

Abstract
The respiratory epithelium is a physical and functional barrier actively involved in the clearance of environmental agents. The alveolar compartment is lined with membranous pneumocytes, known as type I alveolar epithelial cells (AEC I), and granular pneumocytes, type II alveolar epithelial cells (AEC II). AEC II are responsible for epithelial reparation upon injury and ion transport and are very active immunologically, contributing to lung defense by secreting antimicrobial factors. AEC II also secrete a broad variety of factors, such as cytokines and chemokines, involved in activation and differentiation of immune cells and are able to present antigen to specific T cells. Another cell type important in lung defense is the pulmonary macrophage (PuM). Considering the architecture of the alveoli, a good communication between the external and the internal compartments is crucial to mount effective responses. Our hypothesis is that being in the interface, AEC may play an important role in transmitting signals from the external to the internal compartment and in modulating the activity of PuM. For this, we collected supernatants from AEC unstimulated or stimulated in vitro with lipopolysaccharide (LPS). These AEC-conditioned media were used in various setups to test for the effects on a number of macrophage functions: (i) migration, (ii) phagocytosis and intracellular control of bacterial growth, and (iii) phenotypic changes and morphology. Finally, we tested the direct effect of AEC-conditioned media on bacterial growth. We found that AEC-secreted factors had a dual effect, on one hand controlling bacterial growth and on the other hand increasing macrophage activity.
INTRODUCTION
In the respiratory tract, both innate and adaptive immune responses are responsible for the protection and defense against inhaled particles or infectious agents. The respiratory epithelium is considered to be a physical and functional barrier also actively involved in the clearance of environmental agents. Airway epithelial cells produce antibacterial factors, promote inflammatory responses, and regulate gas exchange in the body (1, 2). The alveolar compartment is lined with membranous pneumocytes, known as type I alveolar epithelial cells (AEC I), and granular pneumocytes, type II alveolar epithelial cells (AEC II). AEC I are squamous, large, thin cells that cover 90 to 95% of the alveolar surface. These cells are not only involved in gaseous exchange but also forming a barrier able to sense microbial products and generate inflammatory responses (3, 4). AEC II are cuboidal cells that constitute around 15% of total lung cells and cover about 7% of the total alveolar surface. AEC II are responsible for epithelium reparation upon injury and ion transport. AEC II contribute also to lung defense by secreting antimicrobial products such as complement, lysozyme, and surfactant proteins (SP). SP-A and SP-D (C-type lectins) are responsible for host defense, enhancing the clearance of various microbial pathogens, whereas SP-B and SP-C are responsible for the biophysical reduction of the surface tension during gas exchange (4–6).
Although both AEC I and II are constantly contributing to airway defense, many studies have focused on AEC II, perhaps because these cells are more active immunologically. AEC II secrete a broad variety of factors, such as cytokines and chemokines, involved in activation and differentiation of immune cells and have been described to be able to present antigen to specific T cells (6–11). Together with AEC, other cells, such as macrophages, participate in the defense of the respiratory tract. The lower respiratory tract has two macrophage populations: alveolar macrophages (AM) in the alveoli and interstitial macrophages located in the insterstitium. Both types of macrophages have been described to elicit strong responses against a broad variety of stimuli (12). Although alveolar and interstitial macrophages are morphologically similar, it is possible that their functions are regulated according to their anatomic localization and exposure to different microenvironments in the lungs (12, 13). AM have been described to display a high phagocytic capacity and have a key role in the initiation and resolution of inflammatory responses in the alveoli (13–15). In the maturation process to AM from circulating monocytes, interstitial macrophages are considered to be an intermediate cell type displaying certain characteristics closer to those of monocytes than to AM (16, 17). Both cell types are responsible for maintaining the respiratory tract free from microbes and other particulate agents, but while AM are active on the external airway compartments, the function of interstitial macrophages would be more restricted to the lung tissue and to communicate with other immune cells in the interstitium, constituting an internal link between the innate and the adaptive branches of the immune system.
Much research has been directed to study the behavior of AM, but less is known about interstitial macrophages. In the present work, we aimed to study in closer detail the functional characteristics of interstitial macrophages, named herein pulmonary macrophages (PuM). Considering the architecture of the alveoli, good communication between the external and the internal compartments is crucial to mount effective responses. Our hypothesis is that being in the interface, AEC may play an important role in transmitting signals from the external to the internal compartment and in modulating the activity of PuM. For this, we collected supernatants from AEC unstimulated or stimulated in vitro with lipopolysaccharide (LPS). These AEC-derived media were used in various setups to test for the effect on a number of macrophage functions: (i) migration, (ii) phagocytosis and intracellular control of bacteria growth, and (iii) phenotypic changes and morphology. Finally, we tested the effect on bacteria, namely, direct killing and opsonization. We describe here that AEC-secreted factors had a dual effect, on one hand on the control of bacterial growth and on the other hand in increasing macrophage activity. The observed effects could not be ascribed to the activity of individual factors but rather to the combination of different factors possibly acting in an additive or synergistic manner. We discuss these effects on the context of innate defense in the respiratory tract.
MATERIALS AND METHODS
Mice.
The studies were performed using 8- to 12-week-old female C57BL/6 mice purchased from NOVA-SCB, Sweden, or TLR4−/− mice (18) obtained from the Karolinska Institute, Sweden, with the permission of S. Akira (Osaka University, Japan). All animals were kept at the Animal Department of the Arrhenius Laboratories, Stockholm University, Sweden, and housed in pathogen-free conditions. Experiments were performed in accordance with the guidelines of the Animal Research Ethics Board at Stockholm University. Mice were supervised daily, and sentinel mice were used to assess and ensure pathogen-free conditions in the facility.
Bacteria.
We used Mycobacterium bovis BCG, transformed with a dual reporter plasmid containing the human codon-optimized and enhanced green fluorescent protein (EGFP) and the luxAB genes from Vibrio harveyi (19), called herein GFP-BCG. The use of this construct is convenient, since bacteria can be quantified immediately by luminescence, while the classical evaluation of BCG growth in agar plates takes between 2 to 3 weeks. Bacterial contents are expressed as relative luminescence units (RLU), which correlate with the number of CFU (20, 21). GFP-BCG was grown in Middlebrook 7H9 broth (Difco, Sparks, MD) supplemented with albumin-dextrose-catalase (ADC), 0.5% glycerol, 0.05% Tween 80 (vol/vol), and 50 μg/ml hygromycin for 10 to 15 days. Bacteria were collected at a log phase of growth (absorbance of 1.0 measured at an optical density at 650 nm [OD650]) and frozen in phosphate-buffered saline (PBS) with 10% glycerol and kept at −70°C. Before infection of cell cultures, a vial (108 CFU/ml) was thawed and placed in culture as described above for 4 to 5 days, reaching an early log phase (OD650 of ∼0.3). To determine the RLU, decanal (Sigma-Aldrich) was used as a specific substrate for the bacterial enzyme LuxAB. Decanal was dissolved in 70% ethanol and added to the lysates at a final concentration of 0.01%. The samples were mixed immediately, and luminescence was measured after 15 s in a Turner BioSystems Modulus luminometer.
Isolation of AEC and PuM.
Total pulmonary cells were prepared using Corti's protocol (22) with previously described modifications (20, 21). In short, CD45+ cells were obtained from total lung cells using magnetic-activated cell sorting (MACS; Miltenyi Biotec, Bergisch Gladbach, Germany) and subsequently cultured for 48 h in RPMI (Gibco-Invitrogen, Paisley, United Kingdom) supplemented with 10% fetal calf serum (FCS), 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.02 M HEPES, and 0.05 M 2-mercaptoethanol (Sigma) at 37°C and 5% CO2. Pulmonary macrophages (PuM) were isolated by adhesion to get rid of cellular debris and nonadherent cells, such as dendritic cells (DC). Cells isolated from lung parenchyma and not from bronchoalveolar lavage have been reported previously to be enriched in interstitial macrophages (23–25). On average, 98% of the adherent cells using this methodology were positive for the macrophage marker F4/80, as determined by flow cytometry. Isolated PuM were on average 80% F4/80+ CD11c+ and 30% F4/80+ CD11b+ from cells analyzed separately (Table 2). Alveolar epithelial cells (AEC) were obtained by depleting CD45+ and CD146+ cells from lung preparations using MACS. After 48 h in culture, 92 to 95% of these CD45− CD146− cells exhibited an AEC phenotype, where approximately 22% expressed podoplanin (AEC type I) and approximately 72% expressed CD74 (AEC type II) as determined by flow cytometry.
Table 2.
Effect of AECsup on the surface molecule expression of PuM
| Marker | %a |
||
|---|---|---|---|
| Medium | AECsup | LPS | |
| MHC-II | 58 ± 16 | 47 ± 18 | 75 ± 13 |
| CD80 | 89 ± 4 | 93 ± 2 | 88 ± 1 |
| CD86 | 21 ± 5 | 17 ± 6 | 63 ± 3 |
| CD40 | 12 ± 5 | 16 ± 4 | 82 ± 9 |
| MMR | 18 ± 1 | 38 ± 4 | 15 ± 1 |
| CD11b | 33 ± 8 | 52 ± 0 | 87 ± 7 |
| CD11c | 82 ± 4 | 73 ± 4 | 68 ± 7 |
Data are percentages of events for MHC-II, CD80, CD40, MMR, CD11b, and CD11c of F4/80-positive events. Clear changes in the phenotype marker are shown in bold. The values are given as means ± SD from 2 or 4 independent experiments.
Generation of BMM.
Bone marrow-derived macrophages (BMM) were generated as previously described (26). Briefly, after sacrifice, the femur and tibia of the hind legs were removed. Bone marrow cavities were flushed with cold, sterile PBS, and cells were harvested and frozen at −80°C until further use. After being thawed, the bone marrow cells were cultured in complete RPMI supplemented with 20% L929 cell-conditioned medium (as a source of macrophage colony-stimulating factor) and cultured for 7 days, and medium was replaced every second day. Before use, the BMM were differentiated by incubating cells in complete RPMI or advanced RPMI (AdRPMI; Gibco-Invitrogen, Paisley, United Kingdom) with 10% FCS, 0.02 M HEPES, and 2 mM l-glutamine without L929 cell-conditioned medium for 24 h. For induction of macrophages with an M1 or M2 phenotype, bone marrow cells were cultured until differentiation and then incubated in medium with either 20 ng/ml gamma interferon (IFN-γ) and 10 ng/ml LPS or 10 ng/ml interleukin 4 (IL-4) for 48 h, respectively (27–29).
Preparation of AEC-derived media.
AEC were plated (5 × 104 cells/well) in 96-well flat-bottom plates (Corning Inc., Corning, NY). Cells were either kept unstimulated or stimulated with 10 μg/ml of LPS (Sigma) for 24 h. Cell culture supernatants were collected, pooled, and stored at −70°C until use. These supernatants were named AECsup for medium from untreated AEC and AECLPS for medium from AEC stimulated with LPS. In some experiments, media from total splenocyte (SC) cultures were used and prepared under the same conditions. These are named SCsup for untreated cells.
Phagocytosis and intracellular bacterial growth.
BMM and PuM were cultured in 24- or 48-well plates as described above at 2 × 105 or 1 × 105 cells per well, respectively. Cells were kept with complete RPMI or AdRPMI without antibiotics for 24 h. Before infection, cells were left untreated or treated with AECsup, AECLPS, or SCsup for 20 to 24 h. Next, cells were infected with GFP-BCG at a multiplicity of infection (MOI) of 10:1 (bacteria:cell) for 4 h. We and others have used a 4-h infection time, which may be considered a long period where even bacterial growth can take place. However, since BCG is a slow-growing bacterium with a division time of 18 h, the effect of bacterial growth during the 4-h infection is minimal. After infection, cells were washed three times and treated with gentamicin (100 μg/ml) for 30 to 60 min at 37°C, washed three times, and lysed with 0.2% Triton X-100 (Sigma) to measure phagocytosis. We consider this time point as the starting point (0 h). For the determination of intracellular bacterial growth, cells were kept with or without AEC- or SC-derived medium for 48 h. RLU/106 cells were calculated by measurement of luminescence from lysed cells as described above. To assess for the need of constant presence of AEC-derived factors in the process of intracellular mycobacterial growth control, cells were pretreated only with AECsup and washed before infection. This group is named AECsupPre.
Opsonization and direct bacterial killing.
To evaluate whether AEC-derived media had opsonizing effects, GFP-BCG were incubated with AECsup or normal medium for 30 min at 37°C. Treated and untreated GFP-BCG were used to infect BMM at an MOI of 10:1 (bacteria:cell) for 4 h at 37°C. Phagocytosis by BMM was calculated as RLU/106 cells by measurement of luminescence from lysed cells as described above.
To assess direct bacterial killing by AEC-derived media, 2.5 × 106 CFU/ml of GFP-BCG was incubated with AECsup or gentamicin at concentrations of 16, 50, and 100 μg/ml or left untreated for 4 and 24 h. After this time, GFP-BCG were washed once and kept in complete RPMI without antibiotics for 72 h. Bacterial growth was determined by measurement of RLU as described above.
Transmembrane migration assay.
Total pulmonary cells (106/100 μl) were placed in the upper chamber of a Transwell insert (5-μm pore size, 24-well plate; Corning Costar), and media (AdRPMI with 0.5% FCS and 2 mM L-glutamine) or supernatants from AEC cultures were placed in the lower chamber. After 2 h, cells that had migrated to the lower chamber were removed and analyzed. The relative number of cells migrating was determined on a flow cytometer using Calibrite beads (BD Biosciences), where a fixed number of beads was included in each sample and the number of cells/1,000 beads was evaluated. Data were normalized to the number of cells migrating toward the medium control. To determine the proportion of different cell types migrating, cells were stained with anti-F4/80-allophycocyanin (APC) (Serotec; A3-1), anti-CD11c-R-phycoerythrin (PE) (BD Pharmingen; HL3), anti-CD90.2-fluorescein isothiocyanate (FITC) (BD Pharmingen; 53-2.1), anti-CD19-R-PE (BD Pharminen; ID3), and anti-Ly-6B.2-Alexa 647 (Serotec; 7/4) antibodies and analyzed on a flow cytometer (FACScalibur; BD Biosciences).
In vitro wound healing assay.
Wound healing assay was performed as previously described (30, 31). The J774A1 (referred to as J774 cells in the text) cell line was obtained from the European Type Tissue Culture Collection (CAMR, Salisbury, United Kingdom). PuM were prepared as described above. Cells were cultured to reach around 80% of confluence. After cells were washed, a linear scratch wound was made on the cell monolayer by using a universal pipette tip (P200), after which plates were washed and incubated in either complete RPMI or cell culture supernatants from AEC cultures at the indicated time points. Cells were then fixed with 4% formalin, and the nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen). Cell migration was evaluated by monitoring the number of cells/area using a phase-contrast microscope (Nikon TE 300, ×20 magnification) equipped with a charge-coupled-device (CCD) camera (NIKON digital camera DXM1200F).
Determination of the PuM phenotype.
PuM were treated with AECsup, LPS (10 μg/ml), or kept untreated for 24 h. To determine the phenotype, cells were stained with APC-labeled antibodies to F4/80 (AbD Serotec, Dusseldorf, Germany). Expression of cell surface major histocompatibility complex class II (MHC-II) and costimulatory molecules was determined by using PE-labeled antibodies to MHC-II (I-Ad/I-Ed), CD40, CD80, and CD86 (BD Bioscience Pharmingen, San Diego, CA). CD11c and CD11b were assessed by anti-CD11c-R-PE (BD Pharmingen) and anti-CD11b-FITC (BD Pharmingen) antibodies, respectively. Mannose receptor (MMR) expression was determined by anti-MMR-PE antibody (R&D Systems, Abbington, United Kingdom). All samples were analyzed on a Becton, Dickinson FACScalibur, and data were analyzed using CellQuestPro software (Becton, Dickinson Immunocytometry Systems). All the analyses were performed with an acquisition of 10,000 events.
Actin staining and microscopy.
After BMM or PuM were cultured as described above but on glass cover slides, cells were fixed with 3.7% formaldehyde (Sigma-Aldrich) in PBS for 10 min at 37°C and then permeabilized in a 0.1% Triton X-100 solution for 5 min. Cells were then stained with FITC-labeled phalloidin (Sigma-Aldrich) and DAPI for 45 min and mounted (SlowFade Gold antifade reagent; Invitrogen), and their shape and actin cytoskeleton were visualized using a Zeiss cell observer fluorescence microscope.
Statistical analysis.
Data are presented as the means ± standard deviations (SD) or standard errors of the means (SEM). Differences between treatment groups were analyzed using a Student t test or one-way analysis of variance (ANOVA) followed by Bonferroni's posttest for multiple comparisons. Differences between treatments at different time points were analyzed using a two-way ANOVA followed by Bonferroni's multiple comparison test. Differences were considered significant at a P value of <0.05. All data were analyzed using the GraphPad InStat version 5.0 (GraphPad Software, San Diego, CA).
RESULTS
Treatment with AEC-derived medium increases phagocytosis and intracellular growth control by BMM and PuM.
Since AEC are important in the defense against inhaled pathogens and because of their close vicinity to local macrophages, we considered it important to evaluate the effect of AEC-derived factors on various macrophage functions in vitro. We first studied their effect on phagocytosis and intracellular growth control in two types of macrophages, namely, BMM and PuM. Cells from C57BL/6 and TLR4−/− mice were isolated and kept untreated or treated with supernatants from unstimulated AEC (AECsup) or LPS-induced AEC (AECLPS) for 20 to 24 h. After this, cells were infected with GFP-BCG as described in Materials and Methods, and bacterial loads were monitored in RLU. Phagocytosis was measured at 0 h and intracellular bacterial growth at 48 h after infection. Upon AECsup and AECLPS treatments, BMM increased bacterial uptake 2.0- and 4.3-fold, respectively, compared to the untreated control (Fig. 1a). Bacterial uptake by PuM increased 5.3-fold with AECLPS (Fig. 1a) and 2.4-fold with AECsup (data not shown) treatments. Control of intracellular bacterial growth was also evaluated. The reduction of bacterial growth upon treatment with AECsup was 77% ± 3% in BMM, while upon AECLPS treatment the reduction was 55% ± 10% in BMM and 67% ± 8% in PuM (Fig. 1b). In both cases, reduction in cells treated with AEC-derived supernatants was superior to the reduction in untreated cells. In BMM, AECsup induced stronger reduction in bacterial growth control than AECLPS.
Fig 1.

Phagocytosis and intracellular bacterial growth control by BMM and PuM upon treatment with AEC-derived media. BMM and PuM from TLR4−/− mice were pretreated with either AECsup or AECLPS for 20 to 24 h or left untreated. Cells were infected with GFP-BCG. (a) Phagocytosis. Four hours after infection, phagocytosis was evaluated by determining RLU. Data are shown as RLU/106 cells. (b) Intracellular bacterial growth control. After infection, cells were treated with either AECsup or AECLPS for 48 h or left untreated. Bacterial growth was evaluated by determining RLU. Data are shown as percent reduction of phagocytosed bacteria evaluated as RLU. Values are means ± SD from 3 independent experiments. The differences between groups of BMM were analyzed using a one-way ANOVA followed by Bonferroni's multiple comparison test. *, significantly different from medium control; #, significantly different from AECsup; P < 0.05. The differences between groups of PuM were analyzed using an unpaired Student t test. *, significant differences, P < 0.05.
To assess the need for a constant presence of AEC-derived factors in the process of intracellular growth control, cells were pretreated only with AECsup for 20 to 24 h and washed before infection. This condition was named AECsupPre. The results show that in the absence of AEC-derived medium after infection, the reduction of intracellular bacterial growth was 40% lower than that of pretreated cells (Fig. 2).
Fig 2.
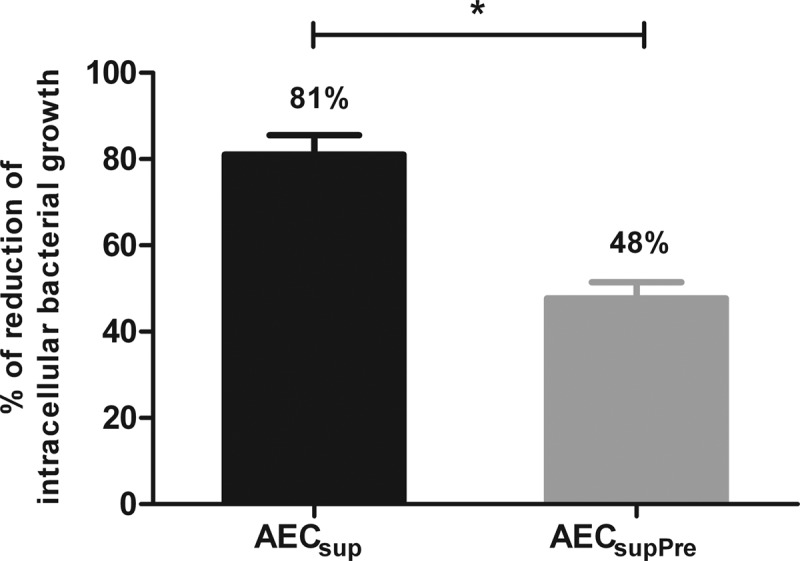
Continuous presence of AEC-derived media is required for optimal control of intracellular bacterial growth. PuM from WT mice were either continuously treated or pretreated only with AECsup and washed before infection (AECsupPre). The bacterial growth was measured by determining RLU. Data are shown as percent reduction of phagocytosed bacteria evaluated as RLU. Values are means ± SD from 3 independent experiments. The differences between groups were analyzed using an unpaired Student t test. *, significant differences, P < 0.05.
Additionally, we tested medium obtained from spleen cells and LPS. Neither SCsup nor LPS affected bacterial uptake or growth control to a level comparable with medium obtained from AEC (data not shown).
Direct effect of AEC-derived medium on opsonization and killing of bacteria.
Since AEC-secreted factors such as SP can act as opsonins and enhance the engulfing of bacteria by phagocytes (32, 33), we evaluated whether AEC-derived media had opsonizing effects in our model. GFP-BCG were treated with AEC-derived media for 30 min, and BMM were infected with the treated GFP-BCG. Our results show that pretreatment of mycobacteria with AECsup did not increase the uptake by BMM (Fig. 3a).
Fig 3.

Opsonization and direct bacterial killing upon treatment with AEC-derived media. (a) Opsonization. GFP-BCG was pretreated with AECsup for 30 min. BMM were infected with either pretreated or untreated GFP-BCG for 4 h. Phagocytosis was evaluated by determining RLU. (b) Direct killing. GFP-BCG was treated with AECsup or gentamicin for 4 and 24 h or kept in medium. Then, bacteria were centrifuged and cultured in medium without antibiotics for 72 h. Bacterial growth was measured by determining RLU. Values are means ± SD from 3 independent experiments. The differences between the groups and time points were analyzed using a two-way ANOVA followed by Bonferroni's multiple comparison test. *, significant differences, P < 0.001.
AEC have also been found to secrete a number of antimicrobial products, such as complement, lysozyme, cathelicidin, β-defensins, and SP, among others (1, 34). We evaluated whether AECsup contained microbial products able to directly kill mycobacteria. GFP-BCG were incubated with AECsup and various concentrations of gentamicin or left untreated for 4 and 24 h. After treatment, GFP-BCG was washed once and kept in complete RPMI without antibiotics for 72 h. Bacterial growth was determined by measurement of RLU. The results show that treatment with AECsup did not inhibit the GFP-BCG growth after 4 h, whereas after 24 h AECsup was as effective as gentamicin in killing mycobacteria (Fig. 3b).
AEC-derived medium increases transmembrane migration of PuM.
Upon activation, AEC secrete a number of chemokines, including keratinocyte cytokine (KC), monocyte chemoattractant protein (MCP-1), and MIP-2α (20). To determine whether AEC promote migration of pulmonary cells and whether a specific cell type is preferentially influenced by AEC-derived factors, we set up a Transwell migration assay, using total lung cells as responder cells, and let these migrate toward supernatants from either AECsup or AECLPS. We observed that more total lung cells migrated toward AEC-derived media than toward medium alone (Fig. 4). Further, there were proportionally more F4/80+/CD11c− cells that migrated toward AECLPS than toward media. The proportion of other cell types migrating toward AEC-derived media was not significantly different from that of the control (Table 1). Determining the net number of each cell type migrating, 3- and 5-fold more F4/80+/CD11c− cells migrated toward AECsup and AECLPS, respectively, than toward control medium. To determine whether media derived from other types of cells were able to induce lung cell migration, we tested the effect of spleen cell-derived media on migration. SCsup did not significantly induce pulmonary cell migration compared with that of the control (data not shown). We also tested the effect of LPS on migration, since impurities in the LPS might induce migration by a TLR4-independent mechanism. Placing medium with LPS in the lower chamber in our system did not affect migration (data not shown).
Fig 4.

Total lung cell migration toward media from unstimulated AEC (AECsup) and LPS-stimulated AEC (AECLPS). Total lung cells from TLR4−/− mice were placed in a Transwell insert in control medium or AEC-derived media in the lower chamber. After 2 h of incubation, cells were removed from the lower chamber and relative cell numbers were determined using a flow cytometer. Values are means ± SD, n = 3. The differences between the groups were analyzed using a one-way ANOVA followed by Bonferroni's multiple comparison test. *, significant differences, P < 0.05. The data are representative of 4 independent experiments.
Table 1.
Phenotypic analysis of cells after migration
| Cell type | % of gated eventsa |
||
|---|---|---|---|
| Medium | AECsup | AECLPS | |
| F4/80+/CD11c+ | 2 ± 1 | 2 ± 1 | 2 ± 1 |
| F4/80+/CD11c− | 6 ± 2 | 12 ± 1 | 16 ± 4* |
| F4/80−/CD11c+ | 1 ± 1 | 2 ± 1 | 2 ± 1 |
| CD90.1 | 20 ± 13 | 32 ± 12 | 26 ± 8 |
| CD19 | 3 ± 2 | 4 ± 1 | 3 ± 2 |
| Ly-6B2 | 24 ± 8 | 36 ± 13 | 31 ± 10 |
Data are percentages of gated events. The values are given as means ± SD from 3 independent experiments. Cells expressing F4/80 are macrophages, F4/80−/CD11c+; dendritic cells, CD90.1; T cells, CD19; B-cells and Ly-6B2; neutrophils. Statistical analysis was performed comparing the proportions of each cell-type after migration.
, significantly different from medium.
AEC-derived medium increases wound healing of macrophages (PuM, J774).
Since AEC-derived media appeared to specifically affect the transmembrane migration of myeloid cells expressing F4/80, we further characterized the effect of this media on macrophage migration using a scratch assay (30, 31). Two different types of macrophages were used, the cell line J774 and primary PuM obtained from TLR4−/− mice. The two cell types exhibited different kinetics in the assay, where J774 cells started migrating at an earlier time point (appearing in the scratch wound after 4 h) than primary PuM (detected only after 24 h). We have not observed a direct effect of the AEC supernatants on cell growth, but the cell line was actively dividing in vitro, and in this case it is obvious that division could mask the migratory effect. In order to avoid such a scenario, we ended the migration assay with the J774 cell line at an earlier time point than with the nondividing PuM, where the differences with the untreated control were clear. For both cell types, AEC-derived media increased migration compared with that of the control (Fig. 5).
Fig 5.

Wound-healing assay performed on J774 cells and PuM from TLR4−/− mice. J774 cells were grown to confluence on tissue culture plates, the monolayers were scratched to create a wound, and the cells were cultured for the indicated time in media from unstimulated (AECsup) or LPS-stimulated AEC (AECLPS). After being cultured, the cells were stained with DAPI and photographed. The number of cells in the scratch wound was evaluated in three different fields within a defined area. Values are means ± SEM, n = 4 experiments. Differences between mean values were calculated using an unpaired Student t test for J774 or one-way ANOVA for PuM, followed by Bonferroni's multiple comparison test. *, significantly different from medium, P < 0.05.
AEC-derived medium affects expression of surface markers on PuM.
Since we have demonstrated a clear effect of AEC-derived medium on macrophage activation, it was of interest to examine whether this activation could be correlated with upregulation of surface marker expression. To address this issue, PuM from wild-type (WT) mice were kept untreated or treated with AECsup for 24 h and stained for fluorescence-activated cell sorting (FACS) analysis. LPS (10 μg/ml) was used as a positive control. The cells were tested for the following costimulatory molecules: CD80, CD86, and CD40. In addition, other common macrophage markers, such as MHC-II, MMR (mannose receptor), CD11b, and CD11c, were analyzed. Treatment with AECsup did not have a major effect on the expression of MHC-II or any of the tested costimulatory molecules. However, treatment with AECsup increased the expression of MMR and CD11b compared with that of untreated PuM (Table 2). BMM were also left untreated and treated with either AECsup or LPS. The FACS analysis shows that BMM treated with AECsup had an increased expression of CD80 and MMR (data not shown).
AEC-derived medium influences the morphology of BMM and PuM.
Phagocytosis and cell migration are dependent in part on activation of the actin cytoskeleton. Furthermore, we noticed morphological changes in cells treated with AEC-derived media, also indicating alterations in the cytoskeleton. We therefore investigated the effects of treatment with AEC-derived media on cell morphology and actin cytoskeleton using microscopy. Depending on the activating conditions, macrophages can develop into M1 (classically activated) (28, 29) or M2 (alternatively activated) (27) phenotypes. To facilitate comparisons with AECsup treatment, we established the model described by Vereyken et al. (29), where BMM were differentiated toward M1 or M2 using IFN-γ and LPS or IL-4, respectively. Upon treatment with IFN-γ and LPS, almost all BMM became round, with a clearly visible actin ring below the surface, whereas IL-4 induced an elongated cell shape (Fig. 6a). Using this model, we studied the effect of AEC-derived media in PuM upon infection with BCG. After infection, cells showed a marked spreading, and only a minor proportion of cells (around 10%) were round M1-like cells (IFN-γ and LPS). In contrast, treating cells after infection with AEC-derived media increased the appearance of elongated cells, resembling the IL-4-differentiated M2-type BMM. This was especially clear in samples treated with AECLPS (Fig. 6b).
Fig 6.

Cytoskeleton staining of M1- or M2-differentiated BMM (a) or BCG-infected PuM (b). BMM were differentiated by adding IFN-γ and LPS (M1) or IL-4 (M2) for 48 h. PuM were infected with BCG for 4 h. After thorough washing and treatment with gentamicin for 30 min, cells were cultured further for 48 h in RPMI or media from untreated AEC (AECsup) or LPS-stimulated AEC (AECLPS). After being cultured, cells were stained with FITC-phalloidin and analyzed by microscopy.
DISCUSSION
Our working hypothesis has been that AEC-derived factors are important in providing micro environmental signals that are able to modify the functional behavior of incoming and tissue-resident cells, thus contributing to the homeostasis and defensive mechanisms of the respiratory tract. We chose to work preferentially with interstitial pulmonary macrophages (PuM) instead of alveolar macrophages (AM) because PuM are important in the defense against microbial aggressions and also because they are in close contact with other immune cells in the lung interstitium. We have used for their purification an established methodology considered to result in high-purity interstitial macrophages (23–25), while high-purity AM are most frequently prepared from the bronchoalveolar lavage (23–25). However, even if it is possible that a minor fraction of AM could be present in our preparations (35), we are confident that the results presented here are compatible with the functionality of PuM. Among other aspects, we have previously shown that PuM are good APC (21) in contrast to what is reported in the literature about AM, which are considered to be poor APC (25). All this may suggest that the PuM in this study are different from AM.
In recent years, it has become clear that resident tissue cells, such as epithelial cells, participate in local immune responses, although the extent of these responses are still not fully understood (7, 9, 36). Also important in defense of the lung integrity are macrophages, a heterogeneous and plastic population of cells whose specific phenotype is largely determined by signals from the surrounding environment (37). Since macrophages are located in a close proximity to the AEC in the alveoli, it is likely that in vivo, AEC play a role in attracting and modulating macrophage activity. In fact, it has been shown that AEC can secrete a variety of active substances, such as growth factors and chemokines (38–41). Infiltration of macrophages/monocytes into the lung is known to happen in homeostasis as well as in several disease settings, such as during ischemia (42), during bleomycin-induced lung injury (43), or after influenza infection (44, 45). Here, we describe that AEC-derived media selectively induced migration of myeloid cells expressing F4/80. Others have shown that AEC-derived media attract human monocytes, T cells, and neutrophils isolated from peripheral blood (11, 46). However, in the current study, our focus was on the responses of local lung cells (PuM) rather than the response of circulating blood cells, which may have a more pronounced migratory capacity. Several factors have been named to be important for monocyte/macrophage migration, and MCP-1 and the IL-8 axis have been shown, by neutralization, to be important in mediating migration toward AEC-derived media (11, 44, 46). In these reports, the AEC-induced migration of blood monocytes could be reduced by 30% to 90% by neutralizing MCP-1. Furthermore, Thorley et al. (11) showed that other factors, such as IL-8 and GRO-α, also are important, as neutralizing these chemokines reduced monocyte migration toward culture medium from primary human type II AEC by 39% and 51%, respectively (11). We have previously published that in our system, AEC secrete high levels of MCP-1 and the murine IL-8 homologs KC and MIP-2 upon stimulation with LPS (20). It is thus likely that these factors play a major role in inducing the macrophage migration in our system.
Once recruited to the tissue, one of the primary functions of macrophages is to phagocytose and destroy invading pathogens. To properly control the growth of intracellular bacteria like BCG, the macrophage needs external stimuli. Here, we show that AEC-derived media increased both macrophage phagocytosis and intracellular bacterial growth control. This is desirable since one of the problems encountered in infection with intracellular pathogens is that the microorganism escapes the immune system by utilizing macrophages to be transported to other places in the body, thus contributing to systemic dissemination of infection (47). That AEC-derived supernatants increase phagocytosis of BCG by PuM is in line with what others have previously described using AM (48). Several factors produced by AEC may increase phagocytosis, e.g., GM-CSF (49) and surfactant proteins (50). Our finding that AECsup induces a stronger intracellular bacterial control than AECLPS was not anticipated. Of the factors that have previously been measured in these supernatants, all of them were increased in the LPS-induced cells compared with that of unstimulated AEC (20). However, we cannot exclude the possibility that some unidentified factors may be present only in the AECsup and not in AECLPS. Another possibility is that the AECLPS activates the BMM in a way that is not as beneficial for the control of intracellular bacterial growth as the AECsup.
It is important to point out that preincubating bacteria with AEC-derived media for 4 h, which is the time required for infection in our experimental model, neither affected phagocytosis nor bacterial survival, indicating that the AEC-secreted factors increased macrophage phagocytosis and killing through a direct effect on the macrophages rather than through indirect mechanisms, such as opsonization or direct killing of the bacteria. However, effective bacterial killing of BCG could be achieved with AEC-derived media but required a longer incubation period (24 h). A plethora of antimicrobial components is synthesized by AEC, including lysozyme (51), secretory leukocyte protease inhibitor (52), and antimicrobial peptides (53–55). It is difficult to speculate by which mechanism the bacterial killing was achieved in our system, although a similarly prolonged incubation time has been shown to be needed for antimicrobial peptides, such as cathelicidins, to achieve an effective killing of BCG (56).
Macrophage differentiation is often accompanied by morphological changes, for instance, regarding the differentiation into classically activated M1 or alternatively activated M2 macrophages. M1 macrophages can display a round appearance, while M2 macrophages are more elongated (27–29). In this study, we also observed alterations in the general shape of the cells, where AEC-derived media induced a more elongated cell shape, similar to that ascribed to M2. These morphological changes, together with the increased expression of MMR, a receptor known to be expressed preferentially by M2 macrophages (13–15), are suggestive of such polarization. Indeed, M1- and M2-type macrophages are thought to have different functions in the body, and the M2 macrophage is typically thought of as less inflammatory and less active in bacterial killing (57, 58). It could be speculated that to promote the induction of this type of a less “aggressive” macrophage is more convenient for keeping the integrity of the tissue. That treatment with AEC-derived factors may induce a differentiation of PuM to an M2-type pathway, while increasing intracellular killing of BCG seems controversial and requires further evaluation.
Taken together, here we demonstrate that factors secreted by AEC have a clear effect both on macrophages and on bacteria. We report that culturing macrophages in AEC-derived media changed macrophage morphology and significantly increased several functional features, including cell migration, phagocytosis, and control of intracellular bacterial growth. In addition, AEC-derived factors promoted effective extracellular bacterial killing. Some of the factors known to be responsible for these effects have been previously characterized (56). We have also tested a number of them, such as M-CSF and MCP-1, and found that these factors were active when used at relatively high concentrations but only in some functions, not in all. For instance, MCP-1 and M-CSF both increased wound healing, while only MCP-1 potently induced migration in the Transwell migration assay (data not shown). All this suggests that AEC secrete many different factors that in an additive or synergistic manner will influence cells residing in that tissue.
Collectively, our data support the theory that AEC are crucial both in the homeostasis of the lungs and as active partners in the defense against infection. AEC are able to control bacterial growth, to attract and activate other cell types exemplified here by their various effects on PuM. This reinforces the importance of AEC and their contribution in making up the mucosal microenvironment, further increasing our understanding of the various partners involved in the defense of mucosal surfaces.
ACKNOWLEDGMENTS
We thank R. Karlsson for his advice in setting up the microscopy experiments and S. Höglund, manager at the Imaging Facility at Wenner-Gren Institute, for her technical assistance. We also thank I. Dellacasa and T. Frisan, Karolinska Institute, for help with the migration assays.
This work was supported by grant FP7 (HEALTH-F3-2008-200732) from the European Commission.
Footnotes
Published ahead of print 12 November 2012
REFERENCES
- 1. Bals R. 2000. Epithelial antimicrobial peptides in host defense against infection. Respir. Res. 1:141–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ryu JH, Kim CH, Yoon JH. 2010. Innate immune responses of the airway epithelium. Mol. Cells 30:173–183 [DOI] [PubMed] [Google Scholar]
- 3. Williams MC. 2003. Alveolar type I cells: molecular phenotype and development. Annu. Rev. Physiol. 65:669–695 [DOI] [PubMed] [Google Scholar]
- 4. Fereol S, Fodil R, Pelle G, Louis B, Isabey D. 2008. Cell mechanics of alveolar epithelial cells (AECs) and macrophages (AMs). Respir. Physiol. Neurobiol. 163:3–16 [DOI] [PubMed] [Google Scholar]
- 5. Castranova V, Rabovsky J, Tucker JH, Miles PR. 1988. The alveolar type II epithelial cell: a multifunctional pneumocyte. Toxicol. Appl. Pharmacol. 93:472–483 [DOI] [PubMed] [Google Scholar]
- 6. Fehrenbach H. 2001. Alveolar epithelial type II cell: defender of the alveolus revisited. Respir. Res. 2:33–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Debbabi H, Ghosh S, Kamath AB, Alt J, Demello DE, Dunsmore S, Behar SM. 2005. Primary type II alveolar epithelial cells present microbial antigens to antigen-specific CD4+ T cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 289:L274–L279 [DOI] [PubMed] [Google Scholar]
- 8. Pechkovsky DV, Goldmann T, Ludwig C, Prasse A, Vollmer E, Muller-Quernheim J, Zissel G. 2005. CCR2 and CXCR3 agonistic chemokines are differently expressed and regulated in human alveolar epithelial cells type II. Respir. Res. 6:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lo B, Hansen S, Evans K, Heath JK, Wright JR. 2008. Alveolar epithelial type II cells induce T cell tolerance to specific antigen. J. Immunol. 180:881–888 [DOI] [PubMed] [Google Scholar]
- 10. Witherden IR, Vanden Bon EJ, Goldstraw P, Ratcliffe C, Pastorino U, Tetley TD. 2004. Primary human alveolar type II epithelial cell chemokine release: effects of cigarette smoke and neutrophil elastase. Am. J. Respir. Cell Mol. Biol. 30:500–509 [DOI] [PubMed] [Google Scholar]
- 11. Thorley AJ, Ford PA, Giembycz MA, Goldstraw P, Young A, Tetley TD. 2007. Differential regulation of cytokine release and leukocyte migration by lipopolysaccharide-stimulated primary human lung alveolar type II epithelial cells and macrophages. J. Immunol. 178:463–473 [DOI] [PubMed] [Google Scholar]
- 12. Lohmann-Matthes ML, Steinmuller C, Franke-Ullmann G. 1994. Pulmonary macrophages. Eur. Respir. J. 7:1678–1689 [PubMed] [Google Scholar]
- 13. Fathi M, Johansson A, Lundborg M, Orre L, Skold CM, Camner P. 2001. Functional and morphological differences between human alveolar and interstitial macrophages. Exp. Mol. Pathol. 70:77–82 [DOI] [PubMed] [Google Scholar]
- 14. Gordon SB, Read RC. 2002. Macrophage defences against respiratory tract infections. Br. Med. Bull. 61:45–61 [DOI] [PubMed] [Google Scholar]
- 15. Laskin DL, Weinberger B, Laskin LD. 2001. Functional heterogeneity in liver and lung macrophages. J. Leukoc. Biol. 70:163–170 [PubMed] [Google Scholar]
- 16. Landsman L, Jung S. 2007. Lung macrophages serve as obligatory intermediate between blood monocytes and alveolar macrophages. J. Immunol. 179:3488–3494 [DOI] [PubMed] [Google Scholar]
- 17. Landsman L, Varol C, Jung S. 2007. Distinct differentiation potential of blood monocyte subsets in the lung. J. Immunol. 178:2000–2007 [DOI] [PubMed] [Google Scholar]
- 18. Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. 1999. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J. Immunol. 162:3749–3752 [PubMed] [Google Scholar]
- 19. Humphreys IR, Stewart GR, Turner DJ, Patel J, Karamanou D, Snelgrove RJ, Young DB. 2006. A role for dendritic cells in the dissemination of mycobacterial infection. Microbes Infect. 8:1339–1346 [DOI] [PubMed] [Google Scholar]
- 20. Chuquimia OD, Petursdottir DH, Rahman MJ, Hartl K, Singh M, Fernandez C. 2012. The role of alveolar epithelial cells in initiating and shaping pulmonary immune responses: communication between innate and adaptive immune systems. PLoS One 7:e32125 doi:10.1371/journal.pone.0032125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rahman MJ, Chuquimia OD, Petursdottir DH, Periolo N, Singh M, Fernandez C. 2011. Impact of Toll-like receptor 2 deficiency on immune responses to mycobacterial antigens. Infect. Immun. 79:4649–4656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Corti M, Brody AR, Harrison JH. 1996. Isolation and primary culture of murine alveolar type II cells. Am. J. Respir. Cell Mol. Biol. 14:309–315 [DOI] [PubMed] [Google Scholar]
- 23. Migliaccio CT, Hamilton RF, Jr, Holian A. 2005. Increase in a distinct pulmonary macrophage subset possessing an antigen-presenting cell phenotype and in vitro APC activity following silica exposure. Toxicol. Appl. Pharmacol. 205:168–176 [DOI] [PubMed] [Google Scholar]
- 24. Zaslona Z, Wilhelm J, Cakarova L, Marsh LM, Seeger W, Lohmeyer J, von Wulffen W. 2009. Transcriptome profiling of primary murine monocytes, lung macrophages and lung dendritic cells reveals a distinct expression of genes involved in cell trafficking. Respir. Res. 10:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kugathasan K, Roediger EK, Small CL, McCormick S, Yang P, Xing Z. 2008. CD11c+ antigen-presenting cells from the alveolar space, lung parenchyma and spleen differ in their phenotype and capabilities to activate naive and antigen-primed T cells. BMC Immunol. 9:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rothfuchs A, Gigliotti GD, Palmblad K, Andersson U, Wigzell H, Rottenberg MR. 2001. IFN-alpha beta-dependent, IFN-gamma secretion by bone marrow-derived macrophages controls an intracellular bacterial infection. J. Immunol. 167:6453–6461 [DOI] [PubMed] [Google Scholar]
- 27. Stein M, Keshav S, Harris N, Gordon S. 1992. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J. Exp. Med. 176:287–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Menzies FM, Henriquez FL, Alexander J, Roberts CW. 2010. Sequential expression of macrophage antimicrobial/inflammatory and wound healing markers following innate, alternative and classical activation. Clin. Exp. Immunol. 160:369–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vereyken EJ, Heijnen PD, Baron de Vries EH, Dijkstra CD, Teunissen CE. 2011. Classically and alternatively activated bone marrow derived macrophages differ in cytoskeletal functions and migration towards specific CNS cell types. J. Neuroinflammation 8:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kheradmand F, Folkesson HG, Shum L, Derynk R, Pytela R, Matthay MA. 1994. Transforming growth factor-alpha enhances alveolar epithelial cell repair in a new in vitro model. Am. J. Physiol. 267:L728–L738 [DOI] [PubMed] [Google Scholar]
- 31. Munugalavadla V, Borneo J, Ingram DA, Kapur R. 2005. p85alpha subunit of class IA PI-3 kinase is crucial for macrophage growth and migration. Blood 106:103–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wright JR. 2005. Immunoregulatory functions of surfactant proteins. Nat. Rev. Immunol. 5:58–68 [DOI] [PubMed] [Google Scholar]
- 33. Sever-Chroneos Z, Krupa A, Davis J, Hasan M, Yang CH, Szeliga J, Herrmann M, Hussain M, Geisbrecht BV, Kobzik L, Chroneos ZC. 2011. Surfactant protein A (SP-A)-mediated clearance of Staphylococcus aureus involves binding of SP-A to the staphylococcal adhesin eap and the macrophage receptors SP-A receptor 210 and scavenger receptor class A. J. Biol. Chem. 286:4854–4870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wu H, Kuzmenko A, Wan S, Schaffer L, Weiss A, Fisher JH, Kim KS, McCormack FX. 2003. Surfactant proteins A and D inhibit the growth of Gram-negative bacteria by increasing membrane permeability. J. Clin. Invest. 111:1589–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bedoret D, Wallemacq H, Marichal T, Desmet C, Quesada Calvo F, Henry E, Closset R, Dewals B, Thielen C, Gustin P, de Leval L, Van Rooijen N, Le Moine A, Vanderplasschen A, Cataldo D, Drion PV, Moser M, Lekeux P, Bureau F. 2009. Lung interstitial macrophages alter dendritic cell functions to prevent airway allergy in mice. J. Clin. Invest. 119:3723–3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sanders CJ, Doherty PC, Thomas PG. 2011. Respiratory epithelial cells in innate immunity to influenza virus infection. Cell Tissue Res. 343:13–21 [DOI] [PubMed] [Google Scholar]
- 37. Guth AM, Janssen WJ, Bosio CM, Crouch EC, Henson PM, Dow SW. 2009. Lung environment determines unique phenotype of alveolar macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 296:L936–L946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sharma AK, Fernandez LG, Awad AS, Kron IL, Laubach VE. 2007. Proinflammatory response of alveolar epithelial cells is enhanced by alveolar macrophage-produced TNF-alpha during pulmonary ischemia-reperfusion injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 293:L105–L113 [DOI] [PubMed] [Google Scholar]
- 39. Thorley AJ, Grandolfo D, Lim E, Goldstraw P, Young A, Tetley TD. 2011. Innate immune responses to bacterial ligands in the peripheral human lung—role of alveolar epithelial TLR expression and signalling. PLoS One 6:e21827 doi:10.1371/journal.pone.0021827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Farberman MM, Hoffmann JW, Ryerse JS, Demello DE. 2004. Diffusible signal to murine alveolar macrophages from lipopolysaccharide- and Escherichia coli-stimulated lung type II epithelial cells. Inflamm. Res. 53:475–483 [DOI] [PubMed] [Google Scholar]
- 41. Rate A, Upham JW, Bosco A, McKenna KL, Holt PG. 2009. Airway epithelial cells regulate the functional phenotype of locally differentiating dendritic cells: implications for the pathogenesis of infectious and allergic airway disease. J. Immunol. 182:72–83 [DOI] [PubMed] [Google Scholar]
- 42. Moldobaeva A, van Rooijen N, Wagner EM. 2011. Effects of ischemia on lung macrophages. PLoS One 6:e26716 doi:10.1371/journal.pone.0026716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liang J, Jung Y, Tighe RM, Xie T, Liu N, Leonard M, Gunn MD, Jiang D, Noble PW. 2012. A macrophage subpopulation recruited by CC chemokine ligand-2 clears apoptotic cells in non-infectious lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. doi:10.1152/ajplung.00256.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Herold S, von Wulffen W, Steinmueller M, Pleschka S, Kuziel WA, Mack M, Srivastava M, Seeger W, Maus UA, Lohmeyer J. 2006. Alveolar epithelial cells direct monocyte transepithelial migration upon influenza virus infection: impact of chemokines and adhesion molecules. J. Immunol. 177:1817–1824 [DOI] [PubMed] [Google Scholar]
- 45. Lin KL, Suzuki Y, Nakano H, Ramsburg E, Gunn MD. 2008. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J. Immunol. 180:2562–2572 [DOI] [PubMed] [Google Scholar]
- 46. Eghtesad M, Jackson HE, Cunningham AC. 2001. Primary human alveolar epithelial cells can elicit the transendothelial migration of CD14+ monocytes and CD3+ lymphocytes. Immunology 102:157–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Krishnan N, Robertson BD, Thwaites G. 2010. The mechanisms and consequences of the extrapulmonary dissemination of Mycobacterium tuberculosis. Tuberculosis (Edinb.) 90:361–366 [DOI] [PubMed] [Google Scholar]
- 48. Kannan S, Huang H, Seeger D, Audet A, Chen Y, Huang C, Gao H, Li S, Wu M. 2009. Alveolar epithelial type II cells activate alveolar macrophages and mitigate P. aeruginosa infection. PLoS One 4:e4891 doi:10.1371/journal.pone.0004891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Berclaz PY, Zsengeller Z, Shibata Y, Otake K, Strasbaugh S, Whitsett JA, Trapnell JC. 2002. Endocytic internalization of adenovirus, nonspecific phagocytosis, and cytoskeletal organization are coordinately regulated in alveolar macrophages by GM-CSF and PU.1. J. Immunol. 169:6332–6342 [DOI] [PubMed] [Google Scholar]
- 50. Chroneos ZC, Sever-Chroneos Z, Shepherd VL. 2010. Pulmonary surfactant: an immunological perspective. Cell. Physiol. Biochem. 25:13–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Singh G, Katyal SL, Brown WE, Collins DL, Mason RJ. 1988. Pulmonary lysozyme—a secretory protein of type II pneumocytes in the rat. Am. Rev. Respir. Dis. 138:1261–1267 [DOI] [PubMed] [Google Scholar]
- 52. Higashimoto Y, Yamagata Y, Iwata T, Ishiguchi T, Okada M, Masuda M, Satoh H, Itoh H. 2005. Adenoviral E1A suppresses secretory leukoprotease inhibitor and elafin secretion in human alveolar epithelial cells and bronchial epithelial cells. Respiration 72:629–635 [DOI] [PubMed] [Google Scholar]
- 53. Hostanska K, Melzer J, Amon A, Saller R. 2011. Suppression of interleukin (IL)-8 and human beta defensin-2 secretion in LPS-and/or IL-1beta-stimulated airway epithelial A549 cells by a herbal formulation against respiratory infections (BNO 1030). J. Ethnopharmacol. 134:228–233 [DOI] [PubMed] [Google Scholar]
- 54. Li Y, Wang Y, Liu X. 2012. The role of airway epithelial cells in response to mycobacteria infection. Clin. Dev. Immunol. 2012:791392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rivas-Santiago B, Schwander SK, Sarabia C, Diamond G, Klein-Patel ME, Hernandez-Pando R, Ellner JJ, Sada E. 2005. Human {beta}-defensin 2 is expressed and associated with Mycobacterium tuberculosis during infection of human alveolar epithelial cells. Infect. Immun. 73:4505–4511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sonawane A, Santos JC, Mishra BB, Jena P, Progida C, Sorensen OE, Gallo R, Appelberg R, Griffiths G. 2011. Cathelicidin is involved in the intracellular killing of mycobacteria in macrophages. Cell. Microbiol. 13:1601–1617 [DOI] [PubMed] [Google Scholar]
- 57. Gordon S, Martinez FO. 2010. Alternative activation of macrophages: mechanism and functions. Immunity 32:593–604 [DOI] [PubMed] [Google Scholar]
- 58. Moreira AP, Hogaboam CM. 2011. Macrophages in allergic asthma: fine-tuning their pro and anti-inflammatory actions for disease resolution. J. Interferon Cytokine Res. 31:485–491 [DOI] [PubMed] [Google Scholar]