

Abstract
Aggregation of TDP-43 proteins to form intracellular inclusions is the primary pathology in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD) with TDP-43 inclusions (FTLD-TDP). Histologically, in the cerebral cortex and limbic regions of affected ALS and FTLD-TDP patients, these pathologies occur as a variety of cytoplasmic, neuritic and intranuclear TDP-43 inclusions. In the spinal cord and lower brainstem of ALS patients, the lesions form cytoplasmic dashes or complex filamentous and spherical profiles in addition to skein-like inclusions (SLI). Ultrastructurally, the morphology of TDP-43 inclusions is heterogeneous but mainly composed of loose bundles of 10–20 nm diameter straight filaments associated with electron dense granular material. All of these TDP-43 inclusions are generally described as disordered amorphous aggregations unlike the amyloid fibrils that characterize protein accumulations in neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease.
We here report that Thioflavin-S positive SLI are present in a subset of ALS cases, while TDP-43 inclusions outside the spinal cord lack the chemical properties of amyloid. Further, we examine the differential enrichment of fibrillar profiles in SLI of ALS cases by TDP-43 immuno-electron microscopy (immuno-EM). The demonstration that pathological TDP-43 can be amyloidogenic in situ suggests the following conclusions: 1) the conformational changes associated with TDP-43 aggregation are more complex than previously thought; 2) Thioflavin-S positive SLI may be composed primarily of filamentous ultrastructures.
Keywords: TDP-43, amyloid, skein, amyotrophic lateral sclerosis, ALS, frontotemporal lobar degeneration, FTLD-TDP
Introduction
Pathological TDP-43 proteins aggregate to form intracellular inclusions that are the signature lesions of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD) with TDP-43 inclusions (now known as FTLD-TDP) [30,27,17]. Histologically and biochemically, these inclusions are dense, insoluble aggregates of TDP-43 proteins that are phosphorylated and ubiquitinated in both sporadic and familial FTLD-TDP and ALS cases. Genetically, mutations in the genes that encode TDP-43 (TARDBP), progranulin (GRN), valosin containing protein (VCP) and the more recently described C9orf72 expansions [7,10,1] are all familial causes of TDP-43 proteinopathies. In the cerebral cortex and limbic regions of FTLD-TDP and in most ALS patients, the inclusions present as a variety of intracytoplasmic, neuritic and intranuclear lesions [17]. In ALS and FTLD-TDP patients with motor neuron disease, these inclusions may take the shape of cytoplasmic dashes, fibrillary, spherical and skein-like lesions (SLI) in affected motor neurons of the spinal cord and lower brainstem [2]. Ultrastructurally, the morphology of TDP-43 inclusions in both ALS and FTLD-TDP are heterogeneous, but generally characterized by bundles of 10–20 nm diameter straight filaments that associate with electron dense granular material in various proportions or just granular and amorphous aggregates [5,19,13]. Although studies have examined SLI using electron microscopy (EM) and immuno-electron microscopy (immuno-EM) with ubiquitin antibodies [21,24], surprisingly, TDP-43 immuno-EM studies have been few [26,25].
Most studies have defined TDP-43 pathology as the formation of disordered accumulations of the protein [15,29], although several algorithms have identified prion-like [16] or amyloid-prone domains [11] in the TDP-43 protein sequence. It is known that amyloids are defined by several salient features including a secondary β-sheet structure detected by infrared spectroscopy, a particular x-ray fiber diffraction pattern, or chemical stains with Congo Red, Thioflavin-S (ThS) or Thioflavin-T dyes [18,34]. In vitro work has demonstrated that C-terminal fragments of TDP-43 with disease causing mutations can indeed form Thioflavin-T positive amyloid fibrils [6,12], but this has not been shown convincingly for wild type TDP-43.
To clarify the amyloid properties of TDP-43 inclusions, we examined TDP-43 inclusions in FTLD-TDP and ALS cases, and we observed that a subset of SLI in the spinal cord have ThS positivity, while TDP-43 inclusions elsewhere lack this chemical property of amyloid. In addition, we report that spinal cord SLI are composed primarily of filamentous material at the ultrastructural level. Hence, these new data suggest that TDP-43 inclusions have heterogeneous properties but those in spinal cord are prominently filamentous and can show properties of amyloid.
Material and methods
Study subjects
69 individuals who underwent postmortem neuropathology assessment in the Center for Neurodegenerative Disease Research (CNDR) in the Perelman School of Medicine at the University of Pennsylvania (Penn) were categorized based on clinical manifestations as ALS [4] or FTLD [28]. Informed consent for autopsy was obtained in all cases from the patient’s family or legal representative in accordance with protocols approved by the Penn Institutional Review Board. The ALS and FTLD patients were longitudinally followed by Penn neurologists and were assigned a neuropathological diagnosis of ALS (n=47) and/or FTLD-TDP (n=22) after review by a neuropathologist (JQT). The FTLD-TDP group included patients closely age matched by age of onset to the ALS patients, with TDP-43 spinal cord pathology and with 3 subtypes of FTLD-TDP pathology, A, B, and C [22]. Genotyping of DNA extracted from brain tissue was performed to detect expansions in C9orf72 for both ALS and FLTD-TDP cases as previously described [3]. In addition, DNA sequencing was performed to screen for mutations in TARDBP in the subset of ALS cases and GRN in FTLD-TDP cases [39,37]. A case was deemed to have family history if one or more family members had ALS, FTLD-TDP or another clinical neurodegenerative disease.
Histochemistry and immunohistochemistry
All histology was performed on 6 μm thick, coronal sections. ThS staining was performed as previously described [5]. This protocol involves the use of 0.0125% ThS dye and differentiation in a 50% ethanol/phosphate buffered saline solution. Sections were aqueous mounted on slides with Vectashield using DAPI mounting medium (Vector Laboratories, Burlingame, CA). Example sections are shown in Supplemental Figure 1.
For immunohistochemistry (IHC), we used the following primary antibodies: phosphorylated TDP-43 monoclonal antibody (mAb) p409/410 at 1:500 (a gift of Dr. Manuela Neumann); phosphorylated tau mAb PHF-1 at 1:1,000 (a gift of Dr. Peter Davies); phosphorylated neurofilament mAb RMO24.9 at 1:1,000 (generated in CNDR, Philadelphia, PA, USA); and α-synuclein mAb Syn303 at 1:4,000 (generated in CNDR, Philadelphia, PA, USA); β-amyloid mAb nab228 at 1:8,000 (generated in CNDR, Philadelphia, PA, USA). Primary Ab binding was visualized with the avidin-biotin complex detection method (VECTASTAIN ABC kit; Vector Laboratories, Burlingame, CA) with ImmPACT diaminobenzidine peroxidase substrate (Vector Laboratories, Burlingame, CA) as the chromogen. Pathology was graded on a semi-quantitative scale (0=none; 0.5=rare, only 1 or 2 confirmed lesions in the section; 1=mild, 2–5 confirmed lesions; 2=moderate, 5 or more confirmed lesions; 3=severe, robust pathology throughout the section).
For combined ThS/TDP-43 IHC, a-protocol was developed based on double-fluorescent IHC with Sudan Black treatment to quench lipofuscin autoflourescence [32]. Slides were microwaved in antigen unmasking solution (1:100, Vector Laboratories Burlingame, CA) for 15 min and thereafter sections underwent the standard ThS protocol (see above). TDP-43 primary antibody p409/410 was incubated for two hr at room temperature. Red fluorescent secondary antibody Alexa Fluor 594 (Molecular Probes, Eugene, OR, USA) was incubated for 2 hr at room temperature. Sudan Black treatment (0.3% Sudan Black in 70% ethanol) was done on a slide by slide basis for 0–30 sec so as not to quench the immunofluorescent or ThS signal. Sections were aqueous mounted as per the ThS protocol.
Digital images were captured using an Olympus BX 51 (Tokyo, Japan) microscope using a digital camera-DP71 and DP controller software (Olympus, Orangeburg, NY, USA).
Electron microscopy
Samples of hippocampus and spinal cord collected at autopsy were fixed overnight in 0.1% glutaraldehyde plus 4% paraformaldehyde in 0.1M PBS, sliced on a vibratome and stored in cryoprotection solution at −20°C. Before IHC, sections were fixed for an additional 15 min in 2.5% glutaraldehyde in 0.1M cacodylate buffer. Immuno-EM was done according to the method of Llewellyn-Smith and Minson[20], using either the Vector ABC system (Vector Laboratories Burlingame, CA) with horseradish peroxidase (HRP) label and 3, 3′ diaminobenzidine (DAB) as the substrate or goat anti-rabbit nanogold (Nanoprobes, Yaphank, NY) as the secondary antibody. The nanogold particles were diameter 1.4 nm. The antibody 1039C to the C-terminal region of TDP-43 (generated in CNDR, Philadelphia, PA, USA) was found to give the best signal compared to other TDP-43 antibodies and was used here at a dilution that preferentially stained cytoplasmic TDP-43 accumulations. Sections stained with HRP/DAB were postfixed for 2 hr in 1% osmium tetroxide plus 1.5% potassium ferrocyanide in 0.05M cacodylate buffer. After nanogold immuno-EM, sections were fixed for 3 hr in 2.5% glutaraldehyde in 0.1M cacodylate buffer, pH7.4, post-fixed for 30 min on ice in 1% OsO4 in 0.1M cacodylate buffer, pH7.4, washed, gold toned 2–6 min according to Marshak [23], post-fixed for an additional 20 min on ice in 1% osmium tetroxide plus 1.5% potassium ferrocyanide in 0.05M cacodylate buffer. Both were dehydrated in ethanol and embedded in the epoxy resin EMbed-812 (Electron Microscopy Sciences, Hatfield, PA). Selected semi-thin sections were re-embedded for thin sectioning. Thin sections were counterstained with bismuth subnitrate (Fig. 4), uranyl acetate and lead citrate (Fig. 5j) or lead citrate alone (Fig. 6and 5a–i,k)
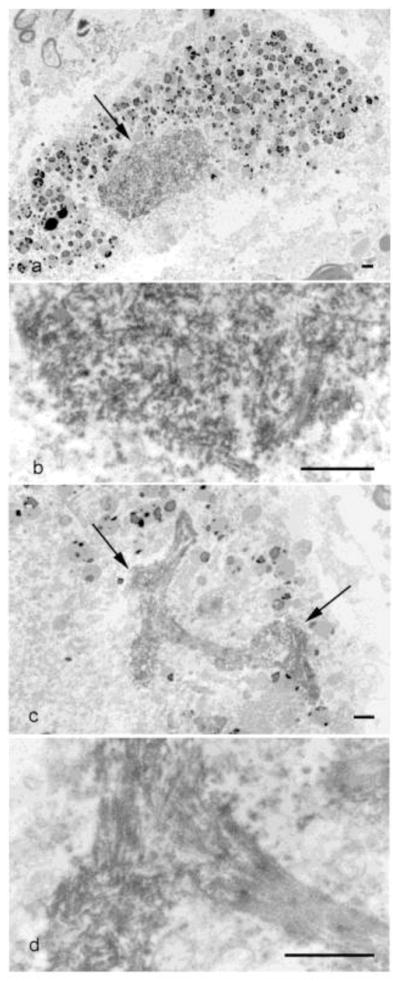
Figure 4. HRP immuno-EM on Semi-thin lumbar spinal cord sections from ALS Case #1.
(a) Granulo-filamentous material is present in a compact inclusion (arrow), surrounded by lipofuscin granules (2600X). (b) Higher magnification of (a) (16,100X). (c). Filamentous material is more prominent in a skein (arrows) (4600X). (d) Higher magnification of (c) (19,400X). (scale bars = 1μm)
Figure 5. TDP-43 nanogold immuno-EM on lumbar spinal cord sections from 3 ALS cases.

Case #1 (a–f) showing an elongated inclusion with bundles of randomly oriented filaments (a–c) and a compact inclusion with granulo-filamentous material (d–f). (a) Semi-thin section of a large motor neuron shows a low power view of the inclusion (arrowhead) (60X). (b) Same cell re-embedded for EM (1300X). (c) Higher magnification of (b). Note bundles of filaments at bottom (arrowheads) and randomly oriented filaments toward top (arrows) (14,600X). (d) Semi-thin section of a large motor neuron with a round inclusion (arrowhead) (60X). (e) Same cell re-embedded for EM (1300X). (f) Higher magnification of boxed area in (e) (16,500X). Note the specificity of the TDP-43-linked nanoparticles by the heaviliy decorated fibrils in the central portion of the panel compared to the tissue in the lower left with minimal particles. Case #2 (g–j) showing a compact inclusion associated with a bundle of filaments (g–i) and bundle of filaments from another section (j). (g) Semi-thin section of a large motor neuron with a compact inclusion (arrowhead) (60X). (h) Same cell re-embedded for EM, showed a tight bundle of filaments (arrowhead) coming out of a round aggregate of granulo-filamentous material (4000X). (i) Higher magnification of boxed area in (h) (22,600X). (j) Higher magnification of a TDP-43 nanogold decorated bundle of filaments from another section (43,400X). Case #3 (k) Cross-section of TDP-43 nanogold decorated filament bundles (5,1400X). (scale bars = 1μm)
Statistical analysis
We compared characteristics of the ThS+ and ThS− ALS groups using Mann Whitney U tests and Chi-squared tests as appropriate. All statistical analyses were performed using SPSS version 19 (SPSS Inc., Chicago, IL).
Results
Histology
Our cohort was composed of 69 subjects, including 47 cases with ALS and 22 with FTLD-TDP (Table 1). Hippocampus, mortor corex, and spinal cord sections from the whole cohort were examined by ThS staining and TDP-43 immunohistochemistry (IHC). All sections showed TDP-43 positive inclusions at varying frequencies (Table 1). ThS positive inclusions were limited to a subset of the SLI in ALS spinal cords and were not observed in ALS hippocampal or motor cortex TDP-43 inclusions, nor in the spinal cords of FTLD-TDP cases (data not shown). In total, ThS positive SLI were confirmed in 28% (n=13) of the ALS cases. The ThS+ ALS cases had a moderate to severe accumulation of TDP-43 spinal cord pathology, with mild to moderate accumulations of ThS+ SLI (see Supplemental Table 1 for individual scores). Interestingly, the ThS+ ALS cases tended to have an overall more severe spinal cord TDP-43 burden (mean 2.6 versus 2.1) than the ThS− ALS cases although this was not statistically significant (Table 1, p=0.07).
Table 1.
Subject characteristics
| Diagnostic group | All ALS (n=47) | ThS+ ALS (n=13) | ThS− ALS (n=34) | FTLD-TDP (n=22) |
|---|---|---|---|---|
| Gender, M:F (%:%) | 33:15 (70:30) | 9:4 (69:31) | 24:11 (71:29) | 12:9 (55:45) |
| Motor symptoms, n (%) | 47 (100) | 13 (100) | 34 (100) | 3 (14) |
| Cognitive deficits, n (%) | 4 (9) | 2 (15) | 2 (6) | 22 (100) |
| Age at onset, y (stdev) | 57 (12) | 56 (10) | 58 (12) | 60 (9) |
| Disease duration, y (stdev) | 3.1 (4.1) | 2.7 (1.8) | 3.3 (2.6) | 6.6 (4.4) |
| Family History1, n (%) | 13 (28) | 5 (38) | 8 (24) | 13 (59) |
| C9orf72 expansion, n (%) | 7 (15) | 3 (23) | 4 (12) | 4 (18) |
| Brain weight, g (stdev) | 1338 (138) | 1367 (129) | 1327 (151) | 1096 (194) |
| PMI, h (stdev) | 13.4 (7.4) | 12.5 (6.5) | 13.2 (6.7) | 12.0 (6.2) |
| TDP-43 pathology2 | mean (stdev) | |||
| Spinal cord | 2.3 (0.7) | 2.6 (0.5) | 2.1 (0.8) | 1.4 (1.1) |
| Motor Cortex | 1.6 (1.0) | 1.7 (1.0) | 1.6 (1.0) | 0.8 (0.9) |
| Hippocampus | 0.8 (0.8) | 0.7 (0.5) | 0.8 (0.9) | 2.3 (0.9) |
Confirmed family history with the same or another neurodegenerative disease (unknown for 9 subjects)
TDP-43 pathology is a mean of the semi-quantitative scores (0–3)
Morphologically, ThS+ SLI show diverse structures similar to those reported for TDP-43 positive SLI (Fig. 1). In some motor neurons, dash-like or dot-like profiles were observed (Fig. 1a). More often, individual or multiple skeins were apparent (Fig. 1b–c). Occasionally, a more compact inclusion was seen (Fig. 1d).
Figure 1. Variety of observed ThS positive inclusions in 4 ALS cases.

ThS positive pathology was observed in lower motor neurons as (a) dash-like or dot-like elements or (b) individual skeins. Multiple skeins could present as (c) a meshwork-like structure or as (d) compact or even Lewy body-like inclusions. (60X, scale bar = 50μm)
To confirm that ThS positivity was associated with TDP-43 inclusions and not other neurodegenerative disease inclusions, IHC was performed on serial sections of lumbar spinal cord (Fig. 2). None of the ThS+ cases had spinal cord pathology that was immuonoreactive for tau, β-amyloid or α-synuclein. In adjacent sections, a subset of the TDP-43 positive skeins and inclusions (Fig. 2a, arrows) were ThS positive (Fig. 2b, arrows). Antibodies to neurofilament (Fig. 2c), tau (Fig. 2d), β-amyloid (data not shown) and α-synuclein (data not shown) did not reveal any ALS-like pathology. We conclude that ThS positive SLI associate with TDP-43 immunoreactivity.
Figure 2.

ThS positivity associates only with TDP-43 positive pathology. Serial sections from an ALS cervical spinal cord with (a) TDP-43 IHC, (b)ThS staining, (c) neurofilament IHC and (d) Tau IHC. Arrows point to ThS and TDP-43 positive skeins, not detected by other antibodies. (20X, scale bar = 100μm)
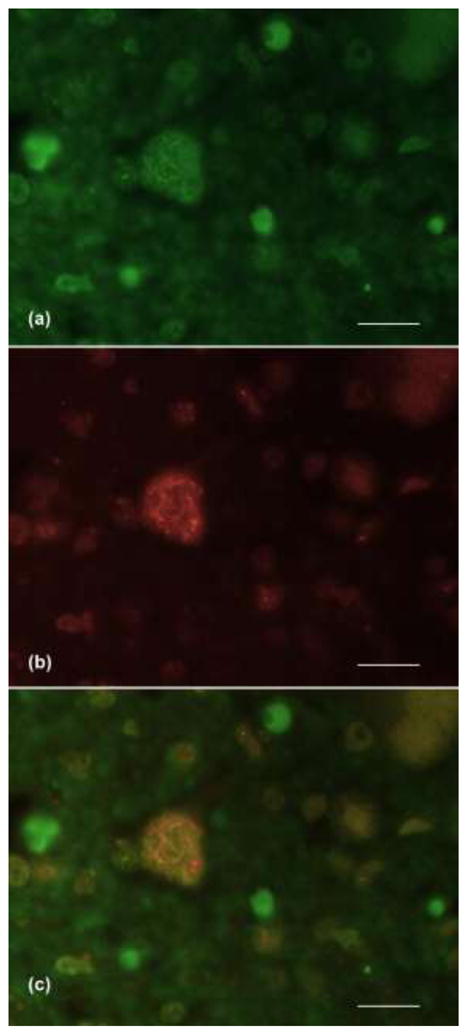
Finally, to directly demonstrate that ThS and TDP-43 staining of SLI co-localize, we next performed ThS histochemistry and TDP-43 IHC on the same section (Fig. 3). These studies showed that the fine skein-like pathology characteristic of ALS spinal cords is detected by both the ThS (Fig. 3a) and TDP-43 (Fig. 3b) signals.
Figure 3.
ThS positivity co-localizes with TDP-43 positive pathology. (a) ThS staining and (b) fluorescent IHC against TDP-43 was performed on the same section from an ALS cervical spinal cord. Overlay of (a) and (b) showed significant co-localization (c). (60X, scale bar = 50μm)
Subject Characteristics
Since ThS positive SLI were present only in a subset of the ALS cases, we next asked whether the ThS+ ALS cases differed clinically or genetically from the ThS− ALS cases (Table 1). Consistent with the continuum of TDP-43 proteinopathies [8], some of the ALS patients had cognitive deficits ranging from impairment to dementia and some of the FTLD-TDP patients developed clinical motor neuron disease. Specifically, 9% (4/47) of the ALS group had cognitive deficits while 14% (3/22) of the FTLD-TDP group presented with motor deficits, but the ThS+ ALS group was not more likely to have cognitive deficits than the ThS-ALS group (p=0.14). In fact, there was no difference between the ThS+ and the ThS-ALS group in mean age of onset, disease duration, brain weight, or post-mortem interval.
A family history of neurodegenerative diseases was present in 28% (13/47) of the ALS group and 59% (13/22) of the FTLD-TDP. All cases with DNA (n=67) were tested for a C9orf72 expansion. Cases of ALS (n=45) were also tested for mutations in TARDBP and no TARDBP mutations were identified. C9orf72 expansion was detected in 15% (7/45) of the ALS group and 18% (4/22) of the FTLD-TDP group. The presence of the C9orf72 expansion did not appear to contribute to the results presented here. Although the presence of the expansion was more frequent (23% versus 12%) and the disease course was slightly faster (mean 2.7 versus 3.3 years) in ThS+ ALS cases, this was not statistically significant (p=0.14 and p=0.65 respectively).
In summary, our cohort included a diversity of ALS and FTLD-TDP cases with widespread TDP-43 pathology. ThS positive SLI were only observed in 28% of examined ALS spinal cords, demonstrating that the amyloid properties of TDP-43 are regionally limited and specific to a subset of spinal cord SLI in ALS.
Ultrastructure
We next examined the ultrastructure of the TDP-43 inclusions found in FTLD-TDP and ALS. Immuno-EM was performed to analyze spinal cord and hippocampal pathology in five individual cases, a subset representing the variety of our cohort (Table 2, Fig. 4–6). None of the TDP-43 inclusions in these cases had tau, β-amyloid or α-synuclein immunoreactivity (data not shown), while Cases #2 and 3 had ThS positive SLI.
Table 2.
Electron microscopy subject characteristics
| Subject | #1 | #2 | #3 | #4 | #5 |
|---|---|---|---|---|---|
| Gender | Male | Male | Male | Female | Male |
| Neuropathological Diagnosis | ALS | ALS | ALS | FTLD-TDP MND | ALS-D |
| C9orf72 Expansion | + | − | + | − | − |
| Age at death (y) | 39 | 57 | 42 | 52 | 66 |
| Disease duration (y) | 2 | 5–6 | 1–2 | 1–2 | 1–2 |
| PMI (h) | 5 | 4 | 6 | 4 | 7 |
| Brain weight (g) | 1406 | 1401 | 1454 | 1203 | 1144 |
| ThS positive SLI | − | + | + | − | − |
Abbreviations: ALS = amyotrophic lateral sclerosis; FTLD-TDP = frontotemporal lobar degeneration with TDP inclusions; MND = motor neuron disease; ALS-D = amyotrophic lateral sclerosis with dementia; PMI = postmortem interval; ThS = ThioflavinS; SLI = skein-like inclusion
Figure 6. TDP-43 nanogold immuno-EM of hippocampal dentate gyrus inclusions in 2 cases.
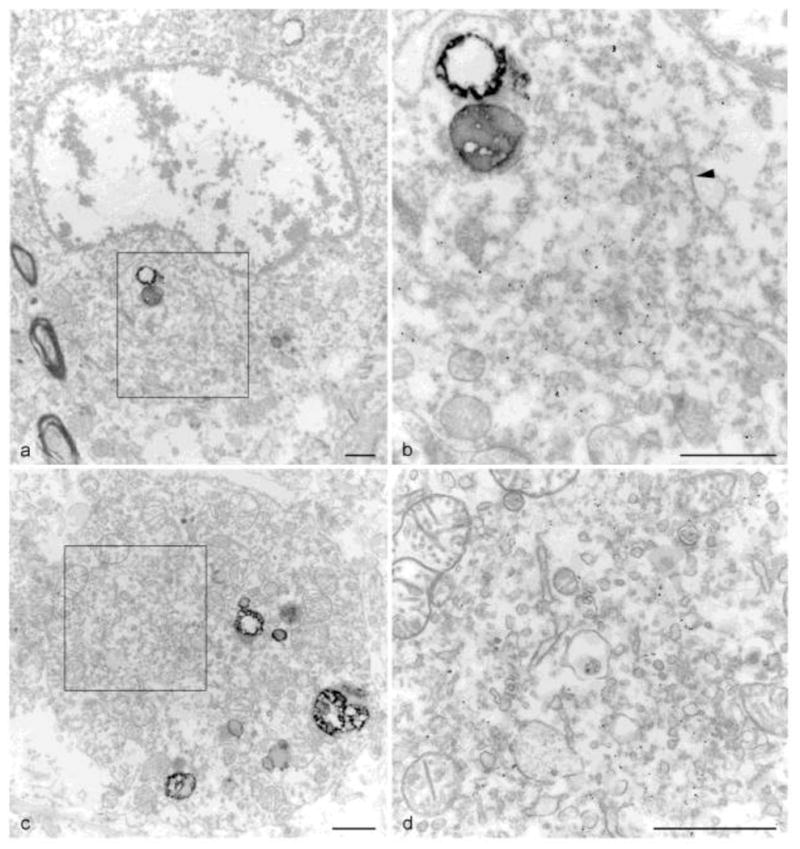
(a–b) FTLD-TDP case with (a) a juxtanuclear inclusion indenting a neuronal nucleus (6700X). (b) Higher magnification of boxed area from (a) showing the predominant granular material with a minor filamentous component (arrowhead) (21,200X). (c–d) ALS case with a similar (c) cytoplasmic inclusion (9700X). (d) Higher magnification of boxed area from (c) showing vesicular materials within the inclusion (26900X). (scale bars = 1μm)
In order to understand the structure of the inclusions, we first examined the spinal cord motor neurons by HRP-immuno-EM on semi-thin sections, we examine the spinal cord by HRP-immuno-EM. This showed both the round or compact inclusions (Fig. 4a, b) and the skeins (Fig. 4c, d) in spinal cord motor neurons appeared to consist of predominantly filamentous material. Round inclusions have a granular profile at lower magnification (Fig. 4a), which are composed of loosely packed and randomly oriented filaments as seen at higher magnification (Fig. 4b). In contrast, filaments in skeins appeared to be more ordered and tightly packed at all magnifications (Fig. 4c, d).
Since the HRP reaction product obscured the fine structure of the inclusions, nanogold immuno-EM was used to better delineate the detailed ultrastructure. TDP-43 nanogold immuno-EM revealed that the variety of observed granulo-filamentous material in lumbar spinal cord sections of ALS cases is TDP-43 positive (Fig. 5). Two different inclusions are presented from Case #1 in semi-thin sections: an elongated inclusion and a compact inclusion (Fig. 5a,d; same case as Fig. 4). The elongated inclusion (Fig. 5a–c) contained loosely packed granular material (arrows in Fig. 5c) and more tightly-bundled filaments (arrowheads in Fig. 5c), both decorated by TDP-43. The second inclusion (Fig. 5d–f) is more granular in nature, but also contains filaments in a more loosely packed configuration. Examination of sections from Case #2 (Fig. 5g–j) revealed a compact inclusion (Fig. 5g) with a nearby filamentous bundle (arrow head in Fig. 5h). TDP-43 labeled both the mixture of granulo-filamentous material seen in the compact inclusion (Fig. 5i) and the densely packed filaments (Fig. 5h arrowhead and Fig. 5j). When TDP-43 inclusions exhibit tightly-bundled filaments, they are best seen in longitudinal views (Fig. 5j), but an inclusion from Case #3 shows a cross-sectional view of bundles of filaments (Fig. 5k). Filaments were approximately 10 to 20 nm in diameter. Intriguingly, SLI in Cases #2 and #3 exhibited ThS positivity at the histological level. Of note, both Cases #1 and #3 had the C9orf72 expansion. We conclude that TDP-43 positive inclusions in ALS motor neurons are composed of both granular and filamentous material as seen by ultrastructural examination.
In contrast, TDP-43 inclusions seen in the dentate gyrus of the hippocampus are predominantly composed of granular material (Fig. 6). This is true in both FTLD-TDP Case #4 (Fig. 6a,b) and ALS Case #5 (Fig. 6c,d). Unlike in the spinal cord inclusions, filaments are rare. The inclusion from Case #4 indents the nucleus above it (Fig. 6a) and while TDP-43 positive profiles are seen throughout the granular material (Fig. 4b), little or no filamentous material is observed to be decorated by TDP-43 (arrowhead in Fig. 6b). The round inclusion from Case #5 is also almost purely granular with numerous membranous organelles. Here TDP-43 decorates non-filamentous material that is surrounded and embedded within more vesicular elements (Fig. 6d).
In conclusion, ultrastructural analysis reveals that TDP-43 inclusions in ALS spinal cord are composed of a mixture of granular and filamentous material while hippocampal TDP-43 inclusions are primarily granular in nature.
Discussion
SLI are primarily observed in the lower motor neurons of ALS and affected FTLD patients [9,17]. In the cerebral cortex and limbic areas, compact inclusions predominate, especially in the smaller neurons of the hippocampal dentate gyrus [26]. In this study, we demonstrate that SLI in ALS lower motor neurons are variably ThS positive, and this may reflect the more frequent presence of filaments in these inclusions. Why was this not observed previously? First, ThS positivity is a rare event. SLI are a subset of TDP-43 positive spinal cord inclusions and ThS positive SLI are a subset of those so the relative scarcity of ThS positive inclusions resulted in them being easily overlooked. In our data, some cases had multiple skeins per section (Fig. 2), while in others only one ThS positive SLI was confirmed for all examined spinal cord sections. Additionally, lipofuscin in the spinal cord accounts for many autofluorescing artifacts (Supplemental Figure 1c,f,i). Both the the scarcity of ThS positive SLI and the prominence of auto-fluorescencel may explain why it was overlooked in previous studies. Second, ThS positivity might be a very late stage conformational event in the biochemical ‘life-cycle’ of TDP-43 accumulation and this heteroogeneity of TDP-43 inclusions in ALS implies that the disease process may play out in different ways in different populations of neurons.
Ultrastructurally, FTLD cytoplasmic and neuritic cerebral and limbic inclusions are composed of granulo-filamentous material as are round spinal cord inclusions [5,13,19,26,33,36]. From our data we conclude that in ALS SLI contain bundles of more parallel oriented filaments, while compact inclusions have looser, more randomly oriented filaments with the presence of more granular material. Differences in preservation, processing techniques, counterstain methods, and the presence of randomly trapped organelles and vesicles, all account for the variation in the cell to cell appearance of inclusions. Nonetheless, our observations indicate that the difference between randomly oriented versus tightly bundled filaments is readily detectable. One interpretation of these data and earlier EM studies is that TDP-43 aggregates appear on a continuum of granular, granulo-filamentous and predominantly filamentous structures as implied by earlier ALS studies using anti-ubiquitin immuno-EM [21,24]. Of course, genetics may also play a role. Cases #1 and #3 both harbor the C9orf72expansion (see Fig. 5). Clearly, more detailed studies will be necessary to fully explain the variety of observed TDP-43 positive ultrastructures.
Several studies have suggested that TDP-43 can be amyloidogenic. The C-terminus region of TDP-43 is predicted to have prion like domain [16,11,12] which implies that TDP-43 C-terminus fragment is required for amyloid fibril formation. In vitro work has supported this hypothesis. Wild-type c-terminal fragments as well as those containing ALS mutations, either G294A [6] or A315T [12] formed ThT positive fibrils. Interestingly, TDP-43 pathology has been shown to be composed predominantly of full-length protein in spinal cord motor neurons, while in the cerebral cortex it is composed mostly of C-terminal fragments [14]. The finding that in human diseases the cortical C-terminal inclusions are granular in nature and ThS negative while many of the full length TDP-43 inclusions in the spinal cord are filamentous with some being ThS positive implies that in vivo the N-terminal portion of TDP-43 plays an under-appreciated role in fibrillization.
Our observations on the potential amyloid properties of TDP-43 inclusions need to be extended to larger numbers of ALS cases in order to validate our conclusions. While we provide in situ evidence that corroborates tissue culture and computational experiments, additional research is needed to verify our findings in post-mortem human tissue from ALS and FTLD-TDP patients. Similarly, more extensive investigations of regions outside the hippocampus, motor cortex and spinal cord should be undertaken to verify the regional spinal cord specificity we observed. Thus, further studies are needed to resolve the nature of an amyloidogenic pathway in the life-cycle of pathological TDP-43, its relationship to other brain amyloids (see Polymenidou et al[31] for a recent review) and the molecular differences in the species of TDP-43 that may underlie regional variation in protein aggregation. Elucidating the mechanisms that determine this heterogeneity could be significant for developing biomarkers and therapeutic interventions for these diseases.
Supplementary Material
Acknowledgments
We thank our many TDP-43 colleagues in the Center for Neurodegerative Disease Research (CNDR) and the Department of Neurology for extensive collaborations that provided essential input at many stages of our research on TDP-43 since 2006. In particular we thank Dr. Edward B. Lee, Ms. Theresa Schuck and Mr. Michael Partain for their input on these studies which were supported by the National Institutes of Health (AG10124, AG17586, K08AG039510, and training grant T32 AG00255). Virgnia M-Y. Lee is the John H. Ware III Chair of Alzheimer’s Research and John Q. Trojanowski is the William Maul Measey-Truman G. Schnabel, Jr., MD Professor of Geriatric Medicine and Gerontology. The authors are especially grateful to the families and patients affected by these diseases, without whose donations, this research would not be possible.
References
- 1.Al-Sarraj S, King A, Troakes C, Smith B, Maekawa S, Bodi I, Rogelj B, Al-Chalabi A, Hortobagyi T, Shaw CE. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 2011;122(6):691–702. doi: 10.1007/s00401-011-0911-2. [DOI] [PubMed] [Google Scholar]
- 2.Braak H, Ludolph A, Thal DR, Del Tredici K. Amyotrophic lateral sclerosis: dash-like accumulation of phosphorylated TDP-43 in somatodendritic and axonal compartments of somatomotor neurons of the lower brainstem and spinal cord. Acta Neuropathol. 2010;120(1):67–74. doi: 10.1007/s00401-010-0683-0. [DOI] [PubMed] [Google Scholar]
- 3.Brettschneider J, Van Deerlin VM, Robinson JL, Kwong L, Lee EB, Ali YO, Safren N, Monteiro MJ, Toledo JB, Elman L, McCluskey L, Irwin DJ, Grossman M, Molina-Porcel L, Lee VM, Trojanowski JQ. Pattern of ubiquilin pathology in ALS and FTLD indicates presence of C9ORF72 hexanucleotide expansion. Acta Neuropathol. 2012 doi: 10.1007/s00401-012-0970-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis and other motor neuron disorders: official publication of the World Federation of Neurology. Research Group on Motor Neuron Diseases. 2000;1 (5):293–299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 5.Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ, Foong C, White CL, 3rd, Schneider JA, Kretzschmar HA, Carter D, Taylor-Reinwald L, Paulsmeyer K, Strider J, Gitcho M, Goate AM, Morris JC, Mishra M, Kwong LK, Stieber A, Xu Y, Forman MS, Trojanowski JQ, Lee VM, Mackenzie IR. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol. 2007;171(1):227–240. doi: 10.2353/ajpath.2007.070182. S0002-9440(10)61957-8 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen AK, Lin RY, Hsieh EZ, Tu PH, Chen RP, Liao TY, Chen W, Wang CH, Huang JJ. Induction of amyloid fibrils by the C-terminal fragments of TDP-43 in amyotrophic lateral sclerosis. J Am Chem Soc. 2010;132(4):1186–1187. doi: 10.1021/ja9066207. [DOI] [PubMed] [Google Scholar]
- 7.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geser F, Lee VM, Trojanowski JQ. Amyotrophic lateral sclerosis and frontotemporal lobar degeneration: a spectrum of TDP-43 proteinopathies. Neuropathology. 2010;30(2):103–112. doi: 10.1111/j.1440-1789.2009.01091.x. NEU1091 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geser F, Martinez-Lage M, Kwong LK, Lee VM, Trojanowski JQ. Amyotrophic lateral sclerosis, frontotemporal dementia and beyond: the TDP-43 diseases. J Neurol. 2009;256(8):1205–1214. doi: 10.1007/s00415-009-5069-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, Janssens J, Bettens K, Van Cauwenberghe C, Pereson S, Engelborghs S, Sieben A, De Jonghe P, Vandenberghe R, Santens P, De Bleecker J, Maes G, Baumer V, Dillen L, Joris G, Cuijt I, Corsmit E, Elinck E, Van Dongen J, Vermeulen S, Van den Broeck M, Vaerenberg C, Mattheijssens M, Peeters K, Robberecht W, Cras P, Martin JJ, De Deyn PP, Cruts M, Van Broeckhoven C. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 2012;11(1):54–65. doi: 10.1016/S1474-4422(11)70261-7. [DOI] [PubMed] [Google Scholar]
- 11.Goldschmidt L, Teng PK, Riek R, Eisenberg D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc Natl Acad Sci U S A. 2010;107(8):3487–3492. doi: 10.1073/pnas.0915166107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo W, Chen Y, Zhou X, Kar A, Ray P, Chen X, Rao EJ, Yang M, Ye H, Zhu L, Liu J, Xu M, Yang Y, Wang C, Zhang D, Bigio EH, Mesulam M, Shen Y, Xu Q, Fushimi K, Wu JY. An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nature structural & molecular biology. 2011;18(7):822–830. doi: 10.1038/nsmb.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y, Beach TG, Buratti E, Baralle F, Morita M, Nakano I, Oda T, Tsuchiya K, Akiyama H. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol. 2008;64(1):60–70. doi: 10.1002/ana.21425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Igaz LM, Kwong LK, Xu Y, Truax AC, Uryu K, Neumann M, Clark CM, Elman LB, Miller BL, Grossman M, McCluskey LF, Trojanowski JQ, Lee VM. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am J Pathol. 2008;173(1):182–194. doi: 10.2353/ajpath.2008.080003. ajpath.2008.080003 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kerman A, Liu HN, Croul S, Bilbao J, Rogaeva E, Zinman L, Robertson J, Chakrabartty A. Amyotrophic lateral sclerosis is a non-amyloid disease in which extensive misfolding of SOD1 is unique to the familial form. Acta Neuropathol. 2010;119(3):335–344. doi: 10.1007/s00401-010-0646-5. [DOI] [PubMed] [Google Scholar]
- 16.King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012 doi: 10.1016/j.brainres.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee EB, Lee VM, Trojanowski JQ. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci. 2012;13(1):38–50. doi: 10.1038/nrn3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levine H, 3rd, Walker LC. Molecular polymorphism of Abeta in Alzheimer’s disease. Neurobiol Aging. 2010;31(4):542–548. doi: 10.1016/j.neurobiolaging.2008.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin WL, Dickson DW. Ultrastructural localization of TDP-43 in filamentous neuronal inclusions in various neurodegenerative diseases. Acta Neuropathol. 2008;116(2):205–213. doi: 10.1007/s00401-008-0408-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Llewellyn-Smith IJ, Minson JB. Complete penetration of antibodies into vibratome sections after glutaraldehyde fixation and ethanol treatment: light and electron microscopy for neuropeptides. J Histochem Cytochem. 1992;40 (11):1741–1749. doi: 10.1177/40.11.1431060. [DOI] [PubMed] [Google Scholar]
- 21.Lowe J. New pathological findings in amyotrophic lateral sclerosis. J Neurol Sci. 1994;124(Suppl):38–51. doi: 10.1016/0022-510x(94)90175-9. [DOI] [PubMed] [Google Scholar]
- 22.Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, Perry RH, Trojanowski JQ, Mann DM, Lee VM. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011;122(1):111–113. doi: 10.1007/s00401-011-0845-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marshak DW. Localization of immunoreactive tyrosine hydroxylase in the goldfish retina with pre-embedding immunolabeling with one-nanometer colloidal gold particles and gold toning. J Histochem Cytochem. 1992;40 (10):1465–1470. doi: 10.1177/40.10.1356122. [DOI] [PubMed] [Google Scholar]
- 24.Mizusawa H, Nakamura H, Wakayama I, Yen SH, Hirano A. Skein-like inclusions in the anterior horn cells in motor neuron disease. J Neurol Sci. 1991;105 (1):14–21. doi: 10.1016/0022-510x(91)90112-k. [DOI] [PubMed] [Google Scholar]
- 25.Mori F, Tanji K, Miki Y, Kakita A, Takahashi H, Wakabayashi K. Relationship between Bunina bodies and TDP-43 inclusions in spinal anterior horn in amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol. 2010;36(4):345–352. doi: 10.1111/j.1365-2990.2010.01081.x. [DOI] [PubMed] [Google Scholar]
- 26.Mori F, Tanji K, Zhang HX, Nishihira Y, Tan CF, Takahashi H, Wakabayashi K. Maturation process of TDP-43-positive neuronal cytoplasmic inclusions in amyotrophic lateral sclerosis with and without dementia. Acta Neuropathol. 2008;116(2):193–203. doi: 10.1007/s00401-008-0396-9. [DOI] [PubMed] [Google Scholar]
- 27.Nakano I. Frontotemporal lobar degeneration (FTLD) concept and classification update. Rinsho Shinkeigaku. 2011;51 (11):844–847. doi: 10.5692/clinicalneurol.51.844. [DOI] [PubMed] [Google Scholar]
- 28.Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J, Benson DF. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51 (6):1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 29.Neumann M, Kwong LK, Sampathu DM, Trojanowski JQ, Lee VM. TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis: protein misfolding diseases without amyloidosis. Arch Neurol. 2007;64(10):1388–1394. doi: 10.1001/archneur.64.10.1388. 64/10/1388 [pii] [DOI] [PubMed] [Google Scholar]
- 30.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi: 10.1126/science.1134108. 314/5796/130 [pii] [DOI] [PubMed] [Google Scholar]
- 31.Polymenidou M, Cleveland DW. Prion-like spread of protein aggregates in neurodegeneration. J Exp Med. 2012;209(5):889–893. doi: 10.1084/jem.20120741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Romijn HJ, van Uum JF, Breedijk I, Emmering J, Radu I, Pool CW. Double immunolabeling of neuropeptides in the human hypothalamus as analyzed by confocal laser scanning fluorescence microscopy. J Histochem Cytochem. 1999;47 (2):229–236. doi: 10.1177/002215549904700211. [DOI] [PubMed] [Google Scholar]
- 33.Sasaki S, Takeda T, Shibata N, Kobayashi M. Alterations in subcellular localization of TDP-43 immunoreactivity in the anterior horns in sporadic amyotrophic lateral sclerosis. Neurosci Lett. 2010;478(2):72–76. doi: 10.1016/j.neulet.2010.04.068. [DOI] [PubMed] [Google Scholar]
- 34.Schmidt ML, Schuck T, Sheridan S, Kung MP, Kung H, Zhuang ZP, Bergeron C, Lamarche JS, Skovronsky D, Giasson BI, Lee VM, Trojanowski JQ. The fluorescent Congo red derivative, (trans, trans)-1-bromo-2,5-bis-(3-hydroxycarbonyl-4-hydroxy)styrylbenzene (BSB), labels diverse beta-pleated sheet structures in postmortem human neurodegenerative disease brains. Am J Pathol. 2001;159(3):937–943. doi: 10.1016/s0002-9440(10)61769-5. S0002-9440(10)61769-5 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sipe JD. Amyloidosis. Annu Rev Biochem. 1992;61:947–975. doi: 10.1146/annurev.bi.61.070192.004503. [DOI] [PubMed] [Google Scholar]
- 36.Thorpe JR, Tang H, Atherton J, Cairns NJ. Fine structural analysis of the neuronal inclusions of frontotemporal lobar degeneration with TDP-43 proteinopathy. J Neural Transm. 2008;115(12):1661–1671. doi: 10.1007/s00702-008-0137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, Clay D, Wood EM, Chen-Plotkin AS, Martinez-Lage M, Steinbart E, McCluskey L, Grossman M, Neumann M, Wu IL, Yang WS, Kalb R, Galasko DR, Montine TJ, Trojanowski JQ, Lee VM, Schellenberg GD, Yu CE. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7(5):409–416. doi: 10.1016/S1474-4422(08)70071-1. S1474-4422(08)70071-1 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilcock DM, Gordon MN, Morgan D. Quantification of cerebral amyloid angiopathy and parenchymal amyloid plaques with Congo red histochemical stain. Nature protocols. 2006;1(3):1591–1595. doi: 10.1038/nprot.2006.277. [DOI] [PubMed] [Google Scholar]
- 39.Yu CE, Bird TD, Bekris LM, Montine TJ, Leverenz JB, Steinbart E, Galloway NM, Feldman H, Woltjer R, Miller CA, Wood EM, Grossman M, McCluskey L, Clark CM, Neumann M, Danek A, Galasko DR, Arnold SE, Chen-Plotkin A, Karydas A, Miller BL, Trojanowski JQ, Lee VM, Schellenberg GD, Van Deerlin VM. The spectrum of mutations in progranulin: a collaborative study screening 545 cases of neurodegeneration. Arch Neurol. 2010;67(2):161–170. doi: 10.1001/archneurol.2009.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.