine proteases [61] and immunogenic surface proteins [62]. It may well be that viral dsRNA itself contributes to the parasite’s virulence, as indicated in the recent finding that dsRNA of Leishmania RNA virus-1 is a key determinant in the clinical outcome of leishmaniasis [63]. Briefly, Ives et al., found that upon parasite death, viral dsRNA is released that subverts the host immune response, promoting parasite metastasis and persistence, leading to severe mucocutaneous infection. TVV has recently been associated with symptomatic T. vaginalis infections [64] and may similarly be activating an innate immune response to promote inflammation. However, a full understanding of how these viruses affect the parasite’s behavior and the host immune response will require genome-wide expression assays such as microarrays, RNA-seq and proteomics.
How TVV is spread and maintained throughout the population is also important to consider if the virus is indeed having an effect on the parasite’s virulence. Similar to other members of the Totiviridae, TVV appears to lack an extracellular transmission cycle and is thought to be passed vertically during mitotic division of the parasite. However, virus particles have been seen at the plasma membrane of the parasite and localized near the Golgi apparatus [65] suggesting horizontal transmission is possible. Several studies have shown a TVV prevalence rate of ~50% [6], while an analysis of 153 T. vaginalis isolates indicated that ~75% of Type 1 isolates harbor TVV, while only 2–3% of Type 2 isolates do [34], suggesting that the Type 1 genotype is more conducive to TVV infection. Ultimately, monitoring the genetic diversity and distribution of distinct parasite populations while continuing to sequence the dsRNA viruses these parasites contain should help elucidate TVV transmission and its potential role as a virulence factor in T. vaginalis.
Concluding remarks
The publication of the first T. vaginalis genome in 2007 has led to significant advances in understanding essential aspects of the parasite’s biology. Gaps in our knowledge remain, but using advances in omics–technology many of these have the potential to be addressed. A key requirement for advancing the T. vaginalis research agenda is renewed funding support for the centralized T. vaginalis–omics database, TrichDB, one of the EuPathDB family of functional genomic databases [66], which has not been financially supported and consequently ‘rebuilt’ for several years. The T. vaginalis community will benefit enormously from efforts to maintain this resource, enabling utilization of–omics data to interrogate the unique and complicated aspects of the parasite’s biology, ultimately leading to practical and effective means of controlling trichomoniasis and its negative impact on global public health.
Figure I.

Cartoon of parabasalid evolutionary relationships.Phylogeny of key parasitic, commensal and free-living parabasalid species based on the protein sequence of the largest subunit of RNA polymerase II (Rbp1). The cartoon represents four of the six resolved groups, the Trichomonadea, Tritrichomonadea, Hypotrichomonidea and Trichonmypha. Adapted from Ref [75].
Acknowledgments
This work was funded in part by US National Institutes of Health grant 5R21AI083954-02. MDC and AG were partially supported by NIH training grant 5T32AI007180-28, and SDW by the MacCracken Program in the Graduate School of Arts and Science at New York University. We thank Steven Sullivan for his comments and proof reading, and apologize to authors whose work could not be cited due to space constraints.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.World Health Organization, D.o.R.H.a.R. WHO Press; 2011. Prevalence and incidence of selected sexually transmitted infections, Chlamydia trachomatis, Neisseria gonorrhoeae, syphilis and Trichomonas vaginalis: methods and results used by WHO to generate 2005 estimates. http://www.who.int/reproductivehealth/publications/rtis/9789241502450/en/index.html. [Google Scholar]
- 2.Van Der Pol B. Editorial Commentary: Trichomonas vaginalis Infection: The Most Prevalent Nonviral Sexually Transmitted Infection Receives the Least Public Health Attention. Clin Infect Dis. 2007;44:23–25. doi: 10.1086/509934. [DOI] [PubMed] [Google Scholar]
- 3.McClelland RS, et al. Infection with Trichomonasvaginalis increases the risk of HIV-1 acquisition. J Infect Dis. 2007;195:698–702. doi: 10.1086/511278. [DOI] [PubMed] [Google Scholar]
- 4.Guenthner PC, et al. Trichomonasvaginalis -induced epithelial monolayer disruption and human immunodeficiency virus type 1 (HIV-1) replication: implications for the sexual transmission of HIV-1. Infect Immun. 2005;73:4155–4160. doi: 10.1128/IAI.73.7.4155-4160.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sorvillo F, et al. Trichomonasvaginalis , HIV, and African-Americans. Emerg Infect Dis. 2001;7:927–932. doi: 10.3201/eid0706.010603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goodman RP, et al. Trichomonasvirus: a new genus of protozoan viruses in the family Totiviridae. Arch Virol. 2011;156:171–179. doi: 10.1007/s00705-010-0832-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Larsen B, Hwang J. Mycoplasma, Ureaplasma, and adverse pregnancy outcomes: a fresh look. Infect Dis Obstet Gynecol. 2010 doi: 10.1155/2010/521921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lumsden WH, RD, Heyworth R, Harrison C. Treatment failure in Trichomonasvaginalis vaginitis. Genitourin Med. 1988;64:217–218. doi: 10.1136/sti.64.4.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carlton JM, et al. Draft genome sequence of the sexually transmitted pathogen Trichomonas vaginalis. Science. 2007;315:207–212. doi: 10.1126/science.1132894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zubacova Z, et al. Comparative analysis of trichomonad genome sizes and karyotypes. Mol Biochem Parasitol. 2008;161:49–54. doi: 10.1016/j.molbiopara.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 11.Pritham EJ, et al. Mavericks, a novel class of giant transposable elements widespread in eukaryotes and related to DNA viruses. Gene. 2007;390:3–17. doi: 10.1016/j.gene.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 12.Kim PG, et al. A scaffold analysis tool using mate-pair information in genome sequencing. J Biomed Biotechnol. 2008 doi: 10.1155/2008/675741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagarajan N, et al. Scaffolding and validation of bacterial genome assemblies using optical restriction maps. Bioinformatics. 2008;24:1229–1235. doi: 10.1093/bioinformatics/btn102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou S, et al. A single molecule scaffold for the maize genome. PLoS genetics. 2009;5:e1000711. doi: 10.1371/journal.pgen.1000711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noel CJ, et al. Trichomonasvaginalis vast BspA-like gene family: evidence for functional diversity from structural organisation and transcriptomics. BMC Genomics. 2010;11:99. doi: 10.1186/1471-2164-11-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horvathova L, et al. Transcriptomic Identification of Iron-Regulated and Iron-Independent Gene Copies within the Heavily Duplicated Trichomonasvaginalis Genome. Genome Biol Evol. 2012;4:905–917. doi: 10.1093/gbe/evs078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen XS, et al. High throughput genome-wide survey of small RNAs from the parasitic protists Giardia intestinalis and Trichomonasvaginalis. Genome Biol Evol. 2009;1:165–175. doi: 10.1093/gbe/evp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen XS, et al. Characterization of RNase MRP RNA and novel snoRNAs from Giardia intestinalis and Trichomonasvaginalis. BMC genomics. 2011;12:550. doi: 10.1186/1471-2164-12-550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piccinelli P, et al. Identification and analysis of ribonuclease P and MRP RNA in a broad range of eukaryotes. Nucleic acids research. 2005;33:4485–4495. doi: 10.1093/nar/gki756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simoes-Barbosa A, et al. Spliceosomal snRNAs in the unicellular eukaryote Trichomonas vaginalis are structurally conserved but lack a 5’-cap structure. RNA. 2008;14:1617–1631. doi: 10.1261/rna.1045408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang PJ, et al. Identification of putative miRNAs from the deep-branching unicellular flagellates. Genomics. 2012;99:101–107. doi: 10.1016/j.ygeno.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 22.Lin WC, et al. Identification of microRNA in the protist Trichomonas vaginalis. Genomics. 2009;93:487–493. doi: 10.1016/j.ygeno.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 23.Lin WC, et al. Malate dehydrogenase is negatively regulated by miR-1 in Trichomonas vaginalis. Parasitol Res. 2009;105:1683–1689. doi: 10.1007/s00436-009-1616-5. [DOI] [PubMed] [Google Scholar]
- 24.Schneider RE, et al. The Trichomonasvaginalis hydrogenosome proteome is highly reduced relative to mitochondria, yet complex compared with mitosomes. Int J Parasitol. 2011;41:1421–1434. doi: 10.1016/j.ijpara.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Miguel N, et al. Proteome analysis of the surface of Trichomonasvaginalis reveals novel proteins and strain-dependent differential expression. Molecular & cellular proteomics : MCP. 2010;9:1554–1566. doi: 10.1074/mcp.M000022-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang KY, et al. A proteome reference map of Trichomonas vaginalis. Parasitol Res. 2009;104:927–933. doi: 10.1007/s00436-008-1274-z. [DOI] [PubMed] [Google Scholar]
- 27.Feschotte C, Pritham EJ. DNA transposons and the evolution of eukaryotic genomes. Annual review of genetics. 2007;41:331–368. doi: 10.1146/annurev.genet.40.110405.090448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fernie AR, et al. Metabolite profiling: from diagnostics to systems biology. Nat Rev Mol Cell Biol. 2004;5:763–769. doi: 10.1038/nrm1451. [DOI] [PubMed] [Google Scholar]
- 29.Mead JR, et al. Use of Trichomonasvaginalis Clinical Isolates to Evaluate Correlation of Gene Expression and Metronidazole Resistance. J Parasitol. 2006;92:196–199. doi: 10.1645/GE-616R.1. [DOI] [PubMed] [Google Scholar]
- 30.Upcroft JA, et al. Genotyping Trichomonasvaginalis. Int J Parasitol. 2006;36:821–828. doi: 10.1016/j.ijpara.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 31.Conrad M, et al. Microsatellite polymorphism in the sexually transmitted human pathogen Trichomonasvaginalis indicates a genetically diverse parasite. Mol Biochem Parasitol. 2011;175:30–38. doi: 10.1016/j.molbiopara.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prokopi M, et al. A preliminary investigation of microsatellite-based genotyping in Trichomonasvaginalis. Trans R Soc Trop Med Hyg. 2011;105:479–481. doi: 10.1016/j.trstmh.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 33.Cornelius DC, et al. Genetic Characterization of Trichomonasvaginalis Isolates by Use of Multilocus Sequence Typing. J Clin Microbiol. 2012;50:3293–3300. doi: 10.1128/JCM.00643-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Conrad MD, et al. Extensive genetic diversity, unique population structure and evidence of genetic exchange in the sexually transmitted parasite Trichomonasvaginalis. PLoS Negl Trop Dis. 2012;6:e1573. doi: 10.1371/journal.pntd.0001573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meade JC, et al. Genetic diversity of Trichomonasvaginalis clinical isolates determined by EcoRI restriction fragment length polymorphism of heat-shock protein 70 genes. Am J Trop Med Hyg. 2009;80:245–251. [PMC free article] [PubMed] [Google Scholar]
- 36.Snipes LJ, et al. Molecular Epidemiology of Metronidazole Resistance in a Population of Trichomonasvaginalis Clinical Isolates. J Clin Microbiol. 2000;38:3004–3009. doi: 10.1128/jcm.38.8.3004-3009.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balmer O, Tanner M. Prevalence and implications of multiple-strain infections. Lancet Infect Dis. 2011;11:868–878. doi: 10.1016/S1473-3099(11)70241-9. [DOI] [PubMed] [Google Scholar]
- 38.Ravel J, et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4680–4687. doi: 10.1073/pnas.1002611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brotman RM, et al. Association between Trichomonas vaginalis and vaginal bacterial communities among asymptomatic reproductive-age women. Sexually Transmitted Diseases. 2012 doi: 10.1097/OLQ.0b013e3182631c79. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nelson DE, et al. Characteristic male urine microbiomes associate with asymptomatic sexually transmitted infection. Plos One. 2010;5:e14116. doi: 10.1371/journal.pone.0014116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dong Q, et al. The microbial communities in male first catch urine are highly similar to those in paired urethral swab specimens. Plos One. 2011;6:e19709. doi: 10.1371/journal.pone.0019709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Price LB, et al. The effects of circumcision on the penis microbiome. Plos One. 2010;5:e8422. doi: 10.1371/journal.pone.0008422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gajer P, et al. Temporal dynamics of the human vaginal microbiota. Sci Transl Med. 2012;4:132ra152. doi: 10.1126/scitranslmed.3003605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jakobsson HE, et al. Short-Term Antibiotic Treatment Has Differing Long-Term Impacts on the Human Throat and Gut Microbiome. Plos One. 2010:5. doi: 10.1371/journal.pone.0009836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wlodarska M, et al. Antibiotic treatment alters the colonic mucus layer and predisposes the host to exacerbated Citrobacter rodentium-induced colitis. Infect Immun. 2011;79:1536–1545. doi: 10.1128/IAI.01104-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cobo ER, et al. Murine models of vaginal trichomonad infections. Am J Trop Med Hyg. 2011;85:667–673. doi: 10.4269/ajtmh.2011.11-0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patton DL, et al. Development of a nonhuman primate model for Trichomonasvaginalis infection. Sex Transm Dis. 2006;33:743–746. doi: 10.1097/01.olq.0000218871.89901.61. [DOI] [PubMed] [Google Scholar]
- 48.Kirkcaldy RD, et al. Trichomonasvaginalis Antimicrobial Drug Resistance in 6 US Cities, STD Surveillance Network, 2009–2010. Emerg Infect Dis. 2012;18:939–943. doi: 10.3201/eid1806.111590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Upcroft JA, et al. Metronidazole resistance in Trichomonasvaginalis from highland women in Papua New Guinea. Sex Health. 2009;6:334–338. doi: 10.1071/SH09011. [DOI] [PubMed] [Google Scholar]
- 50.Crowell AL, et al. In vitro metronidazole and tinidazole activities against metronidazole-resistant strains of Trichomonasvaginalis. Antimicrob Agents Chemother. 2003;47:1407–1409. doi: 10.1128/AAC.47.4.1407-1409.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goldman LM, et al. Treatment of metronidazole-resistant Trichomonas vaginalis. Sex Health. 2009;6:345–347. doi: 10.1071/SH09064. [DOI] [PubMed] [Google Scholar]
- 52.Secor WE. Trichomonasvaginalis : treatment questions and challenges. Expert Rev Anti Infect Ther. 2012;10:107–109. doi: 10.1586/eri.11.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pal D, et al. Giardia, Entamoeba, and Trichomonas enzymes activate metronidazole (nitroreductases) and inactivate metronidazole (nitroimidazole reductases) Antimicrob Agents Chemother. 2009;53:458–464. doi: 10.1128/AAC.00909-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Leitsch D, et al. Down-regulation of flavin reductase and alcohol dehydrogenase-1 (ADH1) in metronidazole-resistant isolates of Trichomonasvaginalis. Mol Biochem Parasitol. 2012;183:177–183. doi: 10.1016/j.molbiopara.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leitsch D, et al. Trichomonasvaginalis : metronidazole and other nitroimidazole drugs are reduced by the flavin enzyme thioredoxin reductase and disrupt the cellular redox system. Implications for nitroimidazole toxicity and resistance. Mol Microbiol. 2009;72:518–536. doi: 10.1111/j.1365-2958.2009.06675.x. [DOI] [PubMed] [Google Scholar]
- 56.Leitsch D, et al. The flavin inhibitor diphenyleneiodonium renders Trichomonasvaginalis resistant to metronidazole, inhibits thioredoxin reductase and flavin reductase, and shuts off hydrogenosomal enzymatic pathways. Mol Biochem Parasitol. 2010;171:17–24. doi: 10.1016/j.molbiopara.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 57.Howden BP, et al. Evolution of multidrug resistance during Staphylococcus aureus infection involves mutation of the essential two component regulator WalKR. PLoS Pathog. 2011;7:e1002359. doi: 10.1371/journal.ppat.1002359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bush K, et al. Tackling antibiotic resistance. Nature reviews Microbiology. 2011;9:894–896. doi: 10.1038/nrmicro2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smillie CS, et al. Ecology drives a global network of gene exchange connecting the human microbiome. Nature. 2011;480:241–244. doi: 10.1038/nature10571. [DOI] [PubMed] [Google Scholar]
- 60.Goodman RP, et al. Clinical isolates of Trichomonasvaginalis concurrently infected by strains of up to four Trichomonasvirus species (Family Totiviridae) Journal of virology. 2011;85:4258–4270. doi: 10.1128/JVI.00220-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Provenzano D, Alderete JF. Analysis of human immunoglobulin-degrading cysteine proteinases of Trichomonasvaginalis. Infect Immun. 1995;63:3388–3395. doi: 10.1128/iai.63.9.3388-3395.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Khoshnan A, Alderete JF. Trichomonas vaginalis with a double-stranded RNA virus has upregulated levels of phenotypically variable immunogen mRNA. Journal of virology. 1994;68:4035–4038. doi: 10.1128/jvi.68.6.4035-4038.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ives A, et al. Leishmania RNA virus controls the severity of mucocutaneous leishmaniasis. Science. 2011;331:775–778. doi: 10.1126/science.1199326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fraga J, et al. Double-stranded RNA viral infection of Trichomonasvaginalis and correlation with genetic polymorphism of isolates. Experimental parasitology. 2011;127:593–599. doi: 10.1016/j.exppara.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 65.Benchimol M, et al. Virus in Trichomonas--an ultrastructural study. Parasitology international. 2002;51:293–298. doi: 10.1016/s1383-5769(02)00016-8. [DOI] [PubMed] [Google Scholar]
- 66.Aurrecoechea C, et al. GiardiaDB and TrichDB: integrated genomic resources for the eukaryotic protist pathogens Giardia lamblia and Trichomonasvaginalis. Nucleic acids research. 2009;37:D526–530. doi: 10.1093/nar/gkn631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lindmark DG, Muller M. Hydrogenosome, a cytoplasmic organelle of the anaerobic flagellate Tritrichomonasfoetus , and its role in pyruvate metabolism. The Journal of biological chemistry. 1973;248:7724–7728. [PubMed] [Google Scholar]
- 68.Cepicka I, et al. Critical taxonomic revision of Parabasalids with description of one new genus and three new species. Protist. 2010;161:400–433. doi: 10.1016/j.protis.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 69.Leterrier M, et al. Trichomonads in pleural effusion: case report, literature review and utility of PCR for species identification. New Microbiol. 2012;35:83–87. [PubMed] [Google Scholar]
- 70.Gookin JL, et al. Molecular characterization of trichomonads from feces of dogs with diarrhea. J Parasitol. 2005;91:939–943. doi: 10.1645/GE-474R.1. [DOI] [PubMed] [Google Scholar]
- 71.Stark D, et al. A review of the clinical presentation of dientamoebiasis. Am J Trop Med Hyg. 2010;82:614–619. doi: 10.4269/ajtmh.2010.09-0478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Reinmann K, et al. Tritrichomonas foetus isolates from cats and cattle show minor genetic differences in unrelated loci ITS-2 and EF-1alpha. Vet Parasitol. 2012;185:138–144. doi: 10.1016/j.vetpar.2011.09.032. [DOI] [PubMed] [Google Scholar]
- 73.Robinson RA, et al. Emerging infectious disease leads to rapid population declines of common British birds. Plos One. 2010;5:e12215. doi: 10.1371/journal.pone.0012215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yubuki N, et al. Cryptic diversity of free-living parabasalids, Pseudotrichomonaskeilini and Lacusteria cypriaca n. g., n. sp., as inferred from small subunit rDNA sequences. J Eukaryot Microbiol. 2010;57:554–561. doi: 10.1111/j.1550-7408.2010.00509.x. [DOI] [PubMed] [Google Scholar]
- 75.Malik SB, et al. Phylogeny of parasitic parabasalia and free-living relatives inferred from conventional markers vs. Rpb1, a single-copy gene. Plos One. 2011;6:e20774. doi: 10.1371/journal.pone.0020774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kulda J. Trichomonads, hydrogenosomes and drug resistance. Int J Parasitol. 1999;29:199–212. doi: 10.1016/s0020-7519(98)00155-6. [DOI] [PubMed] [Google Scholar]
- 77.Upcroft P, Upcroft JA. Drug targets and mechanisms of resistance in the anaerobic protozoa. Clinical microbiology reviews. 2001;14:150–164. doi: 10.1128/CMR.14.1.150-164.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wright JM, et al. Susceptibility in vitro of clinically metronidazole-resistant Trichomonas vaginalis to nitazoxanide, toyocamycin, and 2-fluoro-2’-deoxyadenosine. Parasitology research. 2010;107:847–853. doi: 10.1007/s00436-010-1938-3. [DOI] [PubMed] [Google Scholar]
- 79.Hrdy I, et al. Alternative pathway of metronidazole activation in Trichomonasvaginalis hydrogenosomes. Antimicrob Agents Chemother. 2005;49:5033–5036. doi: 10.1128/AAC.49.12.5033-5036.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Meingassner JG, Thurner J. Strain of Trichomonasvaginalis resistant to metronidazole and other 5-nitroimidazoles. Antimicrob Agents Chemother. 1979;15:254–257. doi: 10.1128/aac.15.2.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Voolmann T, Boreham P. Metronidazole resistant Trichomonasvaginalis in Brisbane. The Medical journal of Australia. 1993;159:490. doi: 10.5694/j.1326-5377.1993.tb137978.x. [DOI] [PubMed] [Google Scholar]
- 82.Yarlett N, et al. Metronidazole-resistant clinical isolates of Trichomonasvaginalis have lowered oxygen affinities. Mol Biochem Parasitol. 1986;19:111–116. doi: 10.1016/0166-6851(86)90115-5. [DOI] [PubMed] [Google Scholar]
- 83.Ellis JE, et al. Influence of oxygen on the fermentative metabolism of metronidazole-sensitive and resistant strains of Trichomonasvaginalis. Mol Biochem Parasitol. 1992;56:79–88. doi: 10.1016/0166-6851(92)90156-e. [DOI] [PubMed] [Google Scholar]
- 84.Wright JM, et al. Hydrogenosomes of laboratory-induced metronidazole-resistant Trichomonasvaginalis lines are downsized while those from clinically metronidazole-resistant isolates are not. J Eukaryot Microbiol. 2010;57:171–176. doi: 10.1111/j.1550-7408.2009.00455.x. [DOI] [PubMed] [Google Scholar]
- 85.Narcisi EM, Secor WE. In vitro effect of tinidazole and furazolidone on metronidazole-resistant Trichomonasvaginalis. Antimicrob Agents Chemother. 1996;40:1121–1125. doi: 10.1128/aac.40.5.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Muller M, et al. Three metronidazole-resistant strains of Trichomonasvaginalis from the United States. Am J Obstet Gynecol. 1980;138:808–812. doi: 10.1016/s0002-9378(16)32741-7. [DOI] [PubMed] [Google Scholar]
- 87.Upcroft P, Upcroft JA. Drug Targets and Mechanisms of Resistance in the Anaerobic Protozoa. Clin Microbiol Rev. 2001;14:150–164. doi: 10.1128/CMR.14.1.150-164.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rasoloson D, et al. Mechanisms of in vitro development of resistance to metronidazole in Trichomonasvaginalis. Microbiology. 2002;148:2467–2477. doi: 10.1099/00221287-148-8-2467. [DOI] [PubMed] [Google Scholar]