

Abstract
Tyrosyl-DNA-phosphodiesterase I (Tdp1) plays a key role in the repair of damaged DNA resulting from the topoisomerase I (Top1) inhibitor camptothecin and a variety of other DNA-damaging anticancer agents. This report documents the design, synthesis, and evaluation of new indenoisoquinolines that are dual inhibitors of both Tdp1 and Top1. Enzyme inhibitory data and cytotoxicity data from human cancer cell cultures were used to establish structure-activity relationship. The potencies of the indenoisoquinolines against Tdp1 ranged from 5 μM to 111 μM, which places the more active compounds among the most potent known inhibitors of this target. The cytotoxicity mean-graph midpoints ranged from 0.02 to 2.34 μM. Dual Tdp1-Top1 inhibitors are of interest because the Top1 and Tdp1 inhibitory activities could theoretically work synergistically to create more effective anticancer agents.
1.Introduction
The phospholipases are a heterogeneous family of enzymes that catalyze the hydrolysis of phosphodiester bonds.1 One member of this family, tyrosyl-DNA-phosphodiesterase I (Tdp1), catalyzes the hydrolysis of 3′-phosphotyrosyl linkers.2 This enzyme is unique because its catalytic site possesses two histidine and two lysine residues but lacks the aspartate residue that is found in the other members of the family.3 When DNA topoisomerase I (Top1) nicks double-stranded DNA, a covalent Top1-DNA complex is made.4 The formed 3′-adducts must be removed by Tdp1 in order to repair damaged DNA in stalled Top1-DNA complexes in which the normal DNA religation reaction has not occurred. The enzyme mechanism is believed to occur in two sequential steps.5, 6 The first step consists of the nucleophilic attack by His263 on the phosphorous atom linked to the oxygen of the Top1 catalytic residue Tyr723 at the 3′ end of DNA (Figure 1). The function of the Lys265 and Lys495 residues found in the catalytic site is to coordinate the oxygen atoms of the phosphate group, which enables the covalent linkage of His263 to the 3′-phosphate end of the DNA. In the second step, this intermediate is hydrolyzed by a His493-activated water molecule. The overall reaction frees the tyrosine residue and affords a DNA strand that has a 3′-phosphate end. The process of DNA restoration is then finished by DNA polymerases and DNA ligases. The role of Tdp1 is to hydrolyze phosphotyrosyl-DNA linkages in denatured or proteolytically degraded Top1-DNA complexes.7
Figure 1.

Mechanism of Tdp1.5
One approach to chemotherapy is based on forming lesions in tumor DNA. Thus, stalled Top1-DNA complexes can also arise when Top1 poisons, such as camptothecins (CPTs) or indenoisoquinolines, are used for cancer therapy.7–9 The function of the CPTs is to stabilize the DNA-Top1 complex and inhibit the religation process. As a consequence, DNA replication forks collide with drug-stabilized complexes causing double-stranded DNA breaks that ultimately result in tumor cell death. It is believed that Tdp1 may be responsible for the drug resistance of some cancers by virtue of repairing DNA lesions caused by CPTs.5, 10 This hypothesis is further supported by the fact that hypersensitivity to CPTs is observed when Tdp1 is inactivated in DNA repair- or checkpoint-deficient yeast.11
Given the relationship between Top1 and Tdp1, inhibitors of Tdp1 could potentiate the effects of Top1 poisons.2, 5 Although the association between these enzymes makes Tdp1 an attractive target for cancer treatment, there are few known inhibitors in the literature.5, 12–15 Moreover, these compounds show weak inhibitory activity with IC50’s usually in the micromolar range. A recent publication reported that indenoisoquinolines bearing three or four methylene units on the amine-substituted side chain are good Tdp1 inhibitors.16 The binding interactions between Tdp1 and indenoisoquinoline 116 (Figure 2) have previously been characterized using surface plasmon resonance (SPR) and fluorescence resonance energy transfer (FRET). During the previous studies, it was concluded that the sulfonate substituent, key for the activity of compounds such as 2, did not provide any advantage when present on the side chain attached to the lactam of indenoisoquinolines. This paper expands the previously reported SAR studies regarding Tdp1 inhibitors and introduces novel indenoisoquinolines that are active against this target. Additionally, compounds with and without dual activity against both Tdp1 and Top1 enzymes are presented.
Figure 2.

Tdp1 inhibitors.
Chemistry
It is known that the Tdp1 active site possesses two lysine (265 and 495) and two histidine (263 and 493) residues. Some Tdp1 inhibitors have functional groups (e.g. carbonyls) that can hydrogen bond with these residues, and a lipophilic core that interacts with the hydrophobic region (Ala520, Phe259, Gly260, Tyr261, etc.) of the active site.5 Indenoisoquinolines with an ester moiety might therefore be inhibitors of the enzyme. The partially negatively charged carbonyl oxygen may interact with some of the charged polar residues in the binding pocket while the indenoisoquinoline aromatic system might sit in the hydrophobic portion of the cavity. In order to build on the previous work,16 an ester moiety was therefore added to 3-amino-6-methyl-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (3)17 as shown in Scheme 1. Briefly, the amino group of 3 was reacted with methyl oxalyl chloride (4) to provide compound 9 that was hydrolyzed, under basic conditions, to yield 14. Compound 9 was inactive but 14 showed modest Tdp1 inhibitory activity with and IC50 of 61.7 μM. Therefore, compounds 5–8 with several methylene units between the acyl chloride and the esters functionalities were reacted with 3 to afford compounds 10–13. The ester 10 was converted to the acid derivative 15.
Scheme 1a.

aReagents and conditions: (a) THF, Et3N; (b) NaOH, MeOH, DMF or NaOH, EtOH.
Compound 1618 was reacted with acyl chlorides 4–8 to produce indenoisoquinolines 17–21, which were converted to the amines 22–26 by deprotection under acidic conditions (Scheme 2). The acids 32–36 were prepared from intermediates 17–21 (Scheme 3) by hydrolysis under basic conditions followed by acid treatment. Also, precursors 3 and 16 were reacted with ethyl glyoxylate using the Borch reduction to give esters 38 and 39 (Scheme 4) that were further hydrolyzed to acids 41 and 43. Compound 40 was prepared by deprotection of 39 (Scheme 4).
Scheme 2a.

aReagents and conditions: (a) CHCl3, Et3N; (b) i. TFA, CHCl3; ii. NH3, MeOH.
Scheme 3a.

aReagents and conditions: (a) NaOH, THF, MeOH; (b) HCl, ethylether, CHCl3.
Scheme 4a.

aReagents and conditions: (a) i. HOAc, MeOH, ii. NaCNBH3;(b) KOH, MeOH, THF, H2O; (c) 2 M HCl, ether, CHCl3.
A set of 3-nitroindenoisoquinolines was prepared because a high-priority goal was to synthesize dual Tdp1-Top1 inhibitors, and it is well established that a 3-nitro substituent increases potency vs Top1.19–21 Compound 51 (Scheme 5) was prepared using the Schiff base-homophthalic anhydride condensation approach.20, 22–24 Briefly, Schiff base 46,25 obtained by the condensation of meta-methoxybenzaldehyde (44) and 3-bromopropylamine hydrobromide (45), was reacted with 5-nitrohomophthalic anhydride (47) to provide acid 48. This compound was subjected to Friedel-Crafts conditions to afford indenoisoquinoline 49, followed by halide displacement with azide to provide 50. Subsequent reduction of 50 under Staudinger conditions gave indenoisoquinoline 51. Precursor 49 was treated with imidazole or morpholine to afford compounds 52 and 53 (Scheme 6). Compounds 54–63 (Figure 3),18, 20, 24 from our existing library of compounds prepared to investigate Top1 inhibition, were also evaluated in order to identify key features for Tdp1 inhibitory activity.
Scheme 5a.

aReagents and conditions: (a) Et3N, CHCl3; (b) CHCl3; (c) i. SOCl2, PhH, ii. AlCl3, PhNO2; (d) NaN3, DMSO, 90 °C; (e) i. P(OEt)3, PhH, ii. HCl, MeOH.
Scheme 6a.

aReagents and conditions: (a) (i) NaI, dioxane, DMF, (ii) imidazole or morpholine, K2CO3.
Figure 3.

Previously reported indenoisoquinolines prepared to study Top1 inhibitory activity.
Additionally, new indenoisoquinolines bearing nitrile and iodo substituents on the A ring were synthesized. There are not many substitutions reported in the literature on this side of the indenoisoquinoline system; thus, the current study provided a prime opportunity to expand the substitution pattern on the A ring. Moreover, given the fact that the methoxy or methylenedioxy substituents have been shown to improve Top1 activity, these substituents were added to some of the new compounds.26 Isochromenone 68 (Scheme 7) was prepared by condensing 6-cyano-3-hydroxypthalide (66), obtained from 3-cyanophthalide (64), with phthalide (67).27 The product was reacted with various substituted aminopropyl compounds to yield indenoisoquinolines 72–74 (Scheme 7). Compound 66 was also condensed with 5,6-methylenedioxyphthalide28 (75, Scheme 8) to provide the methylenedioxy-substituted compound 76, which was reacted N′,N′-dimethylaminopropylamine (71) to yield indenoisoquinoline 77. Compound 85 (Scheme 9) was prepared according to previous procedures from 5-nitrohomophthalic acid (47) and Schiff base 78. 20, 22–24 The nitro group of compound 80 was reduced to aniline 81 by catalytic hydrogenation. The amine functionality of compound 81 was replaced by an iodine atom using Sandmeyer chemistry to provide indenoisoquinoline 82, which was converted to the amino analogue 84 by azide displacement and Staudinger reduction. Reaction with formaldehyde under Borch reduction conditions afforded the dimethylamino analogue 85.
Scheme 7a.

aReagents and conditions: (a) 3-Cl-perBzOH, NBS, hu, CCl4; (b) H2O, reflux; (c) (i) NaOMe, MeOH, EtOAc, (ii) HCl, (iii) pTsOH, PhH; (d) THF, Et3N, reflux; (e) CHCl3, Et3N, reflux.
Scheme 8a.

aReagents and conditions: (a) i. NaOMe, MeOH, EtOAc, ii. HCl, iii. PhH, pTsOH; (b) 71, CHCl3, Et3N.
Scheme 9a.

aReagents and conditions: (a) CHCl3; (b) (i)SOCl2, PhH, (ii) AlCl3, PhNO2; (c) H2, Pd-C, THF, MeOH, EtOAc; (d) (i) NaNO2, HCl, H2O, dioxane, (ii) CuI, KI, H2O; (e) NaN3, DMSO, 90 °C; (f) (i) P(OEt)3, PhH (ii) HCl, MeOH, (iii) K2CO3; (g) (i) CH2(O), MeOH, HOAc, (ii) NaCNBH3.
Finally, to expand the diversity of the set, the effect of combining an aniline group at the 3 position with a methoxy group at the 9 position was investigated. In order to accomplish this, compound 86 (Scheme 10) was prepared following published procedures.20 Reduction of the nitro group of analogue 86 was attempted with several conditions, including Raney nickel as previously done.24 Ultimately, it was discovered that using comparable amounts (by weight) of 5% Pd-C and indenoisoquinolines 86 while hydrogenating at 1 atm in THF provided the most consistent and reproducible yield of intermediate 87. Treatment of compound 87 with sodium azide in DMSO at 100 °C provided analogue 88, which was subsequently reduced with triethyl phosphite to provide compound 89, isolated as the dihydrochloride salt (Scheme 10).
Scheme 10a.

aReagents and conditions: (a) Pd-C, H2 (1 atm), THF; (b) NaN3, DMSO, 100 °C; (c) (i) P(OEt)3, PhH, reflux, (ii) HCl, MeOH, reflux.
Biological Results and Discussion
The IC50 of the oxalic acid derivative 14 vs Tdp1 was 61 ± 7 μM as shown in the titration curve (Figure 4). Surface plasmon resonance studies indicated that the compound binds the protein in a 1:1 ratio (Figure 5). The association and dissociation rates were very fast, but within the limit of detection of the instrument. The experiment also indicated that the compound did not bind to the single-stranded DNA substrate. Based on previous results16 and the activity of compound 14, computational studies using GOLD and Sybyl were performed.29, 30 Docking 14 in the catalytic site of Tdp1 suggested interactions between the oxygen atoms of 14 and the Tdp1 residues Thr261, His263, Thr281, Asn283 and Ser400 (Figure 6). Thus, a series of compounds containing homologous side chains and ester moieties at the 3 position were prepared in order to probe the left side of the binding pocket as it is portrayed in Figure 6.
Figure 4.

Titration curve of compound 14 against Tdp1.
Figure 5.

Surface plasmon resonance for compound 14 with Tdp1. The binding of compound 14 to Tdp1 was examined using SPR spectroscopy. Different concentrations of 14 (50, 25, 12.5 and 6.25 μM) were injected over immobilized Tdp1. The kinetics were fit to a 1:1 binding model yielding the following parameters, ka 7.7e4 1/Ms, kd 0.24 1/s and KD 31 μM.
Figure 6.

Hypothetical binding model of 14 in of the binding pocket of Tdp1.
An analysis of the biological data for the rest of the indenoisoquinolines allowed the determination of some of the important features needed for Tdp1 inhibitory activity. The aminopropyl side chain is clearly important for enzyme inhibition as observed in 22, +(+); 23, ++(+); 40, ++(+); 43 +++; 51, ++; 54, +++; 55, +++; 57, ++; 60, +(+); 62, ++(+); 74, ++; 84, +++; 85, +(+); and 89, +++; (note: the “+ system” is a semiquantitative scale expressing IC50 values at a given range as explained in Table 1, and the IC50 values of the more active Tdp1 inhibitors are reported in Table 2). The indenoisoquinoline 40 was chosen as a representative example of an N-(3-aminopropyl) compound for GOLD docking and molecular mechanics energy minimization studies, which resulted in the hypothetical binding mode displayed in Figure 7. The structure suggests the existence of bonding interactions between the charged ammonium cation of the ligand and the Val401 backbone carbonyl, the Pro461 backbone carbonyl, and the Thr466 side chain oxygen with distances of 2.7 Å, 2.6 Å and 3.0 Å, respectively. These bonding interactions would help to explain the Tdp1 inhibitory activity of the 3-aminopropyl-substituted compounds 40 and 43 vs the inactivity of their N-methylated and Boc-protected counterparts 38, 41, and 42. Additionally, Thr261 with its side chain hydroxyl was within H-bonding distance, 2.7 Å, to the carbonyl oxygen of the ligand 40.
Table 1.
Tdp1 and Top1 Inhibitory Activity
| Compd | Tdp1 | Top1 | Compd | Tdp1a | Top1b |
|---|---|---|---|---|---|
| 1 | ++ | +++ | 40 | ++(+) | ++(+) |
| 2 | +++ | NT | 41 | 0 | 0 |
| 9 | 0 | 0 | 42 | 0 | 0 |
| 10 | 0 | 0 | 43 | +++ | 0 |
| 11 | 0 | (+) | 49 | 0 | +++ |
| 12 | 0 | + | 50 | 0 | + |
| 13 | 0 | 0 | 51 | ++ | ++ |
| 14 | + | ++ | 52 | (+) | +++ |
| 15 | 0 | +(+) | 53 | 0 | ++ |
| 18 | 0 | 0 | 54 | +++ | ++++ |
| 19 | 0 | 0 | 55 | +++ | ++++ |
| 20 | 0 | 0 | 56 | 0 | ++ |
| 22 | +(+) | 0 | 57 | ++ | 0 |
| 23 | ++(+) | ++(+) | 58 | 0 | + |
| 24 | + | (+) | 59 | + | +(+) |
| 25 | + | 0 | 60 | +(+) | ++(+) |
| 26 | + | (+) | 61 | 0 | 0 |
| 29 | + | (+) | 62 | ++(+) | ++(+) |
| 30 | 0 | 0 | 63 | 0 | ++ |
| 31 | + | (+) | 72 | 0 | ++ |
| 32 | 0 | + | 73 | 0 | ++ |
| 33 | 0 | + | 74 | ++ | +++ |
| 34 | 0 | + | 77 | 0 | ++(+) |
| 35 | 0 | 0 | 84 | +++ | +(+) |
| 36 | 0 | + | 85 | +(+) | +++ |
| 38 | 0 | 0 | 89 | +++ | ++(+) |
Tdp1 IC50 was determined by duplicate using a semiquantitative scale: 0, IC50 > 111 μM; +, IC50 between 37–111 μM; ++, IC50 between 12–37 μM; +++, IC50 between 1–12 μM, ++++, IC50 < 1 μM. Active compounds were further evaluated for a more accurate value.
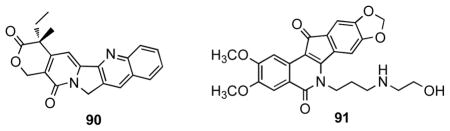
Compound-induced DNA cleavage due to Top1 inhibition is graded by the following semiquantitative scale relative to 1 μM camptothecin (90) or MJ-III-65 (91): 0, no detectable activity; +, weak activity; ++, similar activity to compound 91; +++, greater activity than 91; ++++, equipotent to 90. The (+) ranking indicates the activity lies between two given values.
NT: not tested.
Table 2.
IC50 Values of Tdp1 Active Compounds
| Compd | Tdp1 (μM) | Compd | Tdp1 (μM) |
|---|---|---|---|
| 14 | 61 ± 7 | 54 | 11 ± 1 |
| 22 | 45 ± 10 | 55 | 5.8 ± 0.8 |
| 23 | 18 ± 8 | 57 | 18 ± 1 |
| 40 | 11 ± 5 | 62 | 12 ± 4 |
| 43 | 5.0 ± 1.4 | 84 | 5.2 ± 0.1 |
| 51 | 15 ± 3 | 89 | 6.7 ± 0.8 |
The IC50 values of Tdp1 active compounds were determined by quadruplicate
Figure 7.

Hypothetical binding model of 40 in of the binding pocket of Tdp1.
In general, compounds in which the aminopropyl side chain was Boc-protected were either inactive or had very low Tdp1 inhibitory activity (Table 1). The weakly active Boc-protected compounds were 29 and 31. One N-methylated compound 14 showed weak activity, +, but all of the other analogues containing an N-methyl side chain (9–13, 15, 38, and 41) were inactive. Molecules 15, 30, and 32–36 with an acidic side chain at C-3 on the A ring were inactive but compounds 14 (+) and 43 (+++) were the exceptions. The ester-substituted indenoisoquinolines containing an aminopropyl side chain were all active but the activity decreased when more methylene units were added to the ester side chain, going from +(+) and ++(+) for 22 and 23 to + for 24–26 (Table 1). The ethyl ester 40, which has a short ester side chain, was active.
Given the apparent importance of the aminopropyl side chain for Tdp1 inhibition, ten compounds 54–63 from our existing Top1 inhibitor library were selected for testing in order to further explore this feature (Figure 3).16, 18, 20, 24 These compounds were all active as long as the 3-aminopropyl side chain and the 11-keto functional group were both present. Replacement of the primary amine with bromide, azide, morpholine or imidazole rendered the compounds very weak or inactive, as exemplified by 49, 50, 52, and 53 (Table 1). The primary alcohols 58 and 63 were also inactive, indicating that a primary ammonium ion may be a critical feature that is important for activity as opposed to hydrogen bonding capabilities per se. Compounds 59 and 62, with hydrogen bond acceptors at the 3 position of the A ring, are also Tdp1 inhibitors. It seems that the RXR inhibitory activity of the compounds decreases as the electron withdrawing strength of the substituent at the 3 position increases, as seen with the IC50’s of compounds 60 > 57 > 62, which are >37, 18 and 12 μM, respectively. Compounds 72–74 support the trend that a nitrogen that is more basic than morpholine or imidazole on the aminopropyl side chain is necessary for Tdp1 inhibitory activity. The primary amine 84, +++, was more active than the tertiary amine 85, +(+), suggesting a steric limitation to binding that may also be operating in the 72–74 series.
Molecular modeling indicates that the carbonyl of the five-membered C ring or the amide-carbonyl of the B ring may interact with either the Ser400 or Thr261 side chain hydroxyl groups as seen in Figures 6 and 7. In the case of 14, the lactam carbonyl binds Thr261, whereas in 40, the ketone binds, so the ring systems are “flipped” relative to each other. This suggests that the amino group of the ligand 40 and other N-(3-aminopropyl)indenoisoquinolines can play a major role in orienting the ligand in the binding site of the enzyme.
Compound 61, which contains a hydroxyl group instead of the ketone, was not active, suggesting that a hydrogen bond-accepting carbonyl oxygen is important for activity. The presence of a 9-methoxy group on the D ring of the indenoisoquinolines seems to have a positive effect on the Tdp1 inhibitory activity, as observed for 54 (+++) vs 60 +(+) or 89 (+++) vs 62 ++(+). Also, the position of the methoxy group at C7, C8, or C9 affects the IC50’s as seen with 51 (15 μM), 54 (11 μM) and 55 (5.8 μM). Figure 8 shows a representative gel electrophoresis assay and titration curves for determination of Tdp1 IC50 values for compounds 54 and 55. The 9-methoxy substituent seems to play a bigger role in Tdp1 inhibitory activity than the substituent at the 3 position. Within the 9-methoxy indenoisoquinolines, the electron-withdrawing 3-nitro group decreases the activity slightly when compared with its 3-iodo or 3-amino counterparts. The IC50’s of the active compounds 54 (3-NO2), 84 (3-I), and 89 (3-NH2) are 11 ± 1, 5.2 ± 0.1, 6.7 ± 0.8 (Table 2), respectively. Compound 77, which contains an 8,9-methylenedioxy group, was inactive vs Tdp1.
Figure 8.

(A) Representative gel showing Tdp1 inhibition for compounds 54 and 55. (b) Titration curves for determination of Tdp1 IC50 values for compounds 54 and 55.
The Top1 inhibitory activities of some of the active compounds have previously been analyzed and published.16 Regarding the new compounds, 49–53 were less active compared with 54 and 55. For example, compounds 52 and 53 present Top1 inhibitory activities of +++ and ++, respectively, compared to ++++ observed for both 54 and 55. The 3-cyano-substituted compounds 72–74 showed Top1 inhibitory activities of ++, ++, and +++, respectively. The 3-cyano compound 77 had good Top1 activity of ++(+).
The iodo-substituted compounds 84 and 85 were also active as Top1 inhibitors, although not as active as the previously published analogues such as 54 having a 3-nitro substituent.18 However, the 3-iodo substituent could be easily exchanged, thus expanding the alternatives for substitution. When the nitro group of compound 54 was replaced with an amine to obtain 89, the Top1 inhibitory activity dropped from ++++ to ++. The addition of a methoxy group at the 9 position of compound 60 increased Top1 inhibitory activity as seen with 54 (++++) vs 60 [++(+)]. However, this trend is not observed with the aniline analogues as seen in 62 with an activity of ++(+) vs 89 with an activity of ++(+).
All of the compounds were tested for induction of Top1-DNA cleavage complexes that are stabilized by inhibition of the DNA religation reaction due to intercalation of the drugs between DNA base pairs (Figure 9).31 Camptothecin (90) and the previously synthesized lead compound 9132–34 were included for comparison. The cleavage complexes were monitored using a 32P 3′-end labeled, 117-bp DNA fragment that was reacted with recombinant human Top1 in the presence of increasing concentrations of the indenoisoquinolines while separation of the DNA fragments was carried out on a denaturing gel. The sequence preferences for trapping the Top1-DNA cleavage complexes by the indenoisoquinolines are similar to each other, but the pattern is different from camptothecin, indicating that the indenoisoquinolines target the genome differently from camptothecin. However, the indenoisoquinolines 52, 77, and 85 differ from each other in their abilities to suppress DNA cleavage at high drug concentrations. As is evident from the gel, the indenoisoquinolines 52 and 77, having terminal dimethylamino substituents on the side chain, suppress DNA cleavage at a high concentration of 100 μM, but compound 52, having an imidazole substituent at the end of the chain, does not. DNA unwinding studies on similar 7-azaindenoisoquinolines have shown that compounds with an N-(3-dimethylaminopropyl) substituent intercalate into free DNA at high drug concentrations, making the DNA a poorer substrate for Top1, but compounds with an N-(3-imidazolylpropyl) substituent do not intercalate into free DNA so DNA cleavage is not suppressed at high drug concentration.35 Although slight suppression is evident at high concentrations of the imidazole analogue 52, it does not resemble the complete suppression observed with the dimethylamino analogues 77 and 85.
Figure 9.

Top1-mediated DNA cleavage induced by 85, 77 and 52. Lane 1: DNA alone; lane 2: Top1 alone; lane 3: 1, 1 μM; lane 4: 5, 1 μM; lane 5–16: 85, 77 and 52 at 0.1, 1, 10 and 100 μM respectively from left to right. Numbers and arrows on the left indicate arbitrary cleavage site positions. The activity of the compounds to produce Top1-mediated DNAcleavage was expressed semiquantitatively as follows: +, weak activity; ++ and +++, moderate activity; ++++, similar activity as 1 μM camptothecin (90).
In order to investigate their potential as anticancer agents, a set of indenoisoquinolines was examined for antiproliferative activity against the human cancer cell lines in the National Cancer Institute screen, in which the activity of each compound was evaluated with approximately 55 different cancer cell lines of diverse tumor origins.36, 37 The GI50 values obtained with selected cell lines, along with the mean graph midpoint (MGM) values, are summarized in Table 3. The MGM is based on a calculation of the average GI50 for all of the cell lines tested in which GI50 values below and above the test range (10−8 to 10−4 molar) are taken as the minimum (10−8 molar) and maximum (10−4 molar) drug concentrations used in the screening test. For comparison purposes, the activities of previously reported compounds in Figure 3 are included on the Table 3. Many of the new compounds display significant potencies against various cell lines with GI50’s in the low micromolar or submicromolar range. All of the compounds in Table 3 except 22, 25, 43, and 63 display some degree of inhibitory activity against both Tdp1 and Top1. The most promising new compounds are 52 and 89, with mean-graph midpoint (MGM) GI50 values of 0.02 μM and 0.04 μM, respectively. Overall, the cytotoxicities do not correlate very well with the potencies vs the isolated enzymes. For example, compounds 23, 40, and 62 all have the same ++(+) potencies vs both enzymes, but their cytotoxicity MGM values range from 1.86 to 0.16 μM. Another example would be 54 (MCM 0.02 μM) vs 55 (MGM 1.41 μM), both of which have +++ potency vs Tdp1 and ++++ potency vs Top1. Compounds 52 (MGM 0.02 μM) and 54 (MGM 0.02 μM) provide another way of stating the case, since they both have the same MGM value but on the basis of the Tdp1 and Top1 inhibitory potencies, 54 would be expected to be more cytotoxic than 52. These differences in cytotoxicities may reflect different uptake, distribution, metabolism, and excretion profiles in the cellular systems as well as off-target effects.
Table 3.
Cytotoxicities of Selected Compounds
| cytotoxicity (GI50 in μM)a | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| lung | colon | CNS | melanoma | ovarian | renal | prostate | breast | ||
| Compd | HOP-62 | HCT-116 | SF-539 | UACC-62 | OVCA R-3 | SN12C | DU-145 | MDA-MB-435 | MGM (μM)b |
| 22 | 0.65 | 0.57 | 6.90 | 1.22 | 4.01 | 2.53 | 1.11 | 3.36 | 1.86 ± 0.38c |
| 23 | 1.56 | 0.54 | 4.03 | 1.69 | 3.22 | 1.46 | 1.57 | 2.33 | 1.86 |
| 25 | 0.79 | 0.38 | 1.47 | 1.22 | 1.47 | 1.02 | 0.54 | 2.05 | 1.09 |
| 26 | 2.32 | 0.51 | 5.65 | 2.43 | 3.52 | 1.82 | 0.77 | 4.3 | 2.34 |
| 40 | 0.93 | 0.44 | 8.91 | 2.25 | 3.12 | 1.75 | 1.68 | 3.05 | 1.69 |
| 43 | 0.65 | 0.33 | 1.79 | 0.72 | 1.6 | 0.49 | 1.51 | 1.14 | 0.87 ± 0.014 |
| 51 | 0.14 | 0.06 | 0.61 | 0.14 | 0.54 | 0.15 | 0.18 | 0.58 | 0.17 ± 0.017 |
| 52 | <0.01 | <0.01 | <0.01 | <0.01 | 0.03 | <0.01 | <0.01 | 0.03 | 0.02 ± 0.0006 |
| 54 | <0.01 | <0.01 | <0.01 | <0.01 | 2.82 | <0.01 | - | 3.31 | 0.02 ± 0.0008 |
| 55 | 1.15 | 0.72 | 1.45 | 2.34 | 2.29 | 7.08 | 1.07 | 1.62 | 1.41 ± 0.43 |
| 60 | - | <0.01 | 0.14 | 0.03 | 0.08 | <0.01 | 0.01 | 0.12 | 0.15 ± 0.10 |
| 62 | 0.27 | 0.18 | 0.34 | 0.30 | 0.13 | 0.23 | 0.12 | 0.23 | 0.16 |
| 63 | 0.39 | 0.19 | 0.39 | 0.31 | 0.87 | 0.47 | 0.43 | 1.97 | 0.89 ± 0.22 |
| 74 | 0.26 | 0.05 | 1.12 | 0.19 | 1.72 | 0.19 | 0.24 | 0.62 | 0.30 ± 0.014 |
| 84 | 0.14 | 0.07 | 0.39 | 0.14 | 1.32 | 0.05 | 0.05 | 0.96 | 0.25 ± 0.004 |
| 89 | <0.01 | <0.01 | 0.03 | 0.10 | 0.02 | <0.01 | <0.01 | 0.12 | 0.04 |
The cytotoxicity GI50 values are the concentrations corresponding to 50% growth inhibition. The compounds were tested at concentrations ranging up to 10 μM
Mean graph midpoint for growth inhibition of all human cancer cell lines successfully tested.
For MGM GI50 values in which a standard error appears, the GI50 values for individual cell lines are the average of two determinations; values without standard error are from one determination.
Conclusions
The key points of this study are:
A 3-aminopropyl side chain on the lactam nitrogen is important for Tdp1 inhibitory activity, since compounds that lack this structural feature are either inactive or have very low activity.
The 11-keto group appears to be important for Tdp1 inhibitory activity since its reduction in 57 to an alcohol 61 abolished the activity.
Consistent with previous results,35 indenoisoquinolines 77 and 85 with acyclic 3-aminopropyl side chains suppress Top1-mediated DNA cleavage at high drug concentrations, but the 3-imidazolylpropyl compound 52 does not.
Instillation of a 9-methoxy substituent into 60 (MGM 0.15 μM) results in an increase in cytotoxicity (see 54, MGM 0.02 μM), while an 8-methoxy has little effect (see 51, MGM 0.17 μM) and a 7-methoxy is detrimental (see 55, MGM 1.41 μM). These trends are not reflected in their activities vs the isolated enzymes (compare 54 and 55, Table 3).
With the exception of 14 and 43, instillation of C-3 side chains of various lengths ending in a carboxylate generally resulted in compounds that are inactive vs Tdp1 and have low activity or no activity vs Top1 (see 32–36, Table 1). However, the corresponding esters 22–26 retained low to moderate activity vs Tdp1 and had mixed effects on Top1 compared with the corresponding acids (Table 1).
Within the similar series of compounds 54 (3-NO2), 89 (3-NH2), and 84 (3-I), the substituent at C-3 has no effect on Tdp1 inhibitory activity, but it does have an effect on Top1 inhibitory activity, with the potency ranking being NO2 > NH2 > I. This is reflected in the relative cytotoxicities of these compounds.
There is increasing interest in obtaining drugs that act on more than one target, and in the present case there is a clear rationale for dual inhibition. However, the poor correlation of cytotoxicity with activities against the two isolated enzymes observed here does not allow a convincing argument to me made that inhibition of Tdp1 increases the cytotoxicity resulting from Top1 inhibition.
The indenoisoquinolines 54 and 89 both have significant dual enzyme inhibitory activities and are cytotoxic enough in human cancer cell cultures to warrant further preclinical development.
Experimental Section
General
NMR spectra were obtained at 300 or 500 (1H) and 75 or 125 (13C) MHz using Bruker ARX300 or Bruker DX-2 500 [QNP probe or multinuclear broadband observe (BBO) probe, respectively] spectrometers. Column chromatography was performed with 230–400 mesh silica gel. The melting points were determined using capillary tubes with a Mel-Temp apparatus and are uncorrected. IR spectra were obtained using a Perkin-Elmer 1600 series FTIR spectrometer on salt plates or as KBr pellets. ESI-MS analyses were recorded in a FinniganMAT LCQ Classic mass spectrometer. APCI-MS analyses were performed in an Agilent 6320 ion trap mass spectrometer. EI/CI-MS analyses were obtained in a Hewlett-Packard Engine mass spectrometer. All mass spectral analyses were performed at the Campus-Wide Mass Spectrometry Center of Purdue University. Microanalyses were performed at Midwest Microlab. HPLC analyses were carried out on a Waters 1525 binary HPLC pump/Waters 2487 dual λ absorbance detector system using a 5 μM C18 reverse phase column. All reported yields refer to pure isolated compounds. Chemicals and solvents were of reagent grade and used as obtained from commercial sources without further purification. The purities of all of the tested compounds were >95% as estimated by HPLC or determined by elemental analysis. For HPLC, the peak area of the major product was ≥ 95% of the combined total peak areas when monitored by a UV detector at 254 nm.
Methyl 2-[(6-Methyl-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]-2-oxoacetate (9)
3-Amino-6-methyl-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione10 (3, 100 mg, 0.36 mmol) was dissolved in tetrahydrofuran (10 mL) and the reaction mixture cooled to 0 °C. Methyl 2-chloro-2-oxoacetate (4, 0.1 mL, 1.09 mmol) was added dropwise to the indenoisoquinoline solution and the reaction mixture was stirred for 30 min. Triethylamine (0.1 mL) was slowly added and the reaction mixture was stirred for another 30 min, keeping the temperature at 0 °C. The reaction mixture was diluted with water (30 mL) and chloroform (20 mL) was added. The organic phase was separated and washed with water (30 mL) and brine (30 mL). The organic phase was concentrated and the compound purified by silica gel column chromatography, eluting with chloroform-methanol, 50:1. The product was obtained as a brick-red solid (53 mg, 39%): mp 287–289 °C. IR (Film) 3336, 1642, 1514, 1480, 1406, 1358, 1333, 1231, 1045, 815, 759 cm−1; 1H NMR (300 MHz, CDCl3) δ 11.08 (s, 1 H), 8.67 (d, J = 1.9 Hz, 1 H), 8.40 (d, J = 8.7 Hz, 1 H), 8.06 (dd, J = 8.8 Hz, J = 2.1 Hz, 1 H), 7.82 (d, J = 7.2 Hz, 1 H), 7.50-7.43 (m, 3 H), 3.93 (s, 3 H), 3.87 (s, 3 H); ESIMS m/z (rel intensity) 363 (MH+, 100); HRESIMS calcd for C20H14N2O5 363.0910 (MH+), found 363.0907 (MH+); HPLC purity: 98.3% (MeOH-H2O, 90:10), 95.2% (MeOH-H2O, 85:15).
Methyl 3-[(6-Methyl-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]-3-oxopropanoate (10)
3-Amino-6-methyl-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (3, 150 mg, 0.54 mmol) was dissolved in tetrahydrofuran (20 mL) and the solution was cooled to 0 °C. Methyl malonyl chloride (5, 0.1 mL, 0.93 mmol) was added and the reaction mixture was stirred for 5 min at 0 °C. Triethylamine (0.2 mL) was added and the reaction mixture was stirred for 2 h at 0 °C. The solvent was removed under vacuum and the residue was purified by silica gel column chromatography, eluting with chloroform-methanol, 95:5, and then chloroform-ethyl acetate-methanol, 45:45:10. The fractions were combined, the solvent was removed under vacuum and the solid was washed with hexane-dichloromethane, 1:1 (25 mL). The product was obtained as a reddish-brown solid (60 mg, 29%): mp 230 °C (dec). IR (Film) 3309, 3071, 2952, 1744, 1692, 1661, 1574, 1531, 1317, 1276, 1054, 1017, 901 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 10.46 (s, 1 H), 8.42 (d, J = 1.8 Hz, 1 H), 8.31 (d, J = 8.8 Hz, 1 H), 7.79 (dd, J = 8.8 Hz, J = 2.0 Hz, 1 H), 7.72 (d, J = 7.4 Hz, 1 H) 7.45-7.37 (m, 3 H), 3.85 (s, 3 H), 3.66 (s, 3 H), 3.49 (s, 2 H); 13C NMR (125 MHz, DMSO-d6) δ 190.3, 168.4, 164.6, 162.5, 156.0, 137.9, 137.7, 134.6, 133.9, 131.3, 127.8, 125.8, 124.3, 123.7, 122.8, 117.2, 107.0, 52.4, 43.8, 32.5; ESIMS m/z (rel intensity) 775 (2MNa+, 100), 752 (2MH+, 37), 377 (MH+, 10); HRESIMS calcd for C21H16N2O5 377.1137 (MH+), found 377.1140 (MH+); HPLC purity: 95.0% (75:25), 95.0% (MeOH-H2O, 70:30).
General Procedure for the Synthesis of Indenoisoquinolines 11–13
3-Amino-6-methyl-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (3, 160 mg, 0.58 mmol) was dissolved in tetrahydrofuran (15 mL) and the reaction mixture cooled to 0 °C. The desired acyl chloride (6–8, 100–125 μL, 0.81 mmol) was added dropwise to the indenoisoquinoline solution and the reaction mixture was stirred for 10 min. Triethylamine (0.4 mL) was slowly added and the reaction mixture was stirred for 1 h, keeping the temperature at 0 °C. Chloroform (40 mL) was added to the reaction mixture and the organic solution was washed with water (3 × 40 mL) and brine (1 × 50 mL). The organic phase was concentrated and the compounds purified by silica gel column chromatography.
Methyl 4-[(6-Methyl-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]-4-oxobutanoate (11)
The compound was eluted with chloroform-methanol, 20:1. The product was obtained as an orange solid (101 mg, 44%): mp 250–252 °C. IR (Film) 3339, 3314, 2951, 1735, 1720, 1688, 1658, 1643, 1573, 1518, 1432, 1315, 1195, 1157, 1055, 891, 845, 763, 700 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 10.37 (s, 1 H), 8.49 (d, J = 1.9 Hz, 1 H), 8.35 (d, J = 8.7 Hz, 1 H), 7.87 (dd, J = 8.7 Hz, J = 2.1 Hz, 1 H), 7.78 (d, J = 7.5 Hz, 1 H) 7.49-7.37 (m, 3 H), 3.89 (s, 3 H), 3.57 (s, 3 H), 2.61 (m, 4 H); 13C NMR (125 MHz, DMSO-d6) δ 190.9, 173.8, 171.0, 163.1, 156.3, 139.0, 138.3, 135.1, 134.5, 131.8, 127.9, 126.3, 124.8, 124.3, 124.1, 123.3, 117.4, 107.6, 52.3, 33.0, 31.8, 29.3; ESIMS m/z (rel intensity) 413 (MNa+, 100); HRESIMS calcd for C22H18N2O5 413.1113 (MNa+), found 413.1119 (MNa+); HPLC purity: 98.7% (MeOH-H2O, 85:15), 98.8% (MeOH-H2O, 95:5).
Methyl 5-[(6-Methyl-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]-5-oxopentanoate (12)
The compound was eluted with chloroform-methanol, 30:1. The product was obtained as a dark red solid (225 mg, 96%): mp 184–186 °C. IR (Film) 3335, 3119, 2945, 1730, 1689, 1650, 1580, 1525, 1431, 1316, 1274, 1194, 901, 844, 757, 718 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 10.35 (s, 1 H), 8.49 (d, J = 1.9 Hz, 1 H), 8.35 (d, J = 8.7 Hz, 1 H), 7.85 (dd, J = 8.7 Hz, J = 2.1 Hz, 1 H), 7.76 (d, J = 7.5 Hz, 1 H) 7.45-7.32 (m, 3 H), 3.88 (s, 3 H), 3.56 (s, 3 H), 2.46 (m, 4 H), 1.81 (m, 2 H); 13C NMR (125 MHz, DMSO-d6) δ 190.3, 173.4, 171.2, 162.5, 155.6, 138.3, 137.7, 134.5, 133.8, 131.2, 127.3, 125.8, 124.2, 123.7, 123.5, 122.7, 116.9, 107.0, 51.6, 35.6, 32.9, 32.4, 20.6.; ESIMS m/z (rel intensity) 405 (MH+, 100); HRESIMS calcd for C23H20N2O5 405.1450 (MH+), found 405.1446 (MH+); HPLC purity: 96.0% (MeOH-H2O, 80:20).
Methyl 6-[(6-Methyl-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]-6-oxohexanoate (13)
The compound was eluted with chloroform-methanol, 30:1. The product was obtained as a red solid (221 mg, 91.2%): mp 177–179 °C. IR (Film) 3341, 3071, 2950, 2868, 1732, 1692, 1659, 1573, 1522, 1512, 1434, 1316, 1276, 1196, 1055, 902, 842, 762, 750 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 10.20 (s, 1 H), 8.49 (d, J = 2.0 Hz, 1 H), 8.36 (d, J = 8.8 Hz, 1 H), 7.88 (dd, J = 8.7 Hz, J = 2.0 Hz, 1 H), 7.79 (d, J = 7.5 Hz, 1 H) 7.51-7.36 (m, 3 H), 3.91 (s, 3 H), 3.55 (s, 3 H), 2.31 (m, 4 H), 1.56 (m, 4 H); 13C NMR (125 MHz, DMSO-d6) δ 190.3, 173.6, 171.6, 162.5, 155.8, 138.5, 137.7, 134.6, 133.9, 131.2, 127.3, 125.9, 124.2, 123.7, 123.5, 122.8, 117.0, 107.1, 51.6, 36.4, 33.4, 32.5, 24.8, 24.4; ESIMS (rel intensity) m/z 419 (MH+, 100); HRESIMS calcd for C24H22N2O5 419.1607 (MH+), found 419.1613 (MH+); HPLC purity: 97.0% (MeOH-H2O, 85:15), 96.9% (MeOH-H2O, 90:10).
(6-Methyl-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)carbamic Acid (14)
3-Amino-6-methyl-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (3, 201 mg, 0.72 mmol) was dissolved in tetrahydrofuran (30 mL) and cooled to 0 °C. Methyl oxalyl chloride (4, 0.1 mL, 1.16 mmol) and triethylamine (0.2 mL) were added dropwise and the reaction mixture was stirred for 2 h at 0 °C. The reaction mixture was diluted with water (100 mL) and extracted with chloroform (4 × 50 mL). The solvent was removed under vacuum and the compound passed through a short silica gel column chromatography, eluting with chloroform-methanol, 9:1. The impure solid, compound 9, was dissolved in a solution of sodium hydroxide (0.1 g, 2.5 mmol) in ethanol (50 mL) and water (1 mL) and the reaction mixture was stirred overnight at room temperature. The solvent was removed under vacuum and the residue purified by silica gel column chromatography, eluting with chloroform-methanol-acetic acid, 90:9:1. Compound 14 was obtained as a reddish-brown solid (103 mg, 44.7%, after 2 steps): mp 360 °C (dec). IR (Film) 3325, 2928, 1692, 1657, 1600, 1580, 1533, 1435, 1319, 1197, 903, 701 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 10.9 (s, 1 H), 8.70 (s, 1 H), 8.35 (d, J = 8.2 Hz, 1 H), 8.02 (s, 1 H), 7.90 (br s, 1 H), 7.45 (m, 3 H), 3.89 (s, 3 H); 13C NMR (125 MHz, DMSO-d6) δ 190.1, 162.4, 162.2, 157.9, 156.3, 137.5, 136.9, 134.6, 134.0, 131.4, 128.4, 126.9, 124.4, 123.5, 123.4, 122.8, 118.5, 106.9; ESIMS m/z (rel intensity) 347 [(M – H+)–]; negative ion; HRESIMS calcd for C19H12N2O5 347.0668 [(M – H+)–], found 347.0664 [(M – H+)–]; HPLC purity: 95.2% (MeOH-H2O, 85:15), 95.7% (MeOH-H2O, 75:25).
3-[(6-Methyl-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]-3-oxopropanoic Acid (15)
Methyl 3-[(6-methyl-5,11-dioxo-6,11-dihydro-5H-indeno[1,2- c]isoquinolin-3-yl)amino]-3-oxopropanoate (10, 158 mg, 0.42 mmol) was dissolved in methanol (20 mL) and dimethylformamide (2 mL). A solution of sodium hydroxide (103 mg) in water (2 mL) was added dropwise and the reaction mixture was stirred at room temperature for 24 h. Concentrated hydrochloric acid (1 mL) was added and the solution diluted with chloroform (30 mL). A dark-red precipitated was formed between the two phases. The solid was filtered and the organic and water layers discarded. The solid was washed with water (10 mL) and ether (2 × 10 mL). The solid was dried and compound 15 was obtained as a reddish-brown solid (93 mg, 61%): mp 262 °C (dec). IR (KBr) 3447, 3323, 1731, 1695, 1573, 1529, 1433, 1317, 1195, 1054, 899, 763 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 10.5 (s, 1 H), 8.45 (s, 1 H), 8.31 (d, J = 8.6 Hz, 1 H), 7.80 (d, J = 7.0 Hz, 1 H), 7.46-7.38 (m, 3 H), 3.86 (s, 3 H), 3.39 (s, 2 H); 13C NMR (125 MHz, DMSO-d6) δ 190.1, 169.5, 165.1, 162.4, 155.7, 138.1, 137.5, 134.5, 133.8, 131.2, 127.6, 125.7, 124.1, 123.6, 122.7, 117.0, 116.9, 106.9, 44.4, 32.4; MALDIMS m/z (rel intensity) 363 (MH+, 100); ESIMS (m/z, relative intensity) 361 [(M – H+)–], 100); HRESIMS calcd for C20H14N2O5 361.0824 [(M – H+)–], found 361.0828 [(M – H+)–]; negative ion; HPLC purity: 99.4% (MeOH, 100), 98.6% (MeOH-H2O, 95:5).
Methyl 2-[(6-(3-((tert-Butoxycarbonyl)amino)propyl)-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]-2-oxoacetate (17)
Compound 1618 (0.3 g, 0.715 mmol) was dissolved in chloroform (100 mL). Triethylamine (0.261 g, 2.14 mmol) was added at room temperature followed by methyl oxaloyl chloride (4, 0.113 g, 0.930 mmol) and the reaction mixture was stirred for 2 h. The reaction mixture was washed with water (2 × 20 mL) and dried over anhydrous sodium sulfate. The solvent was removed under vacuum and the residue purified by silica gel column chromatography, eluting with chloroform-methanol, 9.4:0.4, to furnish the product 17 (0.230 g, 65%) as an orange solid: mp 157–159 °C. IR (KBr) 3339, 2976, 1702, 1662, 1580, 1570, 1534, 1514, 1163, 759, 665 cm−1; 1H NMR (CDCl3, 300 MHz) δ 9.02 (s, 1 H), 8.73 (d, J = 8.7 Hz, 1 H), 8.32 (s, 1 H), 8.10 (dd, J = 1.8, J = 5.6 Hz, 1 H), 7.68 (m, 1 H), 7.48 (m, 1 H), 7.30 (m, 2 H), 5.30 (m, 1 H), 4.58 (t, J = 4.8 Hz, 2 H), 3.99 (s, 3 H), 3.26 (m, 2 H), 2.10 (m, 2 H), 1.45 (s, 9 H); 13C NMR (CDCl3 + CD3OD, 75 MHz) δ 163.2, 160.6, 156.5, 154.6, 136.5, 135.6, 134.5, 133.4, 130.9, 129.2, 126.4, 124.3, 123.5, 123.1, 122.5, 118.3, 108.4, 79.3, 53.6, 42.2, 37.2, 29.5, 28.0; ESIMS m/z (relative intensity) 528 (MNa+, 62); HRESIMS calcd for C27H27N3O7Na 528.1747 (MNa+), found 528.1740 (MNa+).
Methyl 3-[(6-(3-((tert-Butoxycarbonyl)amino)propyl)-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]-3-oxopropanoate (18)
Compound 16 (0.100 g, 0.234 mmol) was dissolved in chloroform (75 mL), triethylamine (0.060 g, 0.591 mmol) was added followed by methyl 3-chloro-3-oxopropionate (5, 0.055 g, 0.290 mmol) at room temperature, and the reaction mixture was stirred for 2 h. The reaction mixture was washed with water (2 × 20 mL), extracted with chloroform (2 × 75 mL), and dried over anhydrous sodium sulfate. The solvent was removed under vacuum and the residue purified by silica gel column chromatography, eluting with chloroform-methanol, 9.3:0.7, to furnish the product 18 (0.075 g, 65%) as a red solid: mp 144–145 °C. IR (KBr) 3325, 2976, 1744, 1696, 1664, 1572, 1532, 1512, 1433, 1168, 761, 665 cm−1; 1H NMR (CDCl3, 300 MHz) δ 9.47 (s, 1 H), 8.65 (d, J = 8.8 Hz, 1 H), 8.35 (s, 1 H), 8.06 (m, 1 H), 7.59 (m, 1 H), 7.50 (m, 1 H), 7.41 (m, 2 H), 5.40 (br s, 1 H), 4.60 (t, J = 6.6 Hz, 2 H), 3.82 (s, 3 H), 3.53 (s, 2 H), 3.50 (m, 2 H), 2.08 (m, 2 H), 1.45 (s, 9 H); 13C NMR (CDCl3, 300 MHz) δ 190.0, 169.8, 163.3, 163.0, 156.1, 154.1, 136.7, 136.5, 134.6, 133.2, 130.6, 128.5, 126.5, 124.2, 123.6, 123.0, 122.3, 118.2, 108.3, 79.2, 52.6, 42.0, 41.7, 37.3, 29.9, 28.4; ESIMS m/z (relative intensity) 542 (MNa+, 100); HRESIMS calcd for C28H29N3O7Na 542.1903 (MNa+), found 542.1906 (MNa+); 95.7% (MeOH-H2O, 85:15).
Methyl 4-[(6-(3-((tert-Butoxycarbonyl)amino)propyl)-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]-4-oxobutanoate (19)
Compound 16 (0.150 g, 0.357 mmol) was dissolved in chloroform (100 mL), triethylamine (0.090 g, 0.892 mmol) was added followed by methyl 4-chloro-4-oxobutanoate (6, 0.082 g, 0.536 mmol) at room temperature, and the reaction mixture was stirred for 2 h. The reaction mixture was washed with water (2 × 20 mL), extracted with chloroform (2 × 75 mL), and dried over anhydrous sodium sulfate. The solvent was removed under vacuum and the residue purified by silica gel column chromatography, eluting with chloroform-methanol, 9.5:0.5, to furnish the product 19 (0.114 g, 60%) as an orange solid: mp 176–178 °C. IR (KBr) 3307, 2917, 1728, 1703, 1718, 1695, 1575, 1165, 666 cm−1; 1H NMR (CDCl3, 300 MHz) δ 8.68 (d, J = 5.3 Hz, 1 H), 8.21 (br s, 1 H), 8.02 (m, 2 H), 7.68 (d, J = 4.8 Hz, 1 H), 7.32 (m, 4 H), 5.41 (br s, 1 H), 4.57 (t, J = 3.2 Hz, 2 H), 3.72 (s, 3 H), 3.20 (m, 2 H), 2.79 (m, 4 H), 2.08 (m, 2 H), 1.45 (s, 9 H); 13C NMR (CDCl3 + DMSO-d6, 125 MHz) δ 190.0, 173.0, 170.0, 162.9, 155.8, 153.6, 137.9, 136.7, 134.5, 133.0, 130.3, 127.4, 126.0, 123.7, 123.4, 122.6, 122.0, 117.2, 108.3, 78.7, 51.4, 41.6, 36.9, 31.1, 29.6, 28.6, 28.1; ESIMS m/z (relative intensity) 556 (MNa+, 12); HRESIMS calcd for C29H31N3O7Na 556.2060, found 556.2069; HPLC purity: 95.0% (MeOH-H2O, 85:15).
Methyl 5-[(6-(3-((tert-Butoxycarbonyl)amino)propyl)-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]-5-oxopentanoate (20)
Compound 16 (0.150 g, 0.357 mmol) was dissolved in chloroform (100 mL), triethylamine (0.108 g, 1.07 mmol) was added followed by methyl 5-chloro-5-oxopentanoate (7, 0.088 g, 0.536 mmol) at room temperature, and the reaction mixture was stirred for 2 h. The reaction mixture was washed with water (2 × 25 mL), extracted with chloroform (2 × 60 mL), and dried over anhydrous sodium sulfate. The solvent was removed under vacuum and the residue purified by silica gel column chromatography, eluting with chloroform-methanol, 9.6:0.4, to afford the product 20 (0.1 g, 50%) as a red solid: mp 118–120 °C. IR (KBr) 3329, 1696, 1679, 1663, 1596, 1528, 1169, 760 cm−1; 1H NMR (CDCl3, 300 MHz) δ 8.52 (d, J = 5.3 Hz, 1 H), 8.21 (br s, 1 H), 8.01 (br s, 1 H), 7.48 (d, J = 4.7 Hz, 1 H), 7.40 (m, 2 H), 7.26 (m, 1 H), 5.48 (t, J = 4.5 Hz, 1 H), 4.51 (t, J = 4.5 Hz, 2 H), 3.69 (s, 3 H), 3.20 (m, 2 H), 2.49 (m, 4 H), 2.10 (m, 4 H), 1.45 (s, 9 H); 13C NMR (CDCl3, 125 MHz) δ 190.2, 173.8, 170.8, 163.2, 156.1, 154.0, 137.1, 136.9, 134.7, 133.3, 130.7, 128.3, 126.6, 124.3, 123.7, 123.1, 122.3, 117.9, 108.6, 79.3, 51.7, 41.9, 37.3, 36.1, 32.9, 30.0, 28.4, 20.6; ESIMS m/z (relative intensity) 570 (MNa+, 100); HRESIMS calcd for C30H33N3O7Na 570.2216 (MNa+), found 570.2209 (MNa+); HPLC purity: 96.4% (MeOH-H2O, 85:15).
Methyl 6-[(6-(3-((tert-Butoxycarbonyl)amino)propyl)-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]-6-oxohexanoate (21)
Compound 16 (0.150 g, 0.357 mmol) was dissolved in chloroform (100 mL), triethylamine (0.090 g, 0.892 mmol) was added followed by methyl 6-chloro-6-oxohexanoate (8, 0.095 g, 0.536 mmol) at room temperature, and the reaction mixture was stirred for 2 h. The reaction mixture washed with water (2 × 30 mL), extracted with chloroform (2 × 75 mL), and dried over anhydrous sodium sulfate. The solvent was removed under vacuum and the residue purified by silica gel column chromatography, eluting with chloroform-methanol, 9.7:0.3, to yield the product 21 (0.116 g, 55%) as a red solid: mp 143–145 °C. IR (KBr) 3332, 2974, 1696, 1662, 1580, 1568, 1527, 1169, 760, 666 cm−1; 1H NMR (CDCl3, 300 MHz) δ 8.46 (d, J = 8.7 Hz, 1 H), 8.28 (br s, 1 H), 8.03 (br s, 1 H), 7.93 (d, J = 5.2 Hz, 1 H), 7.52 (d, J = 5.2 Hz, 1 H), 7.31 (m, 2 H), 7.28 (d, J = 6.3 Hz, 1 H), 5.43 (t, J = 5.4 Hz, 1 H), 4.48 (t, J = 6.6 Hz, 2 H), 3.67 (s, 3 H), 3.18 (m, 2 H), 2.38 (m, 4 H), 2.05 (m, 2 H), 1.76 (m, 4 H), 1.45 (s, 9 H); 13C NMR (CDCl3, 125 MHz) δ 190.3, 174.1, 171.1, 163.1, 156.1, 154.1, 137.2, 136.9, 134.8, 133.3, 130.7, 128.3, 126.7, 124.3, 123.7, 123.1, 122.3, 118.0, 108.6, 79.3, 51.6, 41.9, 37.3, 37.0, 33.6, 30.0, 28.4, 24.8, 24.2; ESIMS m/z (relative intensity) 584 (MNa+, 17); HRESIMS calcd for C31H35N3O7Na 584.2373, found 584.2370.
Methyl 2-[(6-(3-Aminopropyl)-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]-2-oxoacetate (22)
Compound 17 (0.070 g, 0.130 mmol) was treated with trifluoroacetic acid (0.5 mL) in chloroform (5 mL) for 2 h at room temperature. The solvent was removed on a rotary evaporator and the residue was then basified with 2 N NH3 in methanol to get the free amine, which was purified by silica gel column chromatography, eluting with chloroform-methanol, 8.7:1.3, to yield the product 22 (0.035 g, 65%) as a brown solid: mp 185–186 °C. IR (KBr) 3014, 1692, 1581, 1570, 1533, 1307, 1198, 722, 455 cm−1; 1H NMR (CD3OD, 300 MHz) δ 8.55 (s, 1 H), 8.23 (d, J = 3.2 Hz, 1 H), 7.71 (d, J = 3.5 Hz, 1 H), 7.54 (m, 1 H), 7.36 (m, 3 H), 4.52 (m, 2 H), 3.91 (s, 3 H), 3.05 (m, 2 H), 2.28 (m, 2 H); 13C NMR (CD3OD, 125 MHz) δ 191.6, 169.9, 166.8, 164.9, 155.4, 138.8, 137.9, 135.7, 134.9, 132.2, 129.6, 127.2, 125.0, 124.8, 124.1, 123.9, 118.6, 109.6, 52.9, 42.5, 38.2, 28.6; ESIMS m/z (relative intensity) 420 (MH+, 100); HRESIMS calcd for C23H21N3O5 420.1559 (MH+), found 420.1554 (MH+); HPLC purity: 98.2% (1% TFA in MeOH).
Methyl 3-[(6-(3-Aminopropyl)-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]-3-oxopropanoate (23)
Compound 18 (0.080 g, 0.145 mmol) was treated with TFA (0.5 mL) in chloroform (5 mL) for 2 h at room temperature. The solvent was removed on a rotary evaporator and the residue was then basified with 2 N NH3 in methanol to get the free amine, which was purified by silica gel column chromatography, eluting with chloroform-methanol, 8.8:1.2, to yield the product 23 (0.040 g, 62%) as a brown solid: mp 225–227 °C. IR (KBr) 3067, 1739, 1673, 1574, 1535, 1511, 1202, 1134, 722 cm−1; 1H NMR (CD3OD, 300 MHz) 8.52 (d, J = 2.1 Hz, 1 H), 8.29 (d, J = 6.8 Hz, 1 H), 7.43 (m, 5 H), 4.50 (t, J = 2.1 Hz, 2 H), 3.78 (s, 3 H), 3.10 (m, 2 H), 2.22 (m, 2 H); 13C NMR (CD3OD, 125 MHz) δ 191.6, 169.9, 166.8, 164.9, 155.4, 138.8, 137.9, 135.7, 134.9, 132.2, 129.6, 127.2, 125.0, 124.8, 124.1, 123.9, 118.6, 109.6, 52.9, 42.5, 38.2, 28.6; ESIMS m/z (relative intensity) 420 (MH+, 100); HRESIMS calcd for C23H21N3O5 420.1559 (MH+), found 420.1554 (MH+); HPLC purity: 100% (1% TFA in MeOH-H2O, 50:50).
Methyl 4-[(6-(3-Aminopropyl)-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]-4-oxobutanoate (24)
Compound 19 (0.060 g, 0.112 mmol) was treated with trifluoroacetic acid (0.5 mL) in chloroform (5 mL) for 2 h at room temperature. The solvent was removed on a rotary evaporator and the residue was then basified with 2 N NH3 in in methanol to get the free amine, which was purified by silica gel column chromatography, eluting with chloroform-methanol, 8.8:1.2, to yield the product 24 (0.028 g, 60%) as a brown solid: mp 272–274 °C. IR (KBr) 2952, 1735, 1690, 1656, 1572, 1532, 1510, 1160, 765, 455 cm−1; 1H NMR (CD3OD, 300 MHz) δ 8.28 (s, 1 H), 7.92 (m, 1 H), 7.31 (m, 3 H), 7.20 (s, 2 H), 4.30 (t, J = 4.8 Hz, 2 H), 3.71 (s, 3 H), 2.83 (m, 2 H), 2.68 (m, 4 H), 2.00 (m, 2 H); 13C NMR (CD3OD, 125 MHz) δ 191.8, 174.9, 172.6, 164.4, 155.6, 139.2, 138.2, 135.9, 134.8, 131.9, 129.3, 127.1, 125.0, 124.9, 124.0, 123.8, 122.9, 118.5, 114.8, 109.5, 57.6, 57.4, 57.2, 43.6, 36.3, 32.2, 30.3, 29.7; ESIMS m/z (relative intensity) 434 (MH+, 100); HRESIMS calcd for C24H23N3O5 434.1852 (MH+), found 434.1835 (MH+); HPLC purity: 98.0% (1% TFA in MeOH).
Methyl 5-[(6-(3-Aminopropyl)-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]-5-oxopentanoate (25)
Compound 20 (0.150 g, 0.274 mmol) was treated with trifluoroacetic acid (1.0 mL) in chloroform (10 mL) for 2 h at room temperature. The solvent was removed on a rotary evaporator and the residue was then basified with 2 N NH3 in methanol to get the free amine, which was purified by silica gel column chromatography, eluting with chloroform-methanol, 9.0:1.0, to afford the product 25 (0.092 g, 75%) as a brown solid: mp 215–217 °C. IR (KBr) 3075, 1729, 1687, 1673, 1572, 1532, 1433, 1202, 760, 722, 455 cm−1; 1H NMR (DMSO-d6, 300 MHz) δ 10.2 (s, 1 H), 8.60 (d, J = 1.8 Hz, 1 H), 8.47 (d, J = 8.7 Hz, 1 H), 7.89 (dd, J = 1.8, J = 8.7 Hz, 1 H), 7.74 (m, 3 H), 7.54 (m, 2 H), 7.47 (m, 1 H), 4.53 (t, J = 3.2 Hz, 2 H), 3.59 (s, 3 H), 2.96 (m, 2 H), 2.38 (m, 4 H), 2.13 (m, 2 H), 1.87 (m, 2 H); 13C NMR (DMSO-d6, 75 MHz) δ 190.1, 173.0, 171.0, 162.6, 154.4, 138.3, 136.6, 134.2, 134.0, 131.0, 127.1, 125.8, 123.5, 123.4, 122.6, 116.6, 107.4, 79.1, 51.3, 41.2, 38.6, 36.6, 35.2, 32.6, 27.3, 20.2; ESIMS m/z (relative intensity) 448 (MH+, 100); HRESIMS calcd for C25H25N3O5 448.1867 (MH+), found 448.1877 (MH+); HPLC purity: 100% (1% TFA in MeOH-H2O, 70:30).
Methyl 6-[(6-(3-Aminopropyl)-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]-6-oxohexanoate (26)
Compound 21 (0.030 g, 0.053 mmol) was treated with trifluoroacetic acid (0.25 mL) in chloroform (3 mL) for 2 h at room temperature. The solvent was removed on a rotary evaporator and the residue was then basified with 2 N NH3 in methanol to get the free amine, which was purified by silica gel column chromatography, eluting with chloroform-methanol, 9.2:0.8, to yield the product 26 (0.015 g, 55%) as a red solid: mp 153–155 °C. IR (KBr) 2947, 1739, 1705, 1693, 1661, 1570, 1530, 1431, 1197, 760, 665 cm−1; 1H NMR (CD3OD, 300 MHz) δ 8.51 (d, J = 2.4 Hz, 1 H), 8.34 (d, J = 9.0 Hz, 1 H), 7.70 (dd, J = 2.4, J = 9.0 Hz, 1 H), 7.60 (t, J = 9.0 Hz, 1 H), 7.51 (m, 2 H), 7.31 (m, 1 H), 4.55 (t, J = 4.5 Hz, 2 H), 3.67 (s, 3 H), 3.13 (m, 2 H), 2.39 (m, 4 H), 2.26 (m, 2 H), 1.70 (m, 2 H); 13C NMR (DMSO-d6, 125 MHz) δ 190.0, 173.3, 171.3, 162.3, 154.4, 138.3, 136.6, 134.2, 133.9, 130.9, 127.0, 125.6, 123.5, 123.4, 123.3, 122.5, 116.5, 107.2, 51.2, 42.1, 38.2, 36.0, 33.0, 30.9, 24.4, 24.0; ESIMS (m/z, relative intensity) 462 (MH+, 100); HRESIMS calcd for C26H27N3O5 462.2023 (MH+), found 462.2031 (MH+); HPLC purity: 98.5% (1% TFA in MeOH-H2O, 70:30).
General Procedure for Synthesis of Indenoisoquinolines 27–31
The esters 17–21 (0.1 g) were dissolved in methanol (10 mL) and tetrahydrofuran (10 mL). An aqueous NaOH solution (4 N, 5 mL) was added at room temperature and the reaction mixture was stirred at room temperature for 6 h. The solvent was removed on a rotary evaporator and the residue washed with 1 N HCl (30 mL), extracted with chloroform (2 × 50 mL), dried over anhydrous sodium sulfate, concentrated and purified by silica gel column chromatography, eluting with chloroformmethanol 9.6:0.4 to 9:1, to afford acids 27–31 in 55–75% yields as orange solids.
2-[(6-(3-((tert-Butoxycarbonyl)amino)propyl)-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]-2-oxoacetic Acid (27)
mp 267–268 °C. IR (KBr) 3295, 1758, 1698, 1646, 1605, 1580, 1535, 1350, 1161, 847, 665 cm−1; 1H NMR (CD3OD + DMSO-d6, 300 MHz) δ 8.87 (s, 1 H), 8.63 (d, J = 8.7 Hz, 1 H), 8.14 (d, J = 9.3 Hz, 1 H), 7.80 (d, J = 7.2 Hz, 1 H), 7.64 (m, 2 H), 7.55 (m, 1 H), 4.60 (t, J = 7.2 Hz, 2 H), 3.21 (m, 2 H), 2.07 (m, 2 H), 1.50 (s, 1 H).
3-[(6-(3-((tert-Butoxycarbonyl)amino)propyl)-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]-3-oxopropanoic Acid (28)
mp 211–212 °C. IR (KBr) 3310, 1768, 1710, 1976, 1522, 1385, 1355, 847, 665 cm−1; 1H NMR (CD3OD, 300 MHz) δ 8.56 (d, J = 2.2 Hz, 1 H), 8.30 (d, J = 5.6 Hz, 1 H), 7.51 (m, 1 H), 7.48 (m, 1 H), 7.44 (m, 1 H), 7.39 (m, 1 H), 7.30 (m, 1 H), 4.52 (t, J = 4.5 Hz, 2 H), 3.52 (s, 2 H), 3.15 (t, J = 5.8 Hz, 2 H), 2.18 (m, 2 H), 1.43 (s, 9 H).
4-[(6-(3-((tert-Butoxycarbonyl)amino)propyl)-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]-4-oxobutanoic Acid (29)
mp 155–157 °C. IR (KBr) 3315, 1763, 1698, 1678, 1543, 1345, 1165, 667 cm−1; 1H NMR (CD3OD, 300 MHz) δ 8.44 (m, 2 H), 7.82 (dd, J = 2.1, J = 5.5 Hz, 1 H), 7.68 (d, J = 5.5 Hz, 1 H), 7.51 (m, 2 H), 7.34 (m, 1 H), 4.45 (m, 2 H), 3.12 (m, 2 H), 2.14 (s, 4 H), 1.96 (m, 2 H), 1.44 (s, 9 H); 13C NMR (CDCl3 + DMSO-d6, 75 MHz) δ 188.4, 172.3, 168.8, 160.8, 154.3, 152.3, 136.8, 135.3, 133.0, 131.8, 129.0, 125.7, 124.0, 122.1, 121.9, 121.2, 120.9, 115.2, 106.2, 40.9, 35.9, 29.7, 29.1, 28.1, 27.3, 26.7; ESIMS (m/z, relative intensity) 542 (MNa+, 100), 420 (loss of Boc, 16); HRESIMS calcd for C28H29N3O7Na 542.1903 (MNa+), found 542.1912 (MNa+).
5-[(6-(3-((tert-Butoxycarbonyl)amino)propyl)-5,11-dioxo-6,11-dihydro-5H-indeno[ 1,2-c]isoquinolin-3-yl)amino]-5-oxopentanoic Acid (30)
mp 198–200 °C. IR (KBr) 3307, 1696, 1690, 1663, 1569, 1528, 1401, 1366, 1250, 1167, 759 cm−1; 1H NMR (CD3OD, 300 MHz) δ 8.52 (d, J = 5.3 Hz, 1 H), 8.21 (br s, 1 H), 8.01 (br, s, 1 H), 7.48 (d, J = 4.7 Hz, 1 H), 7.40 (m, 2 H), 7.26 (m, 1 H), 4.51 (t, J = 4.5 Hz, 2 H), 3.20 (m, 2 H), 2.49 (m, 4 H), 2.10 (m, 4 H), 1.45 (s, 9 H); 13C NMR (CD3OD + DMSO-d6, 75 Hz) δ 191.5, 176.3, 173.2, 164.1, 158.0, 155.6, 139.3, 138.2, 135.9, 134.8, 131.9, 129.0, 127.0, 125.0, 124.8, 124.2, 123.8, 118.4, 109.1, 79.8, 79.7, 36.9, 34.2, 30.9, 29.0, 21.9; ESIMS (m/z, relative intensity) 556 (MNa+, 100); HRESIMS calcd for C29H31N3O7 556.2059 (MNa+), found 556.2063 (MNa+).
6-[(6-(3-((tert-Butoxycarbonyl)amino)propyl)-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]-6-oxohexanoic Acid (31)
mp 206–208 °C. IR (KBr) 3312, 1702, 1695, 1670, 1573, 1534, 1376, 1250, 760, 665 cm−1; 1H NMR (CDCl3 + CD3OD, 300 MHz) δ 8.55 (d, J = 6.2 Hz, 1 H), 8.18 (s, 1 H), 8.01 (m, 1 H), 7.50 (d, J = 3.5 Hz, 1 H), 7.40 (d, J = 3.5 Hz, 1 H), 7.32 (m, 1 H), 7.28 (d, J = 3.5 Hz, 1 H), 4.50 (m, 2 H), 3.12 (m, 2 H), 2.29 (m, 4 H), 1.96 (m, 2 H), 1.65 (m, 4 H), 1.37 (s, 9 H); 13C NMR (DMSO-d6, 75 Hz) δ 190.0, 174.5, 171.5, 162.2, 155.8, 154.5, 138.3, 136.7, 134.3, 133.9, 131.0, 127.1, 125.7, 123.5, 123.3, 123.2, 122.7, 116.6, 107.3, 77.8, 42.5, 37.5, 36.2, 33.5, 29.6, 28.3, 24.6, 24.9; ESIMS (m/z, relative intensity) 570 (MNa+, 23), 448 (loss of Boc, 100); HRESIMS calcd for C30H33N3O7Na 570.2216 (MNa+), found 570.2207 (MNa+).
General Procedure for Synthesis of Indenoisoquinolines 32–36
Acids 27–31 (0.050 g) were treated with HCl in diethyl ether (2 M, 2 mL) at room temperature for 5 h. The solvent was removed on a rotary evaporator to yield the solid hydrochloride salts, which were washed with 5% MeOH in chloroform to remove impurities and afford the pure products 32–36 in quantitative yields.
2-[(6-(3-Aminopropyl)-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]-2-oxoacetic Acid Hydrochloride (32)
mp 251–252 °C. IR (KBr) 2955, 1742, 1733, 1641, 1578, 1535, 1431, 1195, 665 cm−1; 1H NMR (DMSO-d6, 300 MHz) δ 11.04 (s, 1 H), 8.75 (dd, J = 1.7, J = 8.9 Hz, 1 H), 8.49 (dd, J = 4.0, J = 8.8 Hz), 8.11 (m, 2 H), 7.69 (m, 1 H), 7.54 (m, 3 H), 4.54 (m, 2 H), 3.01 (m, 2 H), 2.07 (m, 2 H); ESIMS (m/z, relative intensity) 392 (MH+, 100); HRESIMS calcd for C21H17N3O5 392.1246 (MH+), found 392.1249 (MH+); HPLC purity: 96.6% (1% TFA in MeOH).
3-[(6-(3-Aminopropyl)-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]-3-oxopropanoic Acid Hydrochloride (33)
mp 205–207 °C. IR (KBr) 2917, 2356, 1670, 1582, 1565, 1538, 1191, 665 cm−1; 1H NMR (DMSO-d6, 300 MHz) δ 10.65 (d, J = 8.9 Hz, 1 H), 8.48 (s, 1 H), 8.40 (d, J = 4.5 Hz, 1 H), 7.88 (m, 4 H), 7.71 (d, J = 4.5 Hz, 1 H), 7.56 (m, 2 H), 7.41 (m, 1 H), 4.54 (m, 2 H), 3.64 (s, 1 H), 3.52 (s, 1 H), 3.00 (m, 2 H), 2.09 (m, 2 H); ESIMS (m/z, relative intensity) 406 (MH+, 100); HRESIMS calcd for C22H19N3O5 406.1403 (MH+), found 406.1400 (MH+); HPLC purity: 95.2% (1% TFA in MeOH).
4-[(6-(3-Aminopropyl)-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]-4-oxobutanoic Acid Hydrochloride (34)
mp 167–169 °C. IR (KBr) 3080, 1671, 1571, 1533, 1508, 1197, 734, 665 cm−1; 1H NMR (CD3OD, 300 MHz) δ 8.68 (d, J = 2.3 Hz, 1 H), 8.55 (d, J = 5.2 Hz, 1 H), 7.78 (dd, J = 2.3, J = 5.4 Hz, 1 H), 7.64 (m, 1 H), 7.52 (m, 2 H), 7.37 (m, 1 H), 4.62 (m, 2 H), 3.06 (m, 2 H), 2.30 (m, 2 H); ESIMS (m/z, relative intensity) 420 (MH+, 100); HRESIMS calcd for C23H21N3O5 420.1559 (MH+), found 420.1565 (MH+);: 96.71% (1% TFA in MeOH).
5-[(6-(3-Aminopropyl)-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]-5-oxopentanoic Acid Hydrochloride (35)
mp 242–244 °C. IR (KBr) 3583, 2348, 1761, 1723, 1695, 1510, 1497, 906, 673 cm−1; 1H NMR (DMSO-d6, 300 MHz) δ 10.33 (s, 1 H), 8.63 (d, J = 2.1 Hz, 1 H), 8.49 (d, J = 8.7 Hz, 1 H), 7.89 (dd, J = 2.1, J = 8.7 Hz, 1 H), 7.84 (br s, 2 H), 7.76 (d, J = 7.2 Hz, 1 H), 7.54 (m, 2 H), 7.40 (m, 1 H), 4.53 (m, 2 H), 3.04 (m, 2 H), 2.40 (m, 2 H), 2.04 (m, 4 H), 2.13 (m, 2 H), 1.85 (m, 2 H); ESIMS (m/z, relative intensity) 434 (MH+, 100); HRESIMS calcd for C24H23N3O5 434.1716, found 434.1710; HPLC purity: 95.3% (1% TFA in MeOH).
6-[(6-(3-Aminopropyl)-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]-6-oxohexanoic Acid Hydrochloride (36)
mp 203–205 °C. IR (KBr) 3583, 2952, 1730, 1719, 1646, 1570, 1528, 1431, 1195, 759, 667 cm−1; 1H NMR (DMSO-d6, 300 MHz) δ 10.30 (d, J = 6.6 Hz, 1 H), 8.85 (d, J = 9.9 Hz, 1 H), 8.51 (m, 1 H), 7.91 (m, 1 H), 7.76 (m, 3 H), 7.51 (m, 3 H), 4.50 (m, 2 H), 3.32 (m, 1 H), 3.22 (m, 1 H), 2.39 (m, 2 H), 2.27 (m, 2 H), 2.11 (m, 1 H), 1.91 (m, 1 H), 1.58 (m, 4 H); ESIMS (m/z, relative intensity) 448 (MH+, 100); HRESIMS calcd for C25H26N3O5 448.1872 (MH+), found 448.1868 (MH+); HPLC purity: 96.5% (1% TFA in MeOH).
Ethyl 2-[(6-Methyl-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]acetate (38)
3-Amino-6-methyl-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (3, 400 mg, 1.45 mmol) and ethyl glyoxylate (37, 0.3 mL, 50% solution in toluene) were dissolved in glacial acetic acid (25 mL). The reaction mixture was stirred overnight at room temperature. Sodium cyanoborohydride (1.00 g) was added in portions. Once the production of bubbles stopped, the reaction mixture was heated at reflux for 30 min. Water (150 mL) was added and the compound extracted with chloroform (3 × 50 mL). The organic layers were combined and washed with water (2 × 150 mL), aqueous sodium bicarbonate (100 mL) and brine (100 mL). The solvent was removed under vacuum and the compound purified by silica gel column chromatography, eluting with chloroform-methanol, 50:1. The product was obtained as a dark solid (351 mg, 67%): mp 207 °C (dec). IR (Film) 3380, 2929, 1735, 1694, 1654, 1560, 1434, 1212, 1017 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 8.21 (d, J = 8.7 Hz, 1 H), 7.69 (d, J = 7.5 Hz, 1 H), 7.43-7.38 (m, 2 H), 7.31 (t, J = 7.4 Hz, 1 H), 7.13 (dd, J = 8.8 Hz, J = 2.5 Hz, 1 H), 7.10 (d, J = 2.5 Hz, 1 H), 6.67 (t, J = 6.4 Hz, 1 H), 4.09 (q, J = 7.1 Hz, 2 H), 3.96 (d, J = 6.3 Hz, 2 H), 3.87 (s, 3 H), 1.12 (t, J = 7.1 Hz, 3 H); 13C NMR (125 MHz, CDCl3) δ 190.7, 170.6, 163.0, 152.2, 146.1, 138.3, 134.8, 132.8, 130.0, 125.0, 124.7, 123.7, 122.9, 121.9, 121.2, 109.0, 107.0, 61.4, 45.3, 32.2, 14.1; ESIMS m/z (rel intensity) 363 (MH+, 100); HRESIMS calcd for C21H18N2O4 363.1345 (MH+), found 363.1349 (MH+);: 99.42% (MeOH-H2O-TFA, 95:5:1), 95.33% (MeOH-TFA, 100:1).
Ethyl 2-[(6-(3-((tert-Butoxycarbonyl)amino)propyl)-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl]amino)acetate (39)
Compound 16 (0.200 g, 0.477 mmol) was dissolved in acetic acid (20 mL) and methanol (1 mL). Ethyl glyoxalate (37, 0.058 g, 0.577 mmol) was added to the reaction mixture, which was stirred at room temperature for 1.5 h. Sodium cyanoborohydride (0.071 g, 1.19 mmol) was added and stirring was continued for another 0.5 h. The solvents were removed on a rotary evaporator. The residue was washed with sodium bicarbonate (2 × 15 mL), extracted with CHCl3 (2 × 60 mL), the combined organic layers were dried over anhydrous sodium bicarbonate, and the mixture was concentrated to get a crude product. The crude product was precipitated from hexane-chloroform (7 + 3 mL) to afford a pure violet solid 39 (0.215 g, 90%): mp 203–204 °C. IR (KBr) 2977, 1739, 1698, 1650, 1619, 1579, 1523, 1390, 1365, 1198, 1171, 1020, 758, 455 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.51 (d, J = 8.7 Hz, 1 H), 7.53 (d, J = 6.4 Hz), 7.39 (m, 4 H), 7.09 (dd, J = 2.6, J = 8.7 Hz, 1 H), 5.44 (m, 1 H), 4.66 (m, 1 H), 4.56 (t, J = 4.5 Hz, 2 H), 4.30 (q, J = 7.1 Hz, 2 H), 4.01 (d, J = 5.1 Hz, 2 H), 3.23 (m, 2 H), 2.06 (m, 2 H), 1.45 (s, 9 H), 1.31 (t, J = 7.1 Hz, 3 H); 13C NMR (CDCl3, 125 MHz) δ 190.9, 170.6, 163.6, 156.1, 151.4, 146.3, 137.6, 134.9, 133.3, 130.1, 125.0, 124.9, 123.9, 123.1, 122.3, 121.7, 109.6, 107.2, 79.2, 61.6, 45.3, 41.7, 28.4, 14.2; ESIMS m/z (rel intensity) 528 (MNa+, 100); HRESIMS calcd for C28H31N3O6Na 528.2111 (MNa+), found 528.2108 (MNa+).
Ethyl 2-[(6-(3-Aminopropyl)-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]acetate Hydrochloride (40)
Ester 39 (0.060 g, 0.011 mmol) was treated with 2 M HCl in diethyl ether (2 mL) at room temperature for 2 h. The solvent was removed on a rotary evaporator to yield the solid hydrochloride salt, which was washed with 5% MeOH in chloroform to remove impurities and afford the pure solid product 40 in quantitative yield: mp 253–254 °C. IR (KBr) 3356, 2972, 1711, 1689, 1634, 1598, 1544, 1354, 1129, 665 cm−1 1H NMR (DMSO-d6, 300 MHz) δ 8.31 (d, J = 8.7 Hz, 1 H), 8.04 (br s, 3 H), 7.64 (m, 1 H), 7.47 (m, 2 H), 7.40 (m, 1 H), 7.21 (dd, J = 2.2, J = 8.7 Hz, 1 H), 7.14 (d, J = 2.2 Hz, 1 H), 4.49 (m, 2 H), 4.13 (q, J = 7.1 Hz, 2 H), 4.00 (s, 2 H), 2.92 (m, 2 H), 2.09 (m, 2 H), 1.20 (t, J = 5.9 Hz, 3 H); 13C NMR (DMSO-d6, 300 MHz) δ 190.7, 190.2, 170.8, 162.8, 160.6, 156.0, 155.5, 137.3, 136.7, 134.3, 130.7, 124.2, 123.1, 118.8, 87.0, 60.4, 56.3, 36.8, 27.6, 18.8, 14.3; ESIMS (m/z, relative intensity) 406 (MH+, 100); HRESIMS calcd for C23H23N3O4 406.1767 (MH+), found 406.1772 (MH+)7: 95.66% (1% TFA in MeOH).
2-[(6-Methyl-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino]acetic Acid (41)
Ethyl 2-((6-methyl-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino)acetate (38, 213 mg, 0.59 mmol) was dissolved in methanol (5 mL) and tetrahydrofuran (5 mL). A solution of potassium hydroxide (112 mg, 2.00 mmol) in water (5 mL) was added to the reaction mixture, which was stirred at room temperature for 10 h. Concentrated hydrochloric acid (1 mL) was added to the reaction mixture and the solvent removed under vacuum. Water (20 mL) was added, and the mixture was sonicated and filtered. The solid was washed with water (20 mL). The residue was then dissolved in a mixture of dimethylformamide (0.5 mL) and methanol (9.5 mL), heated and allowed to reach room temperature. Ethyl ether (25 mL) and hexane (10 mL) were added and the reaction mixture placed inside the refrigerator overnight. The solvent was filtered off and the residue dried. The product was obtained as a black solid (59 mg, 30.0%): mp 205 °C (dec). IR (KBr) 3361, 3032, 1725, 1698, 1650, 1619, 1578, 1527, 1432, 1391, 1318, 1217, 1197, 1054, 1016, 901, 839, 758 cm−1; 1H NMR (300 MHz, CDCl3) δ 12.63 (br s, 1 H), 8.21 (d, J = 8.7 Hz, 1 H), 7.68 (d, J = 7.5 Hz, 1 H), 7.43-7.38 (m, 2 H), 7.30 (t, J = 7.4 Hz, 1 H), 7.14 (dd, J = 8.8 Hz, J = 2.5 Hz, 1 H), 7.09 (d, J = 2.5 Hz, 1 H), 6.56 (br s, 1 H), 3.87 (s, 3 H), 3.86 (s, 2 H); 13C NMR (125 MHz, CDCl3) δ 190.7, 172.5, 162.4, 152.7, 147.9, 138.3, 134.6, 133.8, 130.5, 124.8, 123.8, 123.4, 122.6, 122.1, 121.8, 107.9, 106.5, 44.7, 32.3; negative ion ESIMS m/z (rel intensity) 333 [M-H+]−, 100); HRESIMS calcd for C19H14N2O4Na 357.0851 (MNa+), found 357.0857 (MNa+); Anal. calcd for C19H14N2O4: C, 68.26; H, 4.22; N, 8.38. Found: C, 68.03; H, 4.16; N, 8.35.
2-((6-(3-((tert-Butoxycarbonyl)amino)propyl)-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino)acetic Acid (42)
The ester 39 (0.1 g, 0.02 mmol) was dissolved in MeOH-THF (10 +10 mL) and aq NaOH (4 N, 5 mL) was added at room temperature. The reaction mixture was stirred at room temperature for 4 h. The solvent was removed on a rotary evaporator and the residue washed with 1 N HCl (30 mL) and extracted with CHCl3 (2 × 50 mL). The extract was dried over Na2SO4 and concentrated to afford pure 42 (0.075 g, 80%) which was obtained as a brown solid: mp 190–192 °C. IR (KBr) 3350, 2973, 1692, 1645, 1619, 1578, 1520, 1364, 1164, 666 cm−1; 1H NMR (DMSO-d6, 300 MHz) δ 8.30 (d, J = 8.7 Hz, 1 H), 7.59 (m, 1 H), 7.45 (m, 2 H), 7.39 (m, 1 H), 7.21 (dd, J = 2.4, J = 8.7 Hz, 1 H), 7.12 (d, J = 2.4 Hz, 1 H), 7.07 (m, 1 H), 4.40 (m, 2 H), 3.90 (s, 2 H), 3.09 (m, 2 H), 1.86 (m, 2 H), 1.33 (s, 9 H); ESIMS (m/z, relative intensity) 500 (MNa+, 100); HRESIMS calcd for C26H27N3O6Na 500.1798 (MNa+), found 500.1806 (MNa+); HPLC purity: 95.5% (MeOH-H2O, 75:25).
2-((6-(3-Aminopropyl)-5,11-dioxo-6,11-dihydro-5H-indeno[1,2-c]isoquinolin-3-yl)amino)acetic Acid Hydrochloride (43)
Acid 42 (0.070 g, 0.014 mmol) was treated with HCl in diethyl ether (2 M, 2 mL) at room temperature for 1 h. The solvent was removed on a rotary evaporator to provide the solid hydrochloride salt, which was washed with 10% MeOH in chloroform to remove impurities and afford the pure product 43 in quantitative yield: mp 263–264 °C. IR (KBr) 3376, 2982, 1723, 1695, 1634, 1598, 1566, 1354, 1188, 667 cm−1; 1H NMR (DMSO-d6, 300 MHz) δ 8.34 (d, J = 7.8 Hz, 1 H), 8.28 (s, 1 H), 8.12 (br s, 3 H), 7.64 (m, 1 H), 7.49 (m, 2 H), 7.39 (m, 1 H), 7.19 (dd, J = 2.2, J = 8.7 Hz, 1 H), 7.12 (d, J = 2.2 Hz, 1 H), 4.50 (t, J = 6.3 Hz, 2 H), 3.90 (s, 2 H), 2.90 (m, 2 H), 2.10 (m, 2 H); 13C NMR (DMSO-d6, 125 MHz) δ 190.2, 163.1, 162.3, 136.7, 134.1, 130.7, 125.1, 124.2, 124.1, 123.2, 122.6, 107.8, 79.2, 41.2, 36.5, 27.2; ESIMS (m/z, relative intensity) 378 (MH+, 100); HRESIMS calcd for C21H19N3O4 378.1454 (MH+), found 378.1458 (MH+); HPLC purity: 95.3% (1% TFA in MeOH-H2O, 50:50).
cis-2-(3-Bromopropyl)-3-(3-methoxyphenyl)-7-nitro-1-oxo-1,2,3,4- tetrahydroisoquinoline-4-carboxylic acid (48)
The Schiff base 46 (0,618 g, 2.41 mmol) was diluted in chloroform (50 mL) at 0 °C and 4-nitrohomophthalic anhydride (0.5 g, 2.41 mmol) was added. The red mixture was stirred at 0 °C for 1 h and then at room temperature for 3 h. The creamy orange mixture was filtered and the residue was washed with CHCl3 to provide the product as a pale yellow solid (0.91 g, 82%): mp 122–123 °C. IR (film) 3419, 3010, 1714, 1651, 1530, 1489, 1346, 1266, 1163, 758 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.60 (d, J = 2.4 Hz, 1 H), 8.25 (dd, J = 2.4, J = 8.3 Hz, 1 H), 7.47 (d, J = 8.3 Hz, 1 H), 7.16 (m, 1 H), 6.78 (m, 1 H), 6.64 (s, 1 H), 6.59 (d, J = 7.7 Hz, 1 H), 5.10 (d, J = 2.4 Hz, 1 H), 4.08 (m, 1 H), 3.65 (s, 3 H), 3.33 (m, 2 H), 2.92 (m, 2 H), 2.10 (m, 2 H).
6-(3-Bromopropyl)-5,6-dihydro-8-methoxy-3-nitro-5,11-dioxo-11H-indeno[1,2- c]isoquinoline (49)
Thionyl chloride (10 mL) was slowly added to a solution of cis-4-carboxy-N-(3-chloropropyl)-3,4-dihydro-3-(4-methoxyphenyl)-7-nitro-1(2H)isoquinolone (48, 1.94 g, 4.18 mmol) in benzene (60 mL). The reaction mixture was heated at reflux for 30 min, allowed to cool to room temperature, and concentrated. The residue was diluted with nitrobenzene (80 mL), chilled in an ice bath, and aluminum chloride (3.00 g, 22.5 mmol) was added. The reaction mixture was removed from the bath and heated at 100 °C for 1 h. Water (200 mL) was added, and the solution was extracted with CHCl3 (3 × 70 mL). The combined organic layers were washed with sodium bicarbonate (3 × 75 mL) and brine (75 mL), and dried over sodium sulfate. The solution was concentrated, hexanes (250 mL) were added, and the liquid was decanted. The solid was washed with hexanes (100 mL) and the liquid was decanted again. The solid was purified by silica gel column chromatography, eluting with chloroform-methanol, 20:1, to provide a red-orange solid (1.02 g, 55%): mp 282–284 °C (dec). IR (Film) 1699, 1667, 1612, 1555, 1499, 1332, 1159, 1078, 840, 785, 688, 662 cm−1; 1H NMR (CDCl3, 300 MHz) δ 8.85 (d, J = 2.5 Hz, 1 H), 8.70 (d, J = 8.9 Hz, 1 H), 8.55 (d, J = 8.9 Hz, 1 H), 7.60 (d, J = 8.5 Hz, 1 H), 7.38 (s, 1 H), 7.06 (d, J = 8.3 Hz, 1 H), 4.60 (t, J = 7.3 Hz, 2 H), 3.91 (s, 3 H), 3.75 (t, J = 6.4 Hz, 2 H), 2.36 (t, J = 7.0 Hz, 2 H); EIMS m/z (rel intensity) 444 (M+, 100), 442 (M+, 100), 363 [(M – Br)+, 69]; HRESIMS calcd for C20H15BrN2O5 442.0164 (MH+), found 442.0158 (MH+); Anal. Calcd for C20H15BrN2O5·0.8 H2O: C, 52.49; H, 3.66; N, 6.12; Br, 17.46. Found: C, 52.20; H, 3.54; N, 5.96; Br, 17.46.
6-(3-Azidopropyl)-5,6-dihydro-8-methoxy-3-nitro-5,11-dioxo-11H-indeno[1,2- c]isoquinoline (50)
Sodium azide (0.59 g, 9.07 mmol) and 6-(3-bromopropyl)-5,6-dihydro-8-methoxy-3-nitro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (49; 261 mg, 0.59 mmol) were diluted with DMSO (30 mL), and the mixture was heated at 90 °C for 12 h. The reaction mixture was diluted with chloroform (100 mL), washed with water (100 mL) and sat aq NaCl (30 mL), and dried over sodium sulfate. The solution was concentrated to provide a crude solid that was purified by silica gel column chromatography, eluting with chloroform-methanol, 50:1, to afford an orange solid (92 mg, 40%): mp 310 °C (dec). IR (KBr) 3060, 3015, 2979, 2090, 1691, 1668, 1614, 1560, 1504, 1371, 1344, 1258, 1232, 1082, 1022, 911, 876, 843, 775 cm−1; 1H NMR (CDCl3, 300 MHz) δ 8.88 (d, J = 2.5 Hz, 1 H), 8. 67 (d, J = 8.9 Hz, 1 H), 8.50 (dd, J = 8.9 Hz, J = 2.5 Hz, 1 H), 7.59 (d, J = 8.4 Hz, 1 H), 7.38 (d, J = 2.0 Hz, 1 H), 7.04 (dd, J = 8.1 Hz, J = 1.9 Hz, 1 H), 4.57 (t, J = 7.2 Hz, 2 H), 3.91 (s, 3 H), 3.59 (t, J = 6.4 Hz, 2 H), 2.07 (t, J = 7.0 Hz, 2 H); 13C NMR (125 MHz, DMSO-d6) δ 188.6, 164.6, 162.3, 157.7, 145.7, 138.5, 136.7, 127.9, 127.3, 125.2, 124.5, 124.3, 123.2, 114.7, 113.7, 108.0, 56.6, 49.0, 42.8, 28.3; ESIMS m/z (rel intensity) 405 (MH+, 41), 322 [(M – C3H7N3)+, 100]; HRESIMS m/z calcd for C20H15N5O5 405.1073 (MH+), found, 405.1069 (MH+); HPLC purity: 95.0% (MeOH, 100), 95.2% (MeOH-H2O, 85:15).
6-(3-Aminopropyl)-5,6-dihydro-8-methoxy-3-nitro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline Hydrochloride (51)
Triethylphosphite (1.0 mL) was added to a solution of 6-(3- azidopropyl)-5,6-dihydro-8-methoxy-3-nitro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (50, 0.287 g, 0.710 mmol) in benzene (50 mL), and the reaction mixture was heated at reflux for 16 h. The reaction mixture was allowed to cool to room temperature, HCl in methanol (3 M, 10 mL) was added, and the reaction mixture was heated at reflux for 4 h. The reaction mixture was allowed to cool to room temperature and filtered to provide a red solid (0.180 g, 61%): mp 288–290 °C. IR (KBr) 3060, 3015, 2979, 2090, 1691, 1668, 1614, 1560, 1504, 1371, 1344, 1258, 1232, 1082, 1022, 911, 876, 843, 775 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.84 (d, J = 2.5 Hz, 1 H), 8.69 (d, J = 8.9 Hz, 1 H), 8.53 (dd, J = 8.9 Hz, J = 2.5 Hz, 1 H), 7.87 (br s, 3 H), 7.60 (d, J = 8.4 Hz, 1 H), 7.30 (d, J = 2.0 Hz, 1 H), 7.05 (dd, J = 8.1 Hz, J = 1.9 Hz, 1 H), 4.54 (t, J = 7.2 Hz, 2 H), 3.91 (s, 3 H), 2.94 (t, J = 6.4 Hz, 2 H), 2.10 (t, J = 7.0 Hz, 2 H); 13C NMR (125 MHz, DMSO-d6) δ 188.6, 164.6, 162.6, 157.6, 145.8, 138.3, 136.7, 128.0, 127.3, 125.3, 124.6, 124.2, 123.2, 114.6, 113.8, 108.2, 56.6, 42.3, 37.2, 27.3; ESIMS m/z (rel intensity) 380 (MH+, 72), 363 ([(M – NH3]+, 100); HRESIMS m/z calcd for C20H17N3O5 380.1246 (MH+), found, 380.1243; HPLC purity: 97.2% (MeOH-H2O, 90:10).
6-(3-(1H-Imidazol-1-yl)propyl)-8-methoxy-3-nitro-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (52)
6-(3-Bromopropyl)-8-methoxy-3-nitro-5H-indeno[1,2-c]isoquinoline- 5,11(6H)-dione (49, 120 mg, 0.27 mmol) was dissolved dimethylformamide (2 mL) and dioxane (10 mL). Sodium iodide (208 mg, 1.40 mmol) was added and the reaction mixture was heated at 60 °C for 6 h. Imidazole (202 mg, 297 mmol) and potassium carbonate (225 mg, 1.63 mmol) were added and the reaction mixture was stirred at 90 °C for 12 h. Water (15 mL) was added and a precipitated formed. The solid was filtered and kept. The remaining solution was diluted with water (100 mL) and the aqueous phase extracted with chloroform (3 × 30 mL). The organic extracts were combined, washed with water (3 × 100 mL), and dried over sodium sulfate. The filtered solid from the previous step was combined with the organic extracts, the solvent was removed in vacuo and the compound purified by silica gel column chromatography, eluting with chloroform-methanol, 50:1. The compound was obtained as a dark yellow solid (47 mg, 40%): mp 265–276 °C. IR (Film) 1693, 1673, 1614, 1559, 1336, 1234, 1075, 866, 842, 774 cm−1; 1H NMR (CDCl3, 300 MHz) δ 9.19 (d, J = 2.1 Hz, 1 H), 8.89 (d, J = 8.9 Hz, 1 H), 8.48 (dd, J = 8.9 Hz, J = 2.2 Hz, 1 H), 7.68-7.60 (m, 2 H), 7.13 (br s, 1 H), 7.06 (br s, 1 H), 6.86-6.83 (m, 2 H), 4.55 (t, J = 7.1 Hz, 2 H), 4.23 (t, J = 6.5 Hz, 2 H), 3.93 (s, 3 H), 2.41 (p, J = 7.1 Hz, 2 H); 13C NMR (CDCl3, 125 MHz) δ 188.6, 164.3, 162.4, 157.6, 145.4, 137.9, 136.5, 136.1, 128.2, 126.8, 125.4, 124.4, 124.2, 122.9, 114.2, 113.7, 107.9, 56.5, 46.5, 41.7, 29.7; ESIMS m/z (rel intensity) 431 (MH+, 100); HRESIMS calcd for C23H18N4O5 431.1355 (MH+), found 431.1352 (MH+); HPLC purity: 96.8% (MeOH-H2O, 90:10).
8-Methoxy-6-(3-morpholinopropyl)-3-nitro-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (53)
6-(3-Bromopropyl)-8-methoxy-3-nitro-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (49, 117 mg, 0.26 mmol) was dissolved in dimethylformamide (2 mL) and dioxane (10 mL). Sodium iodide (213 mg, 1.42 mmol) was added and the reaction mixture was heated at 60 °C for 6 h. Morpholine (0.2 mL, 2.30 mmol) and potassium carbonate (260 mg, 1.88 mmol) were added and the reaction mixture was stirred at 90 °C for 12 h. Water (200 mL) was added and the compound extracted with chloroform (3 × 35 mL). The organic extracts were combined, washed with water (3 × 100 mL) and dried over sodium sulfate. The solvent was removed in vacuo and the compound purified by silica gel column chromatography, eluting with chloroform-methanol, 50:1. The product was obtained as an orange solid (30 mg, 25%): mp 276–278 °C. IR (Film) 3020, 1676, 1614, 1562, 1337, 1215, 1066, 846, 757, 666 cm−1; 1H NMR (CDCl3, 300 MHz) δ 9.18 (d, J = 2.2 Hz, 1 H), 8.90 (d, J = 8.9 Hz, 1 H), 8.47 (dd, J = 9.0 Hz, J = 2.3 Hz, 1 H), 7.50-7.48 (m, 2 H), 7.10 (dd, J = 8.9 Hz, J = 2.1 Hz, 1 H), 4.63 (t, J = 7.6 Hz, 2 H), 4.03 (s, 3 H), 3.72 (m, 4 H), 2.59 (t, J = 6.2 Hz, 2 H), 2.50 (s, 4 H) 2.06 (m, 2 H); 13C NMR (CDCl3, 125 MHz) δ 188.0, 162.5, 157.5, 156.9, 145.3, 137.8, 136.8, 136.7, 128.2, 124.2, 123.1, 118.6, 118.5, 117.9, 106.9, 63.5, 56.4, 53.5, 51.3, 42.0, 23.5; ESIMS m/z (rel intensity) 450 (MH+, 62); HRESIMS calcd for C24H13N3O6 450.1665 (MH+), found 450.1660 (MH+); HPLC purity: 97.7% (MeOH-H2O, 85:15), 97.8% (MeOH-H2O, 90:10).
1-Bromo-6-cyanophthalide (65)
6-Cyanophthalide (64, 2.73 g, 17.2 mmol) was dissolved in carbon tetrachloride (120 mL). 3-Chloroperbenzoic acid (100 mg) and N-bromosuccinimide (3.20 g, 17.9 mmol) were added. Light was applied with a 250 W lamp and the reaction mixture was heated at reflux for 24 h. The solvent was removed and the residue purified by silica gel column chromatography, eluting with ethyl acetate-hexane, 1:7. The product was obtained as a white solid (2.32 g, 56.7%): mp 132–134 °C. IR (Film) 3090, 3064, 3032, 2992, 2235, 1788, 1738, 1428, 1295, 1227, 1119, 990, 717, 664 cm−1; NMR (300 MHz, CDCl3) δ 8.23 (s, 1 H), 8.05 (dd, J = 1.1 Hz, J = 8.1 Hz, 1 H), 7.79 (d, J = 7.9 Hz, 1 H), 7.44 (s, 1 H); 13C NMR (75 MHz, CDCl3) δ 165.1, 152.2, 138.3, 130.0, 125.0, 121.5, 116.8, 115.3, 73.4; CIMS m/z (rel intensity) 238 (MH+, 13), 160 [(MH – Br)+, 100].
7-Cyano-3-hydroxyphthalide (66)
1-Bromo-6-cyanophthalide (65, 1.99 g, 8.36 mmol) was dissolved in hot water (50 mL). The reaction mixture was heated at reflux for 3 h. Ethanol (20 mL) was added and the solution was concentrated to half its volume and heated at reflux for 5 min. Once the solution reached room temperature the reaction mixture was placed in the refrigerator. The desired compound was recrystallized as off-white solid (1.23 g, 84.1%): mp 138–140 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.42 (br s, 1 H), 8.37 (d, J = 1.8 Hz, 1 H), 8.24 (dd, J = 7.8 Hz, J = 1.7 Hz, 1 H), 7.90 (d, J = 7.9 Hz, 1 H), 6.75 (s, 1 H); ESIMS m/z (rel intensity) 174 (MH+, 100).
3-Cyano-5,11-dihydro-5,11-dioxoindeno[1,2-c]isochromene (68)
7-Cyano-3-hydroxyphthalide (66, 1.41 g, 8.06 mmol) and phthalide (67, 1.08 g, 8.06 mmol) were dissolved in ethyl acetate (15 mL). Sodium (0.80 g, 34.8 mmol) was added to methanol (30 mL) at 0 °C and the resulting solution added to the reaction mixture. The reaction mixture was stirred for 30 min at room temperature and then heated at reflux for 6 h. Concentrated hydrochloric acid (15 mL) was added and the solvent removed under vacuum. Benzene (100 mL) and para-toluenesulfonic acid (50 mg) were added, and the reaction mixture was connected to a Dean-Stark trap and heated at reflux for 16 h. Chloroform (150 mL) was added and the reaction mixture was heated at reflux and immediately filtered. The solvent was removed under vacuum and the solid recrystallized from chloroform to remove unreacted phthalide. The compound was purified by silica gel column chromatography, eluting with chloroform-methanol, 50:1. The product was obtained as a yellow solid (1.03 g, 46.8 %): mp 225–227 °C. IR (KBr) 3118, 3082, 2232, 1760, 1747, 1715, 1503, 1385, 1309, 1018, 877, 777, 763, 665 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.59 (s, 1 H), 8.47 (d, J = 8.2 Hz, 1 H), 7.99 (dd, J =8.2 Hz, J = 1.6 Hz,1 H), 7.64 (d, J = 7.9 Hz, 1 H), 7.55-7.50 (m, 3 H); 13C NMR (125 MHz, DMSO-d6) δ 188.8, 172.6, 158.8, 137.9, 135.8, 135.4, 135.1, 134.0, 132.7, 124.1, 123.6, 120.6, 119.2, 117.3, 11.7, 106.5; CIMS m/z (rel intensity) 274 (MH+, 100).
3-Cyano-5,11-dihydro-6-[3-(1H-imidazolyl)propyl]-5,11-dioxo-6,11-dihydro-5H-indeno[ 1,2-c]isoquinoline (72)
3-Cyano-5,11-dihydro-5,11-dioxoindeno[1,2-c]isochromene (68, 200 mg, 0.73 mmol) was dissolved in tetrahydrofuran (10 mL). 3-(1H-Imidazol-1-yl)propyl-1-amine (69, 335 mg, 2.67 mmol) was dissolved in chloroform (5 mL) and the solution added to the reaction mixture. The reaction mixture was stirred for 3 h at room temperature and then heated at reflux for 1 h. The solution turned red and, upon reflux, an orange precipitate formed. The solvent was removed under vacuum and the solid purified by silica gel column chromatography, eluting with chloroform-methanol, 30:1. The desired compound was obtained as a yellow solid (103 mg, 37%): mp 256–258 °C. IR (KBr) 3103, 2952, 2225, 1694, 1671, 1611, 1534, 1509, 1433, 1192, 850, 764, 665 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.80 (d, J = 8.6 Hz, 1 H), 8.63 (d, J = 1.4 Hz, 1 H), 7.88 (dd, J = 8.4 Hz, J = 1.6 Hz, 1 H), 7.67-7.64 (m, 2 H), 7.43 (t, J = 7.6 Hz, 1 H), 7.35 (dt, J = 7.5 Hz, J = 1.2 Hz, 1 H), 7.21 (s, 1 H), 7.07 (s, 1 H), 6.66 (d, J = 7.5 Hz, 1 H), 4.54 (t, J = 7.8 Hz, 2 H), 4.26 (t, J = 6.2 Hz, 2 H), 2.37 (p, J = 8.2 Hz, J = 6.6 Hz, 2 H); ESIMS m/z (rel intensity) 381 (MH+, 100); HRESIMS calcd for C23H16N4O2 381.1352 (MH+), found 381.1345 (MH+); HPLC purity: 95.0% (MeOH-H2O, 85:15), 95.4% (MeOH-H2O, 95:5).
3-Cyano-6,11-dihydro-5,11-dioxo-6-(3-morpholinylpropyl)-5H-indeno[1,2- c]isoquinoline (73)
3-Cyano-5,11-dihydro-5,11-dioxoindeno[1,2-c]isochromene (68, 130 mg, 0.47 mmol) was dissolved in chloroform (50 mL). 3-Morpholinopropyl-1-amine (70, 152 mg, 1.04 mmol) and molecular sieves were added and the reaction was mixture stirred at room temperature for 8 h. The reaction mixture was then heated at reflux for 1 h. The molecular sieves were filtered off, the solvent removed under vacuum and the residue purified by silica gel column chromatography, eluting with chloroform-methanol, 100:1. The desired compound was obtained as an orange solid (113 mg, 60.0%): mp 259–261 °C. IR (Film) 3083, 2950, 2849, 2223, 1698, 1673, 1611, 1509, 1435, 1275, 1185, 1116, 998, 959, 896, 851, 766, 665 cm−1; 1H NMR (CDCl3, 500 MHz) δ 8.80 (d, J = 8.6 Hz, 1 H), 8.62 (d, J = 1.7 Hz, 1 H), 7.88-7.84 (m, 2H), 7.69 (m, 1 H), 7.51-7.48 (m, 2 H), 4.64 (t, J = 7.8 Hz, 2 H), 3.70 (t, J = 4.4 Hz, 4 H), 2.58 (t, J = 6.2 Hz, 1 H), 2.49 (br s, 4 H), 2.05 (m, 2 H); 13C NMR (125 MHz, CDCl3) δ 189.5, 162.0, 157.8, 136.3, 135.3, 135.0, 133.4, 131.9, 124.4, 123.6, 123.4, 123.0, 110.0, 107.2, 66.9, 56.0, 53.9, 43.9, 25.7; ESIMS m/z (rel intensity) 400 (MH+, 100); HRESIMS calcd for C24H21N3O3 400.1661 (MH+), found, 400.1665 (MH+). Anal. calcd for C24H21N3O3·0.5 H2O: C, 70.57; H, 5.43; N, 10.29. Found: C, 70.71; H, 5.22; N, 10.01.
3-Cyano-6,11-dihydro-5,11-dioxo-6-[3-(dimethylaminopropyl]-5H-indeno[1,2-c]isoquinoline (74)
3-Cyano-5,11-dihydro-5,11-dioxoindeno[1,2-c]isochromene (68, 135 mg, 0.49 mmol) was dissolved in a mixture of tetrahydrofuran (10 mL). N′,N′-Dimethylpropane-1,3-diamine (71, 158 mg, 1.51 mmol) was dissolved in chloroform (5 mL). This solution was added to the first solution, and the reaction mixture was stirred for 3 h at room temperature. The mixture was heated at reflux for 1 h. The solvent was removed under vacuum and the solid purified by silica gel column chromatography, eluting with chloroform-methanol, 50:1. The compound was obtained as a yellow solid (96 mg, 55%): mp 210–212 °C. IR (Film) 2817, 2762, 2223, 1698, 1673, 1611, 1611, 1435, 1261, 1191, 1040, 897, 764 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 8.78 (d, J = 8.5 Hz, 1 H), 8.62 (s, 1 H), 7.87-7.84 (m, 2 H), 7.67 (d, J = 6.7 Hz, 1 H), 7.50-7.46 (m, 2 H), 4.59 (t, J = 5.9 Hz, 2 H), 2.53 (t, J = 6.5 Hz, 2 H), 2.32 (s, 6 H), 2.04 (br p, J = 8.5 Hz, 2 H); 13C NMR (125 MHz, DMSO-d6) δ 189.6, 162.0, 157.9, 136.3, 135.3, 135.1, 135.0, 133.6, 133.5, 131.8, 124.4, 123.7, 123.5, 123.0, 118.2, 109.9, 107.3, 56.6, 45.6, 43.8, 29.9; ESIMS m/z (rel intensity) 402 (MH+, 100); HRESIMS calcd for C23H19N3O4 402.1454 (MH+), found 402.1452 (MH+); HPLC purity: 97.4% (MeOH-H2O, 95:5), 97.4% (MeOH).
3-Cyano-5,11-dihydro-8,9-methylenedioxy-5,11-dioxo[1,2-c]isochromene (76)
7-cyano-3-hydroxyphthalide (66, 1.55 g, 8.85 mmol) and 5,6-methylenedioxyphthalide (75, 1.58 g, 8.06 mmol) were dissolved in ethyl acetate (20 mL). Sodium (0.85 g, 34.8 mmol) was dissolved in methanol (35 mL) at 0 °C and the resulting solution added to the reaction mixture. The reaction mixture was stirred for 30 min at room temperature and then heated at reflux for 15 h. Concentrated hydrochloric acid (20 mL) was added and the solvent removed under vacuum. Benzene (100 mL) and para-toluenesulfonic acid (50 mg) were added, and the reaction mixture was connected to a Dean-Stark trap and heated at reflux for 16 h. Chloroform (100 mL) was added, the reaction mixture heated at reflux and immediately filtered. The solvent was removed under vacuum and the solid recrystallized from chloroform-methanol, 25:1, to remove unreacted phthalide. The compound was purified by silica gel column chromatography, eluting with chloroform-methanol, 50:1. The product was obtained as a yellow solid (133 mg, 10 %): mp > 350 °C. Also, 0.81 g of the starting material 75 was recovered. IR (Film) 2233, 1692, 1417, 1313, 1152, 1029, 925, 861, 785, 665 cm−1; 1H NMR (CDCl3-MeOH-d4, 300 MHz) δ 8.44 (s, 1 H), 8.29 (d, J = 8.1 Hz, 1 H), 7.88 (dd, J = 8.1 Hz, J = 1.5 Hz, 1 H), 7.05 (s, 1 H), 6.94 (s, 1 H), 6.09 (s, 2 H); EIMS m/z (rel intensity) 317 (M+, 100); HREIMS calcd for C18H7NO5 317.0324 (M+), found 317.0327 (M+).
3-Cyano-5,11-dihydro-6-[3-(dimethylamino)propyl]-5,11-dioxoindeno[1,2- c]isquinoline (77)
3-Cyano-5,11-dihydro-8,9-methylenedioxy-5,11-dioxo[1,2-c]isochromene (76, 91 mg, 0.28 mmol) was dissolved in tetrahydrofuran (25 mL). N1,N1-Dimethylpropane-1,3-diamine (71, 156 mg, 1.56 mmol) and molecular sieves were added and the reaction mixture stirred at room temperature for 8 h. The reaction mixture was then heated at reflux for 1 h. The molecular sieves were filtered off, the solvent removed in vacuo and the residue purified by recrystallization from chloroform-hexane. The desired compound was obtained as a purple solid (89 mg, 79%): mp 280–282 °C. IR (Film) 2968, 2909, 2825, 2271, 1694, 1672, 1607, 1577, 1529, 1432, 1309, 1282, 1185, 1033, 852, 793 cm−1; 1H NMR (500 MHz, CDCl3-MeOH-d4) δ 8.62 (d, J = 8.5 Hz, 1 H), 8.51 (s, 1 H), 7.86 (d, J = 8.5 Hz, 1 H), 7.44 (s 1 H), 7.08 (s, 1 H), 6.08 (s, 1 H), 4.45 (t, J = 6.9 Hz, 2 H), 2.46 (br s, 2 H), 2.25 (s, 6 H), 1.94 (m, 2 H); 13C NMR (125 MHz, CDCl3) δ 188.7, 162.2, 157.6, 152.0, 150.0, 135.2, 133.6, 131.3, 130.7, 123.8, 122.1, 118.1, 109.1, 106.6, 106.3, 105.4, 102.9, 56.4, 45.4, 43.5, 33.0, 26.8; ESIMS m/z (rel intensity) 401 (M+, 100); HPLC purity: 100% (MeOH-H2O, 90:10), 96.7% (MeOH-H2O, 85:15).
6-(3-Bromopropyl)-9-methoxy-3-nitro-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (80)
5-Nitrohomophthalic anhydride (47, 5.02 g, 24.2 mmol) was added to solution of 3-bromo-N-(4-methoxybenzylidene)propyl-1-amine (78, 6.21 g, 24.2 mmol) in chloroform (115 mL), and the reaction mixture was allowed to stir at room temperature for 1.5 h and then placed inside the freezer overnight. The precipitate was filtered and washed with chloroform (50 mL) and dried to provide intermediate 79 (5.13 g), which was used without further purification. Thionyl chloride (26 mL) was added to a suspension of 79 in benzene (200 mL) and the reaction mixture was heated at reflux until complete dissolution. The solvent was removed under vacuum and the residue diluted with nitrobenzene (225 mL) and chilled in an ice bath. Aluminum chloride (4.50 g, 33.8 mmol) was added and the reaction mixture was heated at 100 °C for 2 h. Cool water (200 mL) was added, the organic phase separated and the aqueous phase was extracted with CHCl3 (3 × 70 mL). The combined organic layer was washed with a solution of NaHCO3 (4.0 g) in water (75 mL). The solution was concentrated and hexanes were added until the precipitation of a red solid was observed. The solid was filtered, washed with hexanes (100 mL), and purified by flash silica gel column chromatography, eluting with chloroform-methanol, 12:1. The product was obtained as an orange solid (0.79 g, 11.2%): mp 282–284 °C. IR (Film) 3097, 1809, 1745, 1685, 1604, 1510, 1337, 1231, 1127, 1082, 851 cm−1; 1H NMR (300 MHz, CDCl3) δ 9.17 (d, J = 2.3 Hz, 1 H), 8.81 (d, J = 8.9 Hz, 1 H), 8.47 (dd, J = 9.0 Hz, J = 2.3 Hz, 1 H), 7.81 (d, J = 8.5 Hz, 1 H), 7.28 (d, J = 2.5 Hz, 1 H), 6.94 (dd, J = 8.5 Hz, J = 2.5 Hz, 1 H), 4.51 (t, J = 6.0 Hz, 2 H), 3.84 (s, 3 H), 3.75 (dd, J = 7.5 Hz, J = 6.6 Hz, 2 H), 2.30 (br p, J = 8.5 Hz, 2 H); EIMS m/z (rel intensity) 442 (M+, 100); HREIMS calcd for C20H15N2O5Br 442.0164 (M+), found, 442.0158 (M+).
3-Amino-6-(3-bromopropyl)-9-methoxy-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (81)
6-(3-Bromopropyl)-9-methoxy-3-nitro-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (80, 1.00 g, 2.26 mmol) was dissolved in tetrahydrofuran (60 mL), methanol (20 mL), and ethyl acetate (30 mL) and transferred to a Parr shaker flask. The reaction vessel was purged with argon for 10 min and then palladium-charcoal (10%, 20 mg) was added. The reaction mixture was shaken for 24 h under an atmosphere of hydrogen (60 psi). The solid was filtered off, the solvent removed under vacuum, and the compound purified by silica gel column chromatography, eluting with ethyl acetate. The product was obtained as a brown solid (0.55 g, 59%): mp 330 °C (dec). IR (Film) 3367, 1696, 1655, 1506, 1480, 1431, 1293, 1229, 1016, 834, 787, 665 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 8.35 (d, J = 8.6 Hz, 1 H), 7.67–7.62 (m, 2 H), 7.06 (d, J = 2.5 Hz, 1 H), 7.0.1-6.94 (m, 2 H), 4.51 (t, J = 6.0 Hz, 2 H), 3.84 (s, 3 H), 3.75 (dd, J = 7.5 Hz, J = 6.6 Hz, 2 H), 2.30 (br p, J = 8.5 Hz, 2 H); EIMS m/z (rel intensity) 413 (M+, 46), 333 ([M-HBr)]+, 100); HRESIMS m/z calcd for C20H17N2O3Br 413.0501 (MH+), found, 413.0505 (MH+).
6-(3-Bromopropyl)-3-iodo-9-methoxy-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (82)
3-Amino-6-(3-bromopropyl)-9-methoxy-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (81, 420 mg, 1.01 mmol) was dissolved in dioxane (10 mL). Concentrated hydrochloric acid (0.65 mL, 3.3 mmol) was added and the reaction mixture was stirred for 10 min and then cooled down to 0 °C. A solution of sodium nitrite (114 mg, 1.65 mmol) in water (5 mL) was slowly added with stirring while the temperature was −20 °C. The reaction mixture was stirred for 30 min and then slowly added to a solution of copper (I) iodide (200 mg) and potassium iodide (300 mg) in water (15 mL). The reaction mixture was stirred at room temperature for 8 h and then heated at reflux for 1 h. The reaction mixture was diluted with water (200 mL) and extracted with chloroform (3 × 100 mL). The combined organic extracts were washed with an aqueous solution of sodium thiosulfate (2 × 200 mL), aqueous sodium bicarbonate (2 × 200 mL), water (100 mL) and brine (100 mL). The organic solution was dried over sodium sulfate and the solvent removed under vacuum. The residue was purified by silica gel column chromatography, eluting with chloroform-methanol, 30:1. Compound 82 was obtained as a dark red solid (402 mg, 76%): mp 207–209 °C. IR (Film) 2922, 1765, 1655, 1599, 1532, 1478, 1455, 1430, 1379, 1295, 1225, 1047, 969, 812, 788, 691, 666, 592 cm−1; 1H NMR (CDCl3, 500 MHz) δ 8.60 (d, J = 1.5 Hz, 1 H), 8.32 (d, J = 8.6 Hz, 1 H), 7.93 (dd, J = 8.5 Hz, J = 1.5 Hz, 1 H), 7.64 (d, J = 8.4 Hz, 1 H), 7.15 (d, J = 2.3 Hz, 1 H), 6.82 (dd, J = 8.3 Hz, J = 2.4 Hz, 1 H), 4.58 (t, J = 7.2 Hz, 2 H), 3.89 (s, 3 H), 3.64 (t, J = 6.2 Hz, 2 H), 2.44 (t, J = 7.2 Hz, 2 H); 13C NMR (125 MHz, CDCl3) δ 189.3, 162.5, 162.0, 156.6, 142.4, 137.6, 137.0, 131.4, 127.6, 124.7, 124.1, 123.9, 115.8, 111.1, 107.6, 91.2, 55.8, 44.2, 31.1, 30.2; ESIMS m/z (rel intensity) 523 (MH+, 60), 403 [(MH+ – C3H6Br)+, 100]; HRESIMS m/z calcd for C20H15NO3IBr 523.9358 (MH+), found, 523.9362 (MH+).
6-(3-Azidopropyl)-3-iodo-9-methoxy-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (83)
Sodium azide (0.59 g, 9.07 mmol) and 6-(3-bromopropyl)-3-iodo-9-methoxy-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (82, 261 mg, 0.59 mmol) were diluted with DMSO (30 mL) and the mixture was heated at 90 °C for 12 h. The reaction mixture was diluted with CHCl3 (100 mL), washed with water (100 mL) and sat aq NaCl (30 mL), and dried over sodium sulfate. The solution was concentrated to provide a crude solid that was purified by silica gel column chromatography, eluting with chloroform-methanol, 50:1, to afford an orange solid (0.16 g, 83%): mp 267–269 °C (dec). IR (KBr) 2090, 1688, 1662, 1598, 1532, 1478, 1424, 1298,1164, 1014, 819, 791, 694 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.61 (d, J = 1.8 Hz, 1 H), 8.34 (d, J = 8.6 Hz, 1 H), 7.92 (dd, J = 8.6 Hz, J = 1.8 Hz, 1 H), 7.59 (d, J = 8.4 Hz, 1 H), 7.17 (d, J = 2.5 Hz, 1 H), 6.85 (dd, J = 8.3 Hz, J = 2.8 Hz, 1 H), 4.53 (t, J = 7.2 Hz, 2 H), 3.89 (s, 3 H), 3.63 (t, J = 6.1 Hz, 2 H), 2.11 (t, J = 7.2 Hz, 2 H); 13C NMR (125 MHz, CDCl3) δ 188.9, 162.5, 161.8, 156.5, 142.2, 137.7, 136.9, 131.3, 127.6, 124.6, 123.9, 123.8, 115.8, 110.9, 107.4, 91.0, 55.7, 49.2, 42.7, 28.3; ESIMS m/z (rel intensity) 509 (MNa+, 100); HRESIMS m/z calcd for C20H15N4O3I 487.0267 (MH+), found, 487.0271 (MH+).
6-(3-Aminopropyl)-5,6-dihydro-9-methoxy-3-iodo-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (84)
Triethylphosphite (0.2 mL) was added to a solution of 6-(3-azidopropyl)-3-iodo-9-methoxy-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (83, 0.186 g, 0.459 mmol) in benzene (30 mL), and the reaction mixture was heated at reflux for 16 h. The reaction mixture was allowed to cool to room temperature, HCl in methanol (prepared from 0.5 mL of acetyl chloride in 9.5 mL of methanol) was added, and the reaction mixture was heated at reflux for 4 h. The reaction mixture was allowed to cool to room temperature and filtered to provide a red solid (0.159 g, 83%): mp 267–269 °C (dec). IR (KBr) 3399, 2937, 1692, 1658, 1600, 1532, 1478, 1429, 1298, 1228, 909, 823, 733 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.65 (d, J = 1.5 Hz, 1 H), 8.37 (d, J = 8.5 Hz, 1 H), 7.94 (dd, J = 8.6 Hz, J = 1.6 Hz, 1 H), 7.64 (d, J = 8.4 Hz, 1 H), 7.21 (d, J = 2.4 Hz, 1 H), 6.84 (dd, J = 8.3 Hz, J = 2.4 Hz, 1 H), 4.58 (t, J = 7.2 Hz, 2 H), 3.89 (s, 3 H), 2.89 (t, J = 6.3 Hz, 2 H), 2.00 (t, J = 7.3 Hz, 2 H); ESIMS m/z (rel intensity) 461 (MH+, 100); HRESIMS m/z calcd for C20H17N2O3I 461.0362 (MH+), found, 461.0371 (MH+); HPLC purity: 98.9% (MeOH-H2O, 90:10), 96.7% (MeOH-H2O, 80:20).
3-Iodo-9-methoxy-6-(3-(Dimethylamino)propyl)-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (85)
3-Amino-6-(3-aminopropyl)-9-methoxy-5H-indeno[1,2-c]isoquinoline-5,11(6H)-dione (84, 111 mg, 1.93 mmol) was dissolved in methanol (10 mL) and acetic acid (5 mL). Aqueous formaldehyde (37%, 0.15 mL) was added and the reaction mixture was stirred at room temperature for 2 h. The reaction mixture was cooled to 0 °C and sodium cyanoborohydride (300 mg) was slowly added. The reaction mixture was stirred for 1 h at room temperature. Water (30 mL) was added and the reaction mixture extracted with chloroform (2 × 25 mL). The organic extracts were combined and washed with brine (30 mL). The solvent was removed in vacuo and the compound purified by silica gel column chromatography, eluting with chloroform. The product was obtained as a red solid (65 mg, 55%) mp > 350 °C. IR (Film) 3054, 2987, 1698, 1649, 1531, 1477, 1430, 1301, 1265, 740 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.65 (d, J = 1.8 Hz, 1 H), 8.38 (d, J = 8.6 Hz, 1 H), 7.85 (dd, J = 2.0, J = 8.6 Hz, 1 H), 7.67 (d, J = 8.3 Hz, 1 H), 7.21 (d, J = 2.5 Hz, 1 H), 6.83 (dd, J = 2.5, J = 8.4 Hz, 1 H), 4.54 (t, J = 8.1 Hz, 2 H), 3.89 (s, 3 H), 2.50 (d, J = 6.3 Hz, 2 H), 2.02 (m, 2 H); 13C NMR (125 MHz, CDCl3-MeOH-d4) δ 189.5, 162.6, 162.2, 142.4, 137.7, 136.9, 131.6, 127.6, 124.6, 124.3, 123.8, 115.9, 111.2, 107.7, 91.0, 56.2, 55.7, 44.7, 43.0, 26.4; ESIMS m/z (rel intensity) 489 (MH+, 100); HRESIMS m/z calcd for C22H21N2O3I 489.0675 (MH+), found, 489.0670 (MH+); HPLC purity: 97.1% (MeOH-H2O, 95:5); 97.5% (MeOH-H2O, 85:15).
3-Amino-6-(3-chloropropyl)-5,6-dihydro-9-methoxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (87)
6-(3-Chloropropyl)-5,6-dihydro-9-methoxy-3-nitro-5,11-dioxo-11H-indeno[ 1,2-c]isoquinoline (86)20 (0.220 g, 0.552 mmol) and 5% Pd/C (0.200 g) were diluted with THF (125 mL). The solution was degassed and allowed to stir at room temperature under a hydrogen atmosphere for 1 h. The solution was filtered, the filterpad was washed with chloroform (125 mL), and the filtrate was concentrated to provide a crude purple solid. The solid was purified by silica gel flash column chromatography, eluting with chloroform to provide a purple solid (0.080 g, 39%): mp 232–235 °C (dec). IR (KBr) 3357, 1644, 1544, 1510, 1272, 1052 cm−1; 1H NMR (DMSO-d6) δ 8.33 (d, J = 8.6 Hz, 1 H), 7.42 (dd, J = 8.5 Hz, J = 6.8 Hz, 1 H), 7.32 (m, 2 H), 7.14 (dd, J = 6.7 Hz, J = 1.0 Hz, 1 H), 7.09 (dd, J = 8.7 Hz, J = 2.4 Hz, 1 H), 5.74 (s, 2 H), 4.68 (t, J = 7.1 Hz, 2 H), 3.97 (s, 3 H), 3.73 (t, J = 6.6 Hz, 2 H), 2.21 (pent, J = 7.1 Hz, 2 H); ESIMS m/z (rel intensity) 369/371 (MH+, 100/32). Anal. calcd for C20H17ClN2O3: C, 65.13; H, 4.65; N, 7.60. Found: C, 64.85; H, 4.74; N, 7.42.
3-Amino-6-(3-azidopropyl)-5,6-dihydro-9-methoxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (88)
Sodium azide (0.063 g, 0.968 mmol) and 3-amino-6-(3-chloropropyl)-5,6-dihydro-9-methoxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (87, 0.119 g, 0.323 mmol) were diluted with DMSO (25 mL) and the mixture was heated at 100 °C for 16 h. The reaction mixture was diluted with chloroform (50 mL), washed with water (3 × 25 mL), brine (25 mL), and dried over sodium sulfate. The solution was concentrated to provide a crude solid that was purified by silica gel column chromatography, eluting with a gradient of chloroform to 1% MeOH-chloroform, to provide a solid that was washed with diethyl ether to afford a purple solid (0.102 g, 84%): mp 220–223 °C. IR (KBr) 3434, 3349, 2096, 1651, 1509, 1271 cm−1; 1H NMR (DMSO-d6) δ 8.34 (d, J = 8.7 Hz, 1 H), 7.42 (dd, J = 8.5 Hz, J = 6.7 Hz, 1 H), 7.33 (m, 2 H), 7.15 (dd, J = 6.7 Hz, J = 1.1 Hz, 1 H), 7.09 (dd, J = 8.7 Hz, J = 2.5 Hz, 1 H), 5.74 (s, 2 H), 4.65 (m, 2 H), 3.97 (s, 3 H), 3.46 (t, J = 6.7 Hz, 2 H), 1.99 (m, 2 H); ESIMS m/z (rel intensity) 376 (MH+, 55). Anal. calcd for C20H17N5O3: C, 63.99; H, 4.56; N, 18.66. Found: C, 63.78; H, 4.38; N, 18.30.
3-Amino-6-(3-aminopropyl)-5,6-dihydro-9-methoxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline Dihydrochloride (89)
Triethyl phosphite (0.07 mL) was added to a solution of 3-amino-6-(3-azidopropyl)-5,6-dihydro-9-methoxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (88, 0.061 g, 0.163 mmol) in benzene (20 mL) and the reaction mixture was heated at reflux for 16 h. The reaction mixture was allowed to cool to room temperature, 3 M HCl in methanol (10 mL) was added, and the reaction mixture was heated at reflux for 3 h. The reaction mixture was allowed to cool to room temperature, concentrated, and the precipitate was washed with chloroform (50 mL) and filtered to provide an orange solid (0.054 g, 78%): mp 246–248 °C (dec). IR (KBr) 3445, 2929, 1651, 1544, 1505, 1479, 1266 cm−1; 1H NMR (DMSO-d6) δ 8.51 (d, J = 8.7 Hz, 1 H), 8.03 (bs, 2 H), 7.77 (d, J = 2.2 Hz, 1 H), 7.49 (m, 2 H), 7.36 (d, J = 7.6 Hz, 1 H), 7.19 (dd, J = 6.8 Hz, J = 0.8 Hz, 1 H), 4.61 (t, J = 7.0 Hz, 2 H), 4.01 (s, 3 H), 2.86 (m, 2 H), 2.09 (m, 2 H); ESIMS m/z (rel intensity) 350 (MH+, 90). Anal. calcd for C20H21Cl2N3O3·1.0 H2O: C, 54.55; H, 5.26; N, 9.54. Found: C, 54.87; H, 5.10; N, 9.21.
Molecular Modeling
The Tdp1 crystal structure (PDB: 1RFF) was prepared by removing one of the monomers along with all crystallized waters, the polydeoxyribonucleotide 5′-D(*AP*GP*TP*T)-3′, the Top1-derived peptide residues 720–727 (mutation L724Y), and all metal ions. The Lys265, Lys495 and His493 residues were protonated. Missing hydrogens were added as needed. GOLD docking was performed using the centroid x = 8.0128, y = 46.5888, z = −1.5534. The hydrogen bond length was set to 4 Å while the van der Waals parameter was set to 10 Å. The top ligand binding-pose (highest GOLD score) was selected and merged with the prepared protein. The ligand was surrounded by a sphere of 6 Å of radius and minimized by the conjugate gradient method using the MMFF94s force field and MMFF94 charges with Sybyl software. The calculation was terminated when the gradient reached a value of 0.05 kcal/(mol·Å).
Topoisomerase I-Mediated DNA Cleavage Reactions
Top1 reactions were performed as recently described.16 Briefly, a 3′-[32P]-end-labeled 117-bp DNA oligonucleotide (Integrated DNA Technologies) was incubated at 2 nM with recombinant Top1 in 20 μL of reaction buffer [10 mM Tris-HCl (pH 7.5), 50 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, and 15 μg/mL BSA] at 25 °C for 20 min in the presence of various concentrations of compounds. The reactions were terminated by adding SDS (0.5% final concentration) followed by the addition of two volumes of loading dye (80% formamide, 10 mM sodium hydroxide, 1 mM sodium EDTA, 0.1% xylene cyanol, and 0.1% bromphenol blue). Reactions were subjected to 20% denaturing PAGE. Gels were dried and visualized by using a Typhoon 8600 and ImageQuant software (Molecular Dynamics).
Gel-based Assay Measuring the Inhibition of Recombinant Tdp1
Tdp1 reactions were performed as recently described.16 Briefly, a 5′-[32P]-labeled single-stranded DNA oligonucleotide containing a 3′-phosphotyrosine (N14Y) incubated with 5 pM recombinant Tdp1 in the absence or presence of inhibitor for 15 min at room temperature in a buffer containing 50 mM Tris HCl, pH 7.5, 80 mM KCl, 2 mM EDTA, 1 mM DTT, 40 μg/ml BSA and 0.01% Tween-20. Reactions were terminated by the addition of 1 volume of gel loading buffer [99.5% (v/v) formamide, 5 mM EDTA, 0.01% (w/v) xylene cyanol, and 0.01% (w/v) bromophenol blue]. Samples were subjected to a 16% denaturing PAGE and dried gels were exposed to a PhosphorImager screen (GE Healthcare). Gel images were scanned using a Typhoon 8600 (GE Healthcare) and densitometric analyses were performed using the ImageQuant software (GE Healthcare).
Surface Plasmon Resonance Analysis
Binding experiments were performed as recently described.16 Briefly, Tdp1 was amine coupled to a CM5 sensor chip (GE Healthcare, Piscataway NJ). In order to protect the amine groups with the active site from modification, 1 mM Tdp1 was incubated with 2 mM of a 14 base oligonucleotide containing at phosphate group at the 3′ end (GATCTAAAAGACTT) in 10 mM sodium acetate pH 4.5 for 20 min. The CM5 chip surface was activated for 7 min with 0.1 M NHS and 0.4 M EDC at a flow rate of 20 mL/min and Tdp1-oligonucleotide mixture was injected until approximately 4000 RU’s was attached. Activated amine groups were quenched with an injection of 1 M solution of ethanolamine pH 8.0 for 7 min. Any bound oligonucleotide was removed by washing the surface with 1 M NaCl. A reference surface was prepared in the same manner without coupling of Tdp1. Compound 70 was diluted into running buffer [10 mM Hepes, 150 mM NaCl, 0.01% tween 20 (v/v), 5% DMSO (v/v) pH 7.5] and injected over all flow cells at 30 mL/min at 25 °C. Following compound injections, the surface was regenerated with a 30 second injection 1 M NaCl, a 30 s injection of 50% DMSO (v/v) and a 30 s running buffer injection. Each cycle of compound injection was followed by buffer cycle for referencing purposes. A DMSO calibration curve was included to correct for refractive index mismatches between the running buffer and compound dilution series.
Acknowledgments
This work was made possible by the National Institutes of Health (NIH) through support with Research Grant UO1 CA89566. This work was also supported by the NIH, National Cancer Institute R25CA128770 (D. Teegarden) Cancer Prevention Internship Program (M.C.-S.) administered by the Oncological Sciences Center and the Discovery Learning Research Center at Purdue University. This research was also supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. This project has been funded in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract no. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Abbreviations Used
- APCIMS
atmospheric pressure chemical ionization mass spectrometry
- BBO
multinuclear broadband observe
- EIMS
electron impact mass spectrometry
- ESIMS
electrospray ionization mass spectrometry
- QNP
quattro nucleus probe
- Tdp1
Tyrosyl-DNA-phosphodiesterase I
- Top1
topoisomerase
Footnotes
The authors declare no competing financial interest.
References
- 1.Interthal H, Pouliott JJ, Champoux JJ. The Tyrosyl-DNA Phosphodiesterase Tdp1 Is a Member of the Phospholipase D Superfamily. Proc Natl Acad Sci US A. 2001;98:12009–12014. doi: 10.1073/pnas.211429198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang SYN, Pommier Y, Marchand C. Tyrosyl-DNA Phosphodiesterase 1 (Tdp1) Inhibitors. Expert Opin Ther Pat. 2011;21:1285–1292. doi: 10.1517/13543776.2011.604314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davies DR, Interthal H, Champoux JJ, Hol WGJ. The Crystal Structure of Human Tyrosyl-DNA Phosphodiesterase, Tdp1. Structure. 2002;10:237–248. doi: 10.1016/s0969-2126(02)00707-4. [DOI] [PubMed] [Google Scholar]
- 4.Pommier Y, Leo E, Zhang HL, Marchand C. DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chem Biol. 2010;17:421–433. doi: 10.1016/j.chembiol.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dexheimer TS, Antony S, Marchand C, Pommier Y. Tyrosyl-DNA Phosphodiesterase As a Target for Anticancer Therapy. Anti-Cancer Agents Med Chem. 2008;8:381–389. doi: 10.2174/187152008784220357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davies DR, Interthal H, Champoux JJ, Hol WGJ. Insights into Substrate Binding and Catalytic Mechanism of Human Tyrosyl-DNA Phosphodiesterase (Tdp1) from Vanadate and Tungstate-inhibited Structures. J Mol Biol. 2002;324:917–932. doi: 10.1016/s0022-2836(02)01154-3. [DOI] [PubMed] [Google Scholar]
- 7.Strumberg D, Pilon AA, Smith M, Hickey R, Malkas L, Pommier Y. Conversion of Topoisomerase 1 Cleavage Complexes on the Leading Strand of Ribosomal DNA into 5′-Phosphorylated DNA Double-strand Breaks by Replication Runoff. Mol Cell Biol. 2000;20:3977–3987. doi: 10.1128/mcb.20.11.3977-3987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pommier Y, Cushman M. The Indenoisoquinoline Noncamptothecin Topoisomerase I Inhibitors: Update and Perspectives. Mol Cancer Ther. 2009;8:1008–1014. doi: 10.1158/1535-7163.MCT-08-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pommier Y, Marchand C. Interfacial Inhibitors: Targeting Macromolecular Complexes. Nat Rev Drug Discovery. 2012;11:25–36. doi: 10.1038/nrd3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beretta GL, Cossa G, Gatti L, Zunino F, Perego P. Tyrosyl-DNA Phosphodiesterase 1 Targeting for Modulation of Camptothecin-Based Treatment. Curr Med Chem. 2010;17:1500–1508. doi: 10.2174/092986710790979971. [DOI] [PubMed] [Google Scholar]
- 11.Pouliot JJ, Robertson CA, Nash HA. Pathways for Repair of Topoisomerase I Covalent Complexes in Saccharomyces cerevisiae. Genes Cells. 2001;6:677–687. doi: 10.1046/j.1365-2443.2001.00452.x. [DOI] [PubMed] [Google Scholar]
- 12.Antony S, Marchand C, Stephen AG, Thibaut L, Agama KK, Fisher RJ, Pommier Y. Novel High-Throughput Electrochemiluminescent Assay for Identification of Human Tyrosyl-DNA Phosphodiesterase (Tdp1) Inhibitors and Characterization of Furamidine (NSC 305831) as an Inhibitor of Tdp1. Nucleic Acids Res. 2007;35:4474–4484. doi: 10.1093/nar/gkm463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weidlich IE, Dexheimer T, Marchand C, Antony S, Pommier Y, Nicklaus MC. Corrigendum to “Inhibitors of Human Tyrosyl-DNA Phospodiesterase (hTdp1) Developed by Virtual Screening Using Ligand-based Pharmacophores” [Bioorg. Med. Chem. 18 (2010) 182–189] Bioorg Med Chem. 2010;18:2346–2346. doi: 10.1016/j.bmc.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weidlich IE, Dexheimer T, Marahiel MA, Antony S, Pommier Y, Nicklaus MC. Virtual Screening Using Ligand-based Pharmacophores for Inhibitors of Human Tyrosyl-DNA Phospodiesterase (hTdp1) Bioorg Med Chem. 2010;18:2347–2355. doi: 10.1016/j.bmc.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 15.Sirivolu VR, Vernekar SKV, Marchand C, Naumova A, Chergui A, Renaud A, Stephen AG, Chen F, Sham YY, Pommier Y, Wang Z. 5- Arylidenethioxothiazolidinones as Inhibitors of Tyrosyl–DNA Phosphodiesterase I. J Med Chem. 2012;55:8671–8684. doi: 10.1021/jm3008773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nguyen TX, Morrell A, Conda-Sheridan M, Marchand C, Agama K, Bermingam A, Stephen AG, Chergui A, Naumova A, Fisher R, O’Keefe BR, Pommier Y, Cushman M. Synthesis and Biological Evaluation of the First Dual Tyrosyl-DNA Phosphodiesterase I (Tdp1)-Topoisomerase I (Top1) Inhibitors. J Med Chem. 2012;55:4457–4478. doi: 10.1021/jm300335n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park EJ, Kondratyuk TP, Morrell A, Kiselev E, Conda-Sheridan M, Cushman M, Ahn S, Choi Y, White JJ, van Breemen RB, Pezzuto JM. Induction of Retinoid X Receptor Activity and Consequent Upregulation of p21(WAF1/CIP1) by Indenoisoquinolines in MCF7 Cells. Cancer Prev Res. 2011;4:592–607. doi: 10.1158/1940-6207.CAPR-10-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park EJ, Kiselev E, Conda-Sheridan M, Cushman M, Pezzuto JM. Induction of Apoptosis by 3-Amino-6-(3-aminopropyl)-5,6-dihydro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline via Modulation of MAPKs (p38 and c-Jun N-terminal Kinase) and c-Myc in HL-60 Human Leukemia Cells. J Nat Prod. 2012;75:378–384. doi: 10.1021/np200791j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morrell A, Antony S, Kohlhagen G, Pommier Y, Cushman M. Synthesis of Nitrated Indenoisoquinolines as Topoisomerase I Inhibitors. Bioorg Med Chem Lett. 2004;14:3659–3663. doi: 10.1016/j.bmcl.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 20.Morrell A, Antony S, Kohlhagen G, Pommier Y, Cushman M. A Systematic Study of Nitrated Indenoisoquinolines Reveals a Potent Topoisomerase I Inhibitor. J Med Chem. 2006;49:7740–7753. doi: 10.1021/jm060974n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morrell A, Placzek M, Parmley S, Antony S, Dexheimer TS, Pommier Y, Cushman M. Nitrated Indenoisoquinolines as Topoisomerase I Inhibitors: A Systematic Study and Optimization. J Med Chem. 2007;50:4419–4430. doi: 10.1021/jm070361q. [DOI] [PubMed] [Google Scholar]
- 22.Cushman M, Cheng L. Stereoselective Oxidation by Thionyl Chloride Leading to the Indeno[1,2-c]isoquinoline System. J Org Chem. 1978;43:3781–3783. [Google Scholar]
- 23.Antony S, Agama KK, Miao ZH, Takagi K, Wright MH, Robles AI, Varticovski L, Nagarajan M, Morrell A, Cushman M, Pommier Y. Novel Indenoisoquinolines NSC 725776 and NSC 724998 Produce Persistent Topolsomerase I Cleavage Complexes and Overcome Multidrug Resistance. Cancer Res. 2007;67:10397–10405. doi: 10.1158/0008-5472.CAN-07-0938. [DOI] [PubMed] [Google Scholar]
- 24.Morrell A, Antony S, Kohlhagen G, Pommier Y, Cushman M. Synthesis of Nitrated Indenoisoquinolines as Topoisomerase I Inhibitors. Bioorg Med Chem Lett. 2004;14:3659–3663. doi: 10.1016/j.bmcl.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 25.D’Hooghe M, Dekeukeleire S, De Kimpe N. Reactivity of N-(Omega-haloalkyl)-Beta-Lactams with Regard to Lithium Aluminium Hydride: Novel Synthesis of 1-(1-Aryl-3-hydroxypropyl)aziridines and 3-Aryl-3-(N-propylamino)propan-1-ols. Org Biomol Chem. 2008;6:1190–1196. doi: 10.1039/b719686e. [DOI] [PubMed] [Google Scholar]
- 26.Nagarajan M, Morrell A, Fort BC, Meckley MR, Antony S, Kohlhagen G, Pommier Y, Cushman M. Synthesis and Anticancer Activity of Simplified Indenoisoquinoline Topoisomerase I Inhibitors Lacking Substituents on the Aromatic Rings. J Med Chem. 2004;47:5651–5661. doi: 10.1021/jm040025z. [DOI] [PubMed] [Google Scholar]
- 27.Morrell A, Antony S, Kohlhagen G, Pommier Y, Cushman M. Synthesis of Benz[d]indeno[1,2-b]pyran-5,11-diones: Versatile Intermediates for the Design and Synthesis of Topoisomerase I Inhibitors. Bioorg Med Chem Lett. 2006;16:1846–1849. doi: 10.1016/j.bmcl.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 28.Mal D, Jana AK, Mitra P, Ghosh K. Benzannulation for the Regiodefined Synthesis of 2-Alkyl/Aryl-1-naphthols: Total Synthesis of Arnottin I. J Org Chem. 2011;76:3392–3398. doi: 10.1021/jo2003677. [DOI] [PubMed] [Google Scholar]
- 29.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved Protein-Ligand Docking Using GOLD. Protein Struct Funct Genet. 2003;52:609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 30.Liu H, Zang C, Emde A, Planas-Silva MD, Rosche M, Kuhnl A, Schulz CO, Elstner E, Possinger K, Eucker J. Anti-tumor Effect of Honokiol Alone and in Combination with Other Anti-cancer Agents in Breast Cancer. Eur J Pharmacol. 2008;591:43–51. doi: 10.1016/j.ejphar.2008.06.026. [DOI] [PubMed] [Google Scholar]
- 31.Dexheimer TS, Pommier Y. DNA Cleavage Assay for the Identification of Topoisomerase I Inhibitors. Nat Protocol. 2008;3:1736–1750. doi: 10.1038/nprot.2008.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cushman M, Jayaraman M, Vroman JA, Fukunaga AK, Fox BM, Kohlhagen G, Strumberg D, Pommier Y. Synthesis of New Indeno[1,2-c]isoquinolines: Cytotoxic Non-Camptothecin Topoisomerase I Inhibitors. J Med Chem. 2000;43:3688–3698. doi: 10.1021/jm000029d. [DOI] [PubMed] [Google Scholar]
- 33.Antony S, Jayaraman M, Laco G, Kohlhagen G, Kohn KW, Cushman M, Pommier Y. Differential Induction of Topoisomerase I-DNA Cleavage Complexes by the Indenoisoquinoline MJ-III-65 (NSC 706744) and Camptothecin: Base Sequence Analysis and Activity against Camptothecin-Resistant Topoisomerase I. Cancer Res. 2003;63:7428–7435. [PubMed] [Google Scholar]
- 34.Antony S, Kohlhagen G, Agama K, Jayaraman M, Cao S, Durrani FA, Rustum YM, Cushman M, Pommier Y. Cellular Topoisomerase I Inhibition and Antiproliferative Activity by MJ-III-65 (NSC 706744), an Indenoisoquinoline Topoisomerase I Poison. Mol Pharmacol. 2005;67:523–530. doi: 10.1124/mol.104.003889. [DOI] [PubMed] [Google Scholar]
- 35.Kiselev E, DeGuire S, Morrell A, Agama K, Dexheimer TS, Pommier Y, Cushman M. 7-Azaindenoisoquinolines as Topoisomerase I Inhibitors and Potential Anticancer Agents. J Med Chem. 2011;54:6106–6116. doi: 10.1021/jm200719v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. J Natl Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 37.Reinhold WC, Sunshine M, Liu H, Varma S, Kohn KW, Morris J, Doroshow J, Pommier Y. CellMiner: A Web-Based Suite of Genomic and Pharrmacologic Tools to Explore Transcript and Drug Patterns in the NCI-60 Cell Line Set. Cancer Res. 2012;72:3499–3511. doi: 10.1158/0008-5472.CAN-12-1370. [DOI] [PMC free article] [PubMed] [Google Scholar]