
Figure 6.
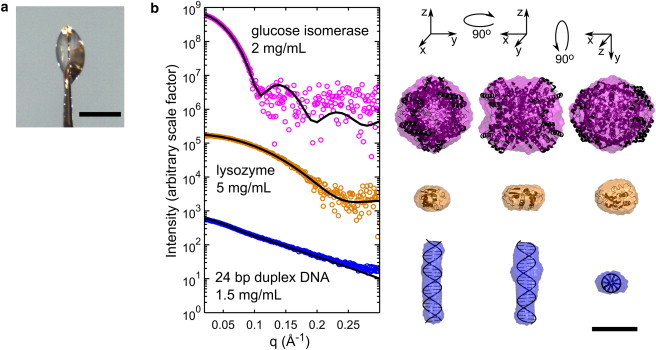
Molecular envelopes from nanoliter volumes. (a) Small, lenticular sample drops were held in a 600 μm diameter polyimide crystallography mount. A representative drop is shown. The scale bar in the image is 500 μm. (b) Cryo-SAXS data were acquired for GI, hen egg white lysozyme, and 24 basepair duplex DNA at the indicated concentrations. Cryo-SAXS profiles for each macromolecule were corrected by applying a constant offset using the BIFT method, as described in the Materials and Methods (this constant offset arises from sample geometry-dependent background subtraction errors at high-q values). Solid lines show the CRYSOL predictions from each atomic structure, based on PDBs 1XIB and 2LYZ, and an ideal 24-bp DNA helix generated using Nucleic Acid Builder (58). No fitting parameters were used except for an overall scale factor for the (arbitrary) intensity. Macromolecule envelopes generated from the cryo-SAXS data were aligned with the atomic structures, and are shown in three orientations. All are scaled according to the 50 Å bar at the lower right. For GI, lysozyme, and DNA, the mean normalized spatial discrepancies were 0.586, 0.459, and 0.542; the x-ray illuminated sample volumes were 16.3, 13.6, and 24.9 nL; the exposure times were 160, 80, and 160 s; and the x-ray doses were 275, 114, and 234 kGy, respectively.