

Abstract
The phosphoinositide 3-kinases (PI3Ks) are heterodimers consisting of the catalytic subunit p110 and the regulatory subunit p85. The PI3K/Akt pathway is strongly deregulated in breast cancer (BC) representing one of the mechanisms of resistance to therapies. Therefore, the identification of inhibitors of PI3K components represents one of the main goals to produce therapeutic agents. Here, we evaluated the efficacy of a phosphopeptide 1257 (P-1257) that targeting p85 strongly inhibits PI3K activity. We tested the effects of P-1257 administration in vitro and in vivo using BC cells expressing different levels of ErbB-2 and resistant or responsive to Trastuzumab. We demonstrated that inhibition of p85 activity by P-1257 induces cell death and sensitizes JIMT-1 and KPL-4 ErbB-2-overexpressing BC cells to Trastuzumab treatment. It is noteworthy that P-1257 delivery in vivo by electroporation or liposomes significantly inhibits the proliferation of tumor cells engrafted at subcutaneous and visceral sites. Overall, our data indicate that the p85 subunit is a valid target for therapeutic approaches and suggest that the structure of the peptide used in our study could be utilized for the development of novel drugs to apply in combination with therapies that fail to cure BCs with high PI3K activity.
Keywords: breast cancer, tumor promotion and progression, cell signaling, growth factors and receptors, oncogenes
The phosphoinositide 3-kinase (PI3K) are heterodimers consisting of two components, the catalytic subunit p110 and the regulatory subunit p85.1 Among this class of enzymes the most studied are class I that are activated by cell surface receptors. After cell stimulation and subsequent activation of receptors tyrosine kinases, PI3K is recruited to the cell membrane by the direct interaction of p85 with the phosphorylated tails of receptors, for example, the epidermal growth factor receptor family members, such as ErbB-3, or with the adaptor proteins associated with receptors, for example, the insulin receptor substrate-1 (IRS-1).1 These enzymes are often deregulated in cancer-promoting cell growth, proliferation and survival.2 Many components of this pathway are mutated, often by activating mutations and gene amplification, or loss of function of PTEN.3, 4 As hyperactivation of PI3K/Akt pathway is found in a large variety of human cancers and is associated with mechanisms of resistance to drugs, in the last years many components of the PI3K/Akt pathway have become targets for therapeutics intervention.5 Some inhibitors of the PI3K/Akt pathway are currently used in clinical trials as anticancer agents, however, when used against breast cancers (BCs), they show modest effects either in vitro or in vivo. All these results are in agreement with a vast literature, which indicates PI3K as one of the major and inter-dependent oncogenic kinases that contribute to resistance to new drugs such as Trastuzumab.6
Overexpression of the ErbB-2 oncogene has been detected in a variety of human tumors. These tumors show poor prognosis and high rates of recurrence and mortality.7, 8, 9, 10 These tumors are currently treated with chemotherapy in combination with Trastuzumab, a humanized monoclonal antibody that binds ErbB2 inducing its endocytosis and degradation.11, 12, 13 However, it must be emphasized that the majority of metastatic ErbB-2-positive BCs become resistant to Trastuzumab suggesting that these tumors possess mechanisms that can induce therapeutic resistance.14, 15, 16, 17, 18 The ErbB-3 receptor possesses six binding sites directed to the N-terminal SRC homology 2 domain (N-SH2) of p85 and the ErbB-2/ErbB-3 heterodimer is one of the strongest stimulators in nature of the PI3K/Akt pathway, which has been reported to abrogate Trastuzumab responsiveness.18, 19, 20, 21, 22 Moreover, BCs overexpressing ErbB-2 or derived from HER2/neu transgenic mice have high levels of ErbB-3.23, 24, 25 In line with these results it has been demonstrated that EbB-3 ablation hinders PI3K-dependent tumorigenesis and favors Tamoxifen responsiveness in hormone-dependent BCs.26, 27 For these reasons, PI3K became one of the major targets for the development of new therapeutics.28, 29, 30 With the aim of inhibiting the PI3K pathway in metastatic ErbB-2-overexpressing BCs, we decided to target p85 by the use of a phosphopeptide 1257 (P-1257) specifically directed to the N-SH2 domain of the molecule. The P-1257, previously described by Suenaga et al.,31 corresponds to one of the ErbB-3 cytoplasmic-binding domain carrying the tyrosine phosphorylation site 1257 and a methionine residue in position 3 after the tyrosine phosphorylation site, indicating that it could inhibit other p85 binding proteins.32 Our current study demonstrates that interference with p85 function by P-1257 inhibits ErbB-3 and IRS-1–p85 interaction, inhibits PI3K activity impacting on cell proliferation in vitro and in vivo, thus exerting strong antitumor activity.
Results
P-1257 inhibits in vitro and in vivo ErbB-3 and IRS-1–p85 interaction
To evaluate the efficacy of P-1257 for therapeutic approaches, we first evaluated its capability to inhibit the interaction of p85 with molecules whose pathways are associated with drug resistance. As negative control, we used a scrambled phosphopeptide (P-scr) designed not to interact with the SH-2 domain or the phosphotyrosine-binding domain of p85. We analyzed the ability of P-1257 to inhibit, in vitro and in vivo, the interaction between ErbB-3 and p85 or IRS-1 and p85. To this end, we chose the JIMT-1 and KPL-4 cell lines, both overexpressing ErbB-2 but resistant to Trastuzumab, BT474 cells overexpressing ErbB-2 and responsive to Trastuzumab treatment, and MCF7 cells expressing low ErbB-2 and unresponsive to Trastuzumab.33, 34 All these cell lines retained PTEN activity.33, 34 Using a GST fusion protein in frame with the N-SH2 domain of p85, we measured the in vitro capability of ErbB-3 to interact with the N-SH2 domain of p85 in the absence or presence of P-1257 or P-scr peptides. Interestingly, we found that P-1257 abolished the in vitro interaction between ErbB-3 and p85 in all four-cell lysates compared with control lysates in the absence or presence of the P-scr (Figures 1a and b, upper, left and right panels). In agreement with previous findings,31 we confirmed that the non-phosphorylated 1257 peptide was unable to interact with the N-SH2 domain of p85 and to inhibit the in vitro ErbB-3–p85 interaction (Figure 1c). The results of GST pull-down were confirmed in vivo on transfection of either the P-1257 or P-scr peptides in JIMT-1, KPL-4, MCF7 and BT474 cells. Immunoprecipitation of p85 from total lysates revealed that the ErbB-3–p85 interaction was abolished or strongly reduced in P-1257 transfected cells, compared with lysates derived from control or P-scr-transfected cells, demonstrating that P-1257 is a strong inhibitor of ErbB-3–p85 interaction (Figures 1a and b, lower, left and right panels). As expected, an unrelated secondary antibody (IgG) was unable to co-immunoprecipitate ErbB-3/p85 (Figures 1a and b, lower, left and right panels). The p85 protein levels found in the immune-complexes demonstrated that equivalent amount of protein was immune-precipitated in each experimental condition (Figures 1a and b, lower panels). As previously suggested,32 the P-1257 peptide could inhibit other N-SH2-p85 binding proteins. To verify this hypothesis, we performed GST pull-down and found that the P-1257 peptide was able to reduce the p85–IRS-1 interaction in MCF7 cells (Figure 1d) suggesting that this peptide possesses a strong ability to inactivate PI3K.
Figure 1.

P-1257 peptide blocks ErbB-3–p85 and IRS-1–p85 interactions in vitro and in vivo. (a and b, upper, left and right panels) Total cell extracts from JIMT-1, KPL-4, BT474 and MCF7 cells were incubated with glutathione-agarose beads bound to GST or GST-N-SH2-p85 domain fusion protein, in the absence or presence of either P-1257 or P-scr. Complexes of GST alone (–) or GST-N-SH2-p85 (p85) interaction were separated by SDS-PAGE and immunoblotted with anti ErbB-3 antibody. (a and b, lower, left and right panels) Total cell extracts from JIMT-1, KPL-4, MCF7 and BT474 cells transiently transfected with either P-scr or P-1257 peptides were immunoprecipitated with anti-p85 antibody or with rabbit IgG as negative control. Immunocomplexes were analyzed by immunoblotting with an anti-ErbB3 antibody. Immunoblotting with anti-p85 antibody show the equivalent amount of total protein immunoprecipitated. (c and d) Total cell extracts from MCF7 cells were incubated with GST alone (–) or GST-N-SH2-p85 (p85) as described above and in the presence of non-phosphorylated 1257 peptide. The interactions were analyzed by immunoblotting with (c) an anti-ErbB3, or (d) with anti-IRS-1 antibody. Each experiment was repeated at least three time and aliquots derived from each experiment were pooled and separated by SDS-PAGE
P-1257 delivery in vitro downregulates Akt and MAPK activity and induces cell death
Having established that the P-1257 peptide inhibits p85 interaction with ErbB-3 and IRS-1 proteins, we evaluated its biological effects in the JIMT-1, KPL-4 and BT474 cell lines. Cells were transfected with either P-1257 or P-scr peptides and through a dose-response experiment we established the functional amount of P-1257 required to inhibit the level of Akt phosphorylation in each cell line, (Supplementary Figure S1). Then, we analyzed the biochemical and biological effects of P-1257 in vitro. As expected, treatment with P-1257 alone abolished Akt phosphorylation in JIMT-1 and KPL-4 cells and surprisingly inhibited ERK1/2 phosphorylation in KPL-4 cells. The combinatory treatment with P-1257 and Trastuzumab inhibited ERK1/2 phosphorylation in both cell lines (Figures 2a and b, left panels). Interestingly, we found that P-1257 caused a significant reduction of ErbB-3 and ErbB-2 expression, suggesting that this peptide, by inhibiting the PI3K/Akt and MAPK pathways, might cause internalization and/or a negative regulation of both proteins at the translational level. Indeed, we previously demonstrated that PI3K inactivation induced negative regulation of ErbB-3 at the translational level in breast carcinoma cells and that this regulation was both rapamycin sensitive and dependent on the eukaryotic translation initiation factor 4E.35 As expected, we found that both pathways were active in control or P-scr transfected cells demonstrating the specificity of P-1257 in inhibiting the PI3K/Akt pathways (Figures 2a and b, left panel). P-1257 induced substantial cell death in both cell lines, which was further increased on Trastuzumab treatment indicating a re-sensitization of the cells to Trastuzumab (JIMT-1 cells P<0.0002 in P-1257 versus P-scr+T treatment or versus the others controls and P<0.01 in P-1257+T versus P-1257 treatment; KPL-4 cells P<0.0006 in P-1257 versus P-scr+T treatment or versus the other controls and P<0.05 in P-1257+T versus P-1257 treatment; Figures 2a and b, right upper panels). Analysis of PARP indicated that cell death was due to apoptosis in both cell lines (Figures 2a and b, right lower panels). Next, we evaluated the biological effects of P-1257 also in Trastuzumab-responsive BT474 cells. Transfection of P-1257 inhibited Akt phosphorylation also in these cells although to a lower extent than in JIMT-1 and KPL-4 cells. The concomitant addition of Trastuzumab abrogated also ERK1/2 phosphorylation (Figure 2c, left panel). As expected, in BT474 control and P-scr transfected cells there was significant inhibition of ERK1/2 phosphorylation on Trastuzumab treatment (Figure 2c, left panel). However, owing to the shortness of this treatment and the high expression of ErbB-2, we were not able to appreciate the downregulation of ErbB-2 in parental and in P-scr transfected BT474 cells (Figure 2c, left panel). In agreement with previous results35 and those obtained in JIMT-1 and KPL-4 cells, we found downregulation of ErbB-3 expression on P-1257 treatment also in BT474 cells. As expected, analysis of cell death in BT474 cells revealed no significative differences among treatments with Trastuzumab, P-scr plus Trastuzumab, and P-1257 in the absence or in the presence of Trastuzumab (Figure 2c, right upper panel). In these cells too, analysis of PARP indicated that cell death on Trastuzumab and/or P-1257 treatments was due to apoptosis (Figure 2c, right lower panel). As the biochemical and biological effects of P-1257 were very strong, we verified the in vitro stability of the peptide. MCF7 cells were transfected with P-1257 and the level of Akt phosphorylation was evaluated. We found that P-1257 inhibited Akt phosphorylation until day 6 post-transfection demonstrating its strong stability in the cells (Figure 3a, upper panel). To further establish the specificity of P-1257 in inhibiting PI3K, we used LY294002, a well-known PI3K inhibitor. Interestingly, we found in both KPL-4 and JIMT-1 cells that LY294002 addition at time 0 and after 48 h from the first treatment almost abolished P-Akt 24 h after each treatment. However, we also found that the phosphorylation status of Akt was recovered at these times (Figures 3b and c, upper panels). Analysis of the cells during LY294002 treatment revealed that the cells were alive suggesting that the effect of LY294002 at least in these cells was transient and not toxic (data not shown).
Figure 2.

P-1257 peptide affecting PI3K/Akt and MAPK pathways favors Trastuzumab responsiveness and induces apoptosis. (a–c, left panels) JIMT-1, KPL-4 and BT474 parental cells, transiently transfected with P-scr or P-1257 peptides, were maintained in the presence or absence of Trastuzumab. Total cell extracts were analyzed by immunoblotting with ErbB-3, ErbB-2, P-Akt and P-ERK1/2 antibodies. Antibodies specific for Hsp-70, total Akt and ERK1/2 were used for loading control. (a–c, right upper panels) cell viability was evaluated by Trypan blue exclusion. Percentage of cell death was evaluated from three independent experiments; bars±S.D. (a) Right panel, JIMT-1 cells: ***P<0.0002; and *P<0.01. (b) Right panel, KPL-4 cells: ***P<0.0006 and *P<0.05. (c) Right panel, the cell death evaluated in BT474 cells was not significantly different between P-1257 and P-1257+T versus T alone or P-scr+T. (a–c, right lower panels) Total cell extracts were analyzed by immunoblotting with anti-intact human PARP antibody and anti-Hsp-70 for loading control
Figure 3.
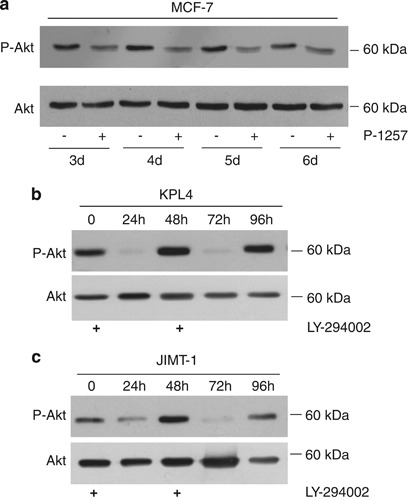
P-1257 inhibits PI3K/Akt until day 6 post-transfection. (a) Total cell extracts from MCF7 cells, control or transfected with P-1257, were separated by SDS-PAGE and analyzed by immunoblotting with anti-total (lower panel) or P-Akt (upper panel) from day 3 to 6 post-transfection. (b and c) Total cell lysates from KPL-4 and JIMT-1 cells untreated or treated with LY294002 at time 0 and after 48 h were separated by SDS-PAGE and immunoblotted with anti-total (lower panel) or P-Akt antibodies (upper panel) at different times as indicated in the figure
In vivo delivery of P-1257 strongly reduces tumor mass of BC engrafts derived from resistant JIMT-1 cells
To verify the possibility of employing P-1257 in cancer therapy, we performed in vivo experiments. JIMT-1 cells (1 × 107 cells per mouse) were injected subcutaneously (s.c.) in the intra-scapular region of nude (nu/nu) mice and tumors were allowed to grow until they reached a volume of 200 mm3. Mice were then divided in two groups and treated by intratumoral injection with either P-1257 or P-scr, as control, followed by electroporation to favor the entry of the phosphopeptides. In addition, half of each group of animals received Trastuzumab. As further controls, we followed tumor growth in: (i) untreated mice (ctrl); (ii) mice treated exclusively by electroporation (ctrl electr); (iii) mice treated exclusively with Trastuzumab (ctrl+T). The experiment was repeated twice and eight mice for each experimental condition were used. P-peptides were administered once a week for three times, as indicated by the arrows in Figure 4. Trastuzumab was administered twice a week by intraperitoneal (i.p.) injection until the animals were killed. Tumor growth was followed by caliper measurements twice a week. As previously observed in vitro, P-1257 strongly inhibited tumor growth compared with controls tumors (ctrl, ctrl electr, ctrl+T, P-scr and P-scr+T: P<0.0005). This effect was augmented in vivo by combined treatment of P-1257 with Trastuzumab when compared with control tumors or P-1257 treatment alone (P<0.00029 and P<0.0003, respectively) indicating additive effects of the combined treatment. Indeed, as previously described,33 we found a slight response to Trastuzumab of JIMT-1-derived tumors, which after 7 weeks (T or P-scr+T) started to grow exponentially as the control tumors (Figure 4a, left panel). Representative tumor masses at week 9 of treatment are shown in Figure 4b. Tumor growth of P-1257 plus or minus Trastuzumab treatment was followed until week 14 and no differences were detected. All control tumors grew exponentially and were followed until established endpoints.
Figure 4.

P-1257 in vivo delivering inhibits tumor growth and favors Trastuzumab responsiveness of JIMT-1-derived tumors. (a, left panel) JIMT-1-derived tumors were allowed to grow until 200 mm3. Then, P-1257 or P-scr peptides were administered by direct intratumor injection followed by electroporation. Treatment was performed once a week for 3 weeks as indicated by the arrows. Half of each group was treated with Trastuzumab. Untreated JIMT-1-derived tumors, tumors electroporated and tumors treated with Trastuzumab alone were used as further controls. Tumor development was followed twice a week by caliper measurements. P-values are reported in the graphs (JIMT-1 tumors, ***P<0,0005 in the treatment of P-1257 versus control tumors; ***P<0.00029 in the treatment of P-1257+T versus control tumors; ***P<0.0003 in the treatment of P-1257+T versus P-1257 alone). (b) Representative tumor masses were reported for each experimental condition
Treatment with P-1257 inhibits growth also of MCF7-derived tumors
The survival of MCF7 cells depends only in part from ErbB-3;34 moreover these cells carry a mutated form of mutated p110 catalytic subunit of PI3 (PIK3CA),36 and express low level of ErbB-2.34 Interestingly, we found that P-1257 was able to inhibit in vitro p85–IRS-1 interaction in these cells; a pathway that is downstream to PI3K.27 Thus, we verified whether P-1257 delivery was able to inhibit growth of MCF7-derived tumors. The cells were injected s.c. in immunocompromised mice (SCID) and treatments were performed on established tumors (200 mm3) by intratumoral injection with either P-1257 or P-scr followed by electroporation and Trastuzumab as described above. We followed tumor growth in untreated mice, mice treated exclusively by electroporation, or exclusively with Trastuzumab. We found that P-1257 inhibited tumor growth compared with all control tumors (ctrl, ctrl electr, ctrl+T, P-scr and P-scr+T) (P<0.05). As expected, P-1257 in combination with Trastuzumab did not further inhibit MCF7 tumor growth (P<0.16; Figure 5 left and right panels). As in this cell line P-1257 inhibits p85–IRS-1 interaction, this result suggests that the PI3K/IRS-1 pathway supports at least in part the survival of MCF7 cells. Representative tumor masses at week 9 of treatment are shown in Figure 5b.
Figure 5.

P-1257 in vivo delivering inhibits tumor growth also of engrafts derived from ErbB-2 low expressing MCF7 cells. (a, left and right panels) MCF7-derived tumors were allowed to grow until 200 mm3. Then P-1257 or P-scr peptides were administered by direct intratumoral injection followed by electroporation and tumor masses were treated with P-peptides and Trastuzumab as described above for JIMT-1-derived tumors. Untreated MCF7-derived tumors, tumors electroporated and tumors treated with Trastuzumab alone were used as further controls. Tumor development was followed twice a week by caliper measurements as reported in the graphs (MCF7 tumors, *P<0.05 in the treatment of P-1257 versus control tumors; *P<0.16 in the treatment of P1257+T versus P-1257). (b) Representative tumor mass were reported for each experimental conditions
Treatment with P-1257 inhibits cell proliferation and induces necrosis in vivo
The strong reduction of tumor growth caused by P-1257 in both BC engrafts prompted us to verify whether the P-1257 treatment induced apoptosis in vivo. To confirm this hypothesis, we analyzed, by TUNEL assay, the ability of P-1257 or Trastuzumab, alone or in combination, to trigger apoptosis in BC engrafts derived from JIMT-1 and MCF7 cells. The results show that P-1257 fails to induce significant apoptosis in vivo suggesting that apoptosis is an early event that can be appreciated in vitro but not in vivo after several weeks of treatment (data not shown). Then, we determined proliferation indexes of JIMT-1 and MCF7 BC engrafts using the Mib-1 (Ki-67) monoclonal antibody. Tumors from untreated mice had significantly higher proliferation index (73%) than those from mice treated with P-1257 and Trastuzumab (mean 29%, with a P<0.0001). Staining with hematoxylin–eosin of BC engrafts from controls and treated tumors revealed necrotic areas in tumors that received the combined therapy (Figure 6b).
Figure 6.

In vivo administration of P-1257 in combination with Trastuzumab inhibits cell proliferation and induces necrosis. (A and B, left panels) JIMT-1 and MCF7: proliferation index, using the anti-ki-67 monoclonal antibody (clone MIB-1), variance of Trastuzumab resistant JIMT-1 and MCF7 BC engrafts as detected by the Bonferroni test. Ki-67 proliferation index, expressed as mean percentage, in engrafts untreated, electroporated and treated with T alone compared with engrafts treated with P-1257 or P-1257 plus T (P<0.0001). (A and B, a/a-d and b/a-d, right panels) Two immunohistochemical explicative panels are provided. Pictures show that the proliferation index is higher (73% a-a; b-a) and the necrosis, as detected by hematoxylin-eosin colorimetric assay, is lower (a-c; b-c) in untreated mice than in 1257+Trastuzumab treated mice (29% A-b; B-b and A-d; B-d), respectively. Scale bar=30 μ
Treatment with P-1257-containing liposomes strongly inhibits tumor growth at visceral site
We have shown that locally administered P-1257 inhibits the proliferation of tumor cells engrafted at s.c. sites. We next asked whether tumor cells engrafted at a visceral site could be successfully targeted by a P-1257 formulation administered via a systemic route. To this end, we injected MCF7 cells stably expressing the luciferase reporter gene in the tail vein of SCID mice. Four-weeks post-injection lung colonization was readily detected (Figure 7a). Animals were then treated once a week for the subsequent 5 weeks by intravenous (i.v.) injection of control or P-1257-loaded liposomes. Half of the mice in each group were also treated with Trastuzumab. One week after the end of the dosage schedule, the animals were injected i.p with 𝒟-luciferin and killed to analyze lung tumors. Results showed that mice treated with control liposomes had large tumors covering the entire surface of the lung 10 weeks post i.v. injection. In contrast, we observed a significant inhibition of tumor growth in the lungs of mice treated with P-1257-loaded liposomes (P<0.0004 in P-1257 versus ctrl or versus ctrl+T treatment). Combining Trastuzumab with P-1257-loaded liposomes did not provide additional benefit (P<0.3; Figures 7a and b, upper panels). Representative lung tumors at 4-week post-injection and at the end of treatment, visualized by light emission, are shown in Figure 7b, lower panels. These results provide a proof of principle that p85 can be subjected to therapeutic targeting in order to curb oncogenic signaling by PI3K.
Figure 7.

Therapy by liposomes containing P-1257 inhibits tumor growth at the visceral site. (a, left panel) Total photon flux of lung metastases derived from MCF7 cells injected i.v in SCID mice 4 weeks post-injection. (a, right panel) At week 4 post-injection the animals were treated once a week for five times with liposomes empty or containing P-1257. Half of each group of mice was treated with Trastuzumab (***P<0.0004 P-1257 versus ctrl or ctrl+T treatment; P<0.3 P-1257+T versus P-1257 treatment). (b) Representative lung engraftments at week 4 post-injection and at the end of treatments (ctrl, ctrl+T, P-1257, P-1257+T) were visualized by light emission
Discussion
It is widely known that signaling downstream of growth factor receptors is involved in the mechanisms that promote tumor progression and resistance to apoptotic stimuli. One of these mechanisms involves the level of ErbB-2 homodimers1 and/or ErbB2/ErbB3 heterodimers,1, 16, 21, 22, 23, 24, 25 whose expression induces strong activation of the MAPK and PI3K/Akt pathways and regulates survival and proliferation of many tumors. Downstream many growth factor receptor signaling there is the PI3K/Akt pathway, which is involved in the resistance to targeted therapies. Although the results are not definitive and not all are as expected, few new drugs that target PI3K appear promising.31 Indeed, the extensive effort dedicated to produce new drugs targeting this pathway underlines the pivotal role of PI3K in tumor progression.
The involvement of PI3K activity in the mechanism of resistance to Trastuzumab in metastatic ErbB-2-overexpressing BCs is becoming apparent.17, 21, 22, 27, 28, 29, 30, 31 Although the role played by ErbB-2 in BC prognosis and therapeutic response is clear, less is known on the mechanisms that determine resistance to Trastuzumab.37, 38 One mechanism of this resistance involves activation of the tyrosine kinase SRC.38 Although this is not the only mechanism, it is clear that the loss of PTEN increases SRC phosphorylation, which in turn confers in vivo resistance to Trastuzumab38 as well as mutations of several components of the pathway.3, 4 Here we have demonstrated that the use of the P-1257 phosphopeptide interfering with the function of the non-catalytic subunit of PI3K, p85, induces cell death. Specifically, we demonstrated that treatment of JIMT-1 and KPL-4 cells with P-1257 inhibits the p85/ErbB-3 and/or IRS-1 complexes formation, inhibits PI3K and MAPK activities, induces apoptosis and overcomes Trastuzumab resistance. The transient inhibition of PI3K and the absence of toxic effects observed in JIMT-1 and KPL-4 cells during LY294002 treatment, a specific inhibitor of PI3K, indicates the specificity of P-1257 in inhibiting an important survival pathway in these cells. This might be explained by different reasons: first, P-1257 being very stable in the cells (up to 6 days) might become available for many interactions further reducing cell proliferation; second, P-1257 treatment reduces both ErbB-2 and ErbB-3 expression on the cell surface and, as a consequence, their proliferation and survival signaling; third, the combined treatment of P-1257 with Trastuzumab in JIMT-1 and KPL-4 cells further increases cell death suggesting that the residual ErbB-2 activity, independent from ErbB-3, could become responsive to Trastuzumab.
The use of peptides in therapy is recent and, although introducing hydrophilic molecules such as phosphopeptides into cells or tumors represents a limiting issue for therapy, there are several methods aimed at increasing uptake efficiency in vivo and sophisticated procedures are being adopted such as electroporation.39 We found that in vivo delivery of P-1257 inhibits cell proliferation, induces necrosis that results in strong reduction of tumor mass formation that is further reduced when administrated in combination to Trastuzumab. Furthermore, P-1257 inhibits tumor growth and reduces metastasis formation also of ErbB-2-negative MCF7 engrafts. This result was surprising because these cells express an activating mutation of the catalytic subunit of PI3K (PIK3CA).40 In this cell line, P-1257 inhibits ErbB-3 and IRS-1/p85 complexes formation suggesting that the inactivation of Akt phosphorylation we observed on the phosphopeptide treatment could be sufficient to inhibit PI3K activity despite the mutation of PIK3CA.
The strong biological effects induced by the P-1257 peptide itself suggest that its mechanism of action may go beyond the inhibition of the interaction between p85 and ErbB-3 or IRS-1. The presence of a methionine residues after the tyrosine 1257 phosphorylation site suggests that the peptide could inhibit the interaction between the N-SH2 domain of p85 and other upstream or downstream molecules whose signaling are strongly related to cell proliferation and survival. This hypothesis supports the notion that this peptide could possess strong potentiality to inhibit the PI3K pathway.
Overall, our data indicate that p85 is a valid target for therapeutic approaches and that the structure of the P1257 peptide supports the possibility of developing novel therapeutic drugs to apply in combination with therapies that fail to cure metastatic BCs.
Materials and Methods
Cell lines
The human BC cell lines KPL4 and JMT-1 were obtained from Dr. G Tortora (33); MCF7 and BT474 were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The cell lines were maintained in RPMI or DMEM medium supplemented with 10% FBS, 1% penicillin/streptomycin and 1% glutamine (Invitrogen, Milan, Italy).
Antibodies
Rabbit anti-Phospho-ser473 (#9271) and total-Akt (#9272), phospho- (#9101L) and total-ERK 1/2 (#9102), PARP (#9542) and IRS-1 (#2382) antibodies were obtained from Cell Signaling, Milan, Italy. Rabbit anti-ErbB-2 (#Ab-1), ErbB-3 (sc-285), mouse anti-Hsp-70 (N27F34), mouse anti-intact human PARP (#42) antibodies were from Oncogene (Cambridge, MA, USA), Santa Cruz Biotechnology (Santa Cruz, CA, USA), Stressgen (New York, NY, USA) and Biosciences (San Jose, CA, USA), respectively. Mouse and rabbit HRP-conjugated secondary antibodies were from Bio-Rad (Milan, Italy). The rabbit anti-p85 (#06-496) was from Upstate Biotechnology (Millipore, Milan, Italy). The Ki-67 antibody (MIB-1) was from Dako (Milan, Italy).
GST pull-down
KPL-4, JIMT-1, MCF-7 and BT474 cell lines were washed twice with cold PBS and proteins extracted in 0,1% Triton X-100 Lysis buffer. After 20 min on ice, lysates were clarified by centrifugation at 14 000 r.p.m for 20 min. Cell extracts (1 mg per sample) were incubated with 60 μl of glutathione-agarose beads bound to GST or to GST-N-SH2-p85 fusion protein in the presence or absence of P-1257 or P-scr. We used 80 μg for BT474 and MCF7, 150 μg for KPL-4 and 180 μg for JIMT-1 cells, respectively, or the P-scr or non-phosphorylated 1257 peptide using the same concentration for each cell line as above reported. The lysates were incubated for 4 h at 4 °C, protein complexes were washed with Lysis buffer and solubilized with Laemmli Buffer 2 × . Samples were separated by 8% SDS-PAGE and transferred to nitrocellulose. The proteins were detected by western blot analysis with anti-ErbB-3 antibody. All the experiments were repeated three times obtaining equivalent results.
Peptide transfection
Peptides: unphosphorylated (1257), phosphorylated RDGGGPGGDpYAAMGACPA (P-1257), and PYQMRpYNADTDGERTTEP (P-scr) were synthesized by INBIOS (Naples, Italy). To inhibit p85–ErbB-3 or p85–IRS-1 interactions, cells (5 × 105) were plated in 60 mm dish and transfected with either P-1257 or P-scr peptides by Lipofectamine 2000 reagent (Invitrogen), following the manufacturer's procedures. The peptides were used at the following concentration (80 μg for BT474 and MCF7, 150 μg for KPL-4 and 180 μg for JIMT-1 cells) as above reported for the GST pull-down experiments.
Treatments, western blots and immunoprecipitation
KPL-4 and JIMT-1 cells, resistant to Trastuzumab,33 and BT474-responsive cells, were plated as above reported in 60 mm dish. After 24 h, the cells were transfected with P-scr or P-1257 peptides (stock solution 5 mg/ml in PBS containing 0.5% glycerol) and 3 h later treated with Trastuzumab at the final concentration of 10 μg/ml or with PBS as control. At the end of treatment, the cells were lysed with RIPA buffer, as previously described,35 to analyze ErbB-3, ErbB-2, Phospho- and Total-Erk 1/2 and Hsp-70 proteins or NP40 buffer to analyze Phosho- and Total Akt. Samples were analyzed by SDS-PAGE and transferred onto nitrocellulose membrane (Bio-Rad). The blots were probed with antibodies of interest, washed and developed with HRP-conjugated secondary antibodies and enhanced chemiluminescence (Lite-blot-Euroclone, Milan, Italy). Same amount of total protein derived from three independent experiments were pooled and analyzed as above described.
KPL-4 and JIMT-1 cells were treated for 24 and 48 h with LY-294002 (Roche, Milan, Italy) at the final concentration of 10 μM.
Cell death and apoptosis
KPL-4, JIMT-1, MCF-7 and BT474 cell lines were plated at concentration of 5 × 105 per 60-mm dish. The next day, the cells were transfected with P-1257 or P-scr peptides and treated with Trastuzumab for 40 hr. Cell vitality was evaluated by Trypan blue exclusion and apoptosis by western blot with anti-PARP antibodies as above reported. The chromogenic in situ detection of apoptosis in formalin-fixed, paraffin-embedded mouse tumor tissues was performed by TdTmediated dUTP-biotin nick end labeling assay (TUNEL Apoptosis Detection Kit, GenScript USA Inc., Piscataway, NJ, USA).
Animal studies and tumor therapy by electroporation
All procedures involving animals and their care were conducted in conformity with the institutional guidelines. Engrafts from JMT-1 or MCF7 cells were obtained on 40 days old nu/nu or SCID female mice (Harlan Laboratories, Udine, Italy), respectively. Hormone-independent JIMT-1 cells (1 × 107) were injected s.c. in the intrascapular region of nu/nu mice and left to growth up to 200 mm3. At day 0, SCID mice were fully anesthetized and 17-h estradiol pellets were s.c. injected. The day after, 5 × 106 exponentially growing MCF7 cells were mixed 1:1 with Matrigel (BD Biosciences, San Jose, CA, USA) and injected s.c. as above described. The Trastuzumab treatment was performed twice a week by i.p. injection of 200 μl of sterile PBS containing 400 μg of Trastuzumab. Tumor development was followed twice a week by caliper. Tumor-bearing nu/nu or SCID mice (8 mice per group), derived from JIMT-1 or MCF7 engrafts, were treated with P-1257 (75 μg diluted in 15 μl of PBS) peptide by intratumoral injection followed by electroporation with a Square Electro Porator (CUY21; Nepagene, Ichikawa, Japan). Treatments were repeated for 3 weeks. Two groups of animals received the peptides in combination with Trastuzumab. As further controls, we monitored tumor growth of mice either untreated, electroporated or treated with Trastuzumab alone.
Liposomes therapy
MCF7 cells were engineered with pGL4.51 (luc2/CMV/Neo) Luc-reporter vector (Promega, Milan, Italy) generating MCF7/luc cells. To induce engrafts at the visceral site, MCF7/luc cells (5 × 106 per mouse) were injected i.v. in SCID mice. After 4 weeks, empty liposomes (negative control) or containing P-1257 were injected weekly i.v. for 5 weeks. Two groups of animals (ctrl or P-1257) were also treated with Trastuzumab by i.p. injection as described above. For each experimental condition six mice were used. At the end of all treatments, the animals were injected i.p. with 𝒟-luciferin (75 mg/kg), then animals were killed and the light emission of lungs was detected with the IVIS Lumina II (Caliper Life Sciences, Waltham, MA, USA) CCD camera system (Xenogen, Alameda, CA, USA) and analyzed with the Living Image 2.20 software package (Xenogen). A parallel gray-scale image of the lungs permits the localization of the photon emission. All the experiments were done at least twice.
Immunohistochemistry
Proliferation index (PI) of untreated and treated tumor was determined with the monoclonal antibody anti-Ki-67 (MIB-1 Dako) by immunohistochemistry on formalin-fixed, paraffin-embedded sections. Tumor sections were stained with a streptavidin-enhanced immunoperoxidase technique (Supersensitive Multilink, Novocastra, Menarini, Florence, Italy) in an automated autostainer (Bond Max, Menarini). The pH 6.0 citrate buffer antigen retrieval protocol was applied for the antibody. Slides were mounted in aqueous mounting medium (glycergel, Dako).
PI in mouse tumor cells was determined counting positive nuclei in eight high-power fields ( × 400 magnification) per section. Negative controls were parallel sections in which the primary antibody was omitted. Evaluation of the immunohistochemistry results was performed from two independent experiments and in blinded manner by two investigators (MM, ADB). Necrotic areas were visualized by staining with hematoxylin–eosin section derived from control ant tumors treated with P-1257 in combination with Trastuzumab.
Statistical analysis
To determine the significant differences between group means in an analysis of variance setting (PI and apoptotic index) the Bonferroni test was used. All results are expressed as mean percentage. Differences were considered statistically significant when P<0.05. All analyses were performed using SPSS, version 17.0 (SPSS, Chicago, IL, USA). The χ2 test was used to analyze the in vitro cell death (*P≤0.05, **P≤0.001, ***P≤0.0005). Statistical significance of the in vivo data were evaluated by Student's t-test (P≤0.05).
Acknowledgments
This work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC: IG and 5 × 1000), Ministero della Salute e Filas Lazio to RF, and Cariplo Foundation (grant no. 2009-2439). Valentina Folgiero was recipient of a fellowship from Federazione Italiana Ricerca sul Cancro (FIRC). We are grateful to Oreste Segatto for critical discussion and suggestions. We thank Silvia Bacchetti, and Silvia Soddu for reading of the manuscript. We thank Gianpaolo Tortora for KPL-4 and JIMT-1 cells and Silvia Giordano for the GST-N-SH2 domain of p85. Dafne Italiano for technical support.
Glossary
- PI3K
phosphoinositide 3-kinase
- p110
the catalytic subunit of PI3K
- p85
the regulatory subunit of PI3K
- IRS-1
insulin receptor substrate
- BC
breast cancer
- P-1257
phosphopeptide 1257
- N-SH2
N-terminal Src homology 2 domain
- P-scr
scrambled phosphopeptide
- PIK3CA
mutated p110 catalytic subunit of PI3K
- nu/nu
nude mice
- SCID
immunocompromised mice
- T
Trastuzumab
- Ctrl
untreated mice
- ctrl electr
mice treated exclusively by electroporation
- ctrl+T
mice treated exclusively with Trastuzumab
- P-1257+T
mice treated with P-1257 in combination with Trastuzumab
- scr+T
mice treated with scramble phosphopeptide in combination with Trastuzumab
- s.c.
subcutaneous injection
- i.p.
intraperitoneal injection
- i.v.
intravenous injection
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on Cell Death and Disease website (http://www.nature.com/cddis)
Edited by G Raschella′
Supplementary Material
References
- Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–644. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA. Targeting signaling in cancer: opportunities, challenges and limitation. Nat Rev Cancer. 2009;9:550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- Yuan TL, Canntley LC. PI3K pathway alteration in cancer: variation on a theme. Oncogene. 2008;27:5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems L, Tamburini J, Chapuis N, Lacombe C, Mayeux P, Bouscary D. PI3K and mTOR signaling pathways in cancer: new data on targeted therapies. Curr Oncol Rep. 2012;14:129–138. doi: 10.1007/s11912-012-0227-y. [DOI] [PubMed] [Google Scholar]
- Lou J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003;4:257–262. doi: 10.1016/s1535-6108(03)00248-4. [DOI] [PubMed] [Google Scholar]
- Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L. Treatment of HER2-positive breast cancer: current status and future prospective. Nat Rev Clin Oncol. 2011;9:16–32. doi: 10.1038/nrclinonc.2011.177. [DOI] [PubMed] [Google Scholar]
- Di Fiore PP, Pierce JH, Kraus MH, Segatto O, King CR, Aaronson SA. erbB-2 is a potent oncogene when overexpressed in NIH73T3 cells. Science. 1987;237:178–182. doi: 10.1126/science.2885917. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplified HER-2/neu oncogene. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, et al. Studies of HER2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–712. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- Segatto O, Lonardo F, Helin K, Wexler D, Fazioli F, Rhee SG, et al. erbB-2 autophosphorylation is required for mitogenic action and high affinity substrate coupling. Oncogene. 1992;7:1339–1346. [PubMed] [Google Scholar]
- Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW, et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421:756–760. doi: 10.1038/nature01392. [DOI] [PubMed] [Google Scholar]
- Molina MA, Codony-Servat J, Albanell J, Rojo F, Arribas J, Baselga J. Trastuzumab (Herceptin), a humanized anti-Her2 receptor monoclonal antibody, inhibits basal and activated Her2 ectodomain cleavage in breast cancer cells. Cancer Res. 2001;61:4744–4749. [PubMed] [Google Scholar]
- Hudis CA. Trastuzumab-mechanism of action and use in clinical practice. N Engl J Med. 2007;357:39–51. doi: 10.1056/NEJMra043186. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpress HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- Marty M, Cognetti F, Maraninchi D, Snyder R, Mauriac L, Tubiana-Hulin M, et al. Randomized phase II Trial of the efficacy and safety of trastuzumab combined with docetaxel in patients with epidermal growth factor-2-positive metastatic breast cancer administrated as first line treatment: the M77001 Study Group. J Clin Oncol. 2005;23:4265–4274. doi: 10.1200/JCO.2005.04.173. [DOI] [PubMed] [Google Scholar]
- Oakman C, Sapino C, Marchiò C, Pestrin M, Biganzoli L, Di Leo A. Chemotherapy with and without trastuzumab. Ann Oncol. 2010;21:vii112–vii119. doi: 10.1093/annonc/mdq283. [DOI] [PubMed] [Google Scholar]
- Normanno N, Bianco C, De Luca A, Maiello MR, Salomon DS. Target-based agents against ErbB receptors and their ligands: a novel approach to cancer treatment. Endocr Relat Cancer. 2003;10:1–21. doi: 10.1677/erc.0.0100001. [DOI] [PubMed] [Google Scholar]
- Yarden Y, Sliwkowski MX. Untangling the ErbB signaling network. Nat. Rev. Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- Wallasch C, Weiss FU, Nierderfellner G, Jallal B, Issing W, Ullrich A. Heregulin-dependent regulation of HER2/neu oncogenic signaling by heterodimerization with HER3. EMBO J. 1995;14:4267–4275. doi: 10.1002/j.1460-2075.1995.tb00101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellyer NJ, Cheng K, Koland JG. ErbB3 (HER3) interaction with the p85 regolatory subunit of phosphoinositide3-kinase. Biochem J. 1988;333:757–763. doi: 10.1042/bj3330757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergina NV, Rausch M, Wang D, Blair J, Hann B, Shokat KM, et al. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 2007;445:347–441. doi: 10.1038/nature05474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12:395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB-2 requires ErbB3 to drive breast cancer tumor cell proliferation. Proc Natl Acad Sci USA. 2003;100:8933–8938. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel PM, Ryan ED, Cardiff RD, Muller WJ. Elevated expression of activated forms of Neu/ErbB-2 and ErbB-3 are involved in the induction of mammary tumors in transgenic mice: implications for human breast cancer. EMBO J. 1999;18:2149–2164. doi: 10.1093/emboj/18.8.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook RS, Garrett RT, Sanchez V, Stanford JC, Young C, Chakrabarty A, et al. ErbB3 ablation impairs PI3K/Akt-dependent mammary tumorigenesis. Cancer Res. 2011;71:3941–3951. doi: 10.1158/0008-5472.CAN-10-3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folgiero V, Avetrani P, Bon G, Di Carlo SE, Fabi A, Nisticò C, et al. Induction of ErbB-3 expression by α6β4 integrin contributes to Tamoxifen resistance in ERβ1-negative breast carcinoma. PLoS ONE. 2008;3:e1592. doi: 10.1371/journal.pone.0001592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. The phosphatydilinositol 3-kinase Akt pathway in human cancer. Nat Rev Cancer. 2002;2:489–51. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Engelman JA, Luo J, Cantley LC. The evolution of phosphatidilinositol 3-kinase of growth and metabolism. Nat Rev Cancer. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–643. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suenaga A, Takada N, Hatakeyama M, Ichikawa M, Yu X, Tomii K, et al. Novel Mechanism of interaction of p85 subunit of phosphatidylinositol 3-kinase and ErbB3 receptor-derived phosphotyrosyl peptides. J Biol Chem. 2005;280:1321–1326. doi: 10.1074/jbc.M410436200. [DOI] [PubMed] [Google Scholar]
- Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, et al. SH2 domains recognize specific phosphopeptide sequences. Cell. 1993;72:767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- Damiano V, Garofalo S, Rosa R, Bianco R, Caputo R, Gelardi T, et al. A novel toll-like receptor agonist cooperates with trastuzumab in trastuzumab-resistant breast tumors through multiple mechanisms of action. Clin Cancer Res. 2009;15:6921–6930. doi: 10.1158/1078-0432.CCR-09-1599. [DOI] [PubMed] [Google Scholar]
- DeFazio A, Chiew YE, Sini RL, Janes PW, Sutherland RL. Expression of c-erbB receptors, heregulin and oestrogen receptor in human breast cancer cell lines. Int J Cancer. 2000;87:487–498. [PubMed] [Google Scholar]
- Folgiero V, Bachelder RE, Bon G, Sacchi A, Falcioni R, Mercurio AM. The alpha6beta4 integrin can regulate ErbB-3 expression: implications for alpha6beta4 signaling and function. Cancer Res. 2007;67:1645–1652. doi: 10.1158/0008-5472.CAN-06-2980. [DOI] [PubMed] [Google Scholar]
- Saal LH, Holm K, Maurer M, Memeo L, Su T, Wang X, et al. PIK3CA mutations correlate with hormone receptor, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast cancer. Cancer Res. 2005;65:2554–2559. doi: 10.1158/0008-5472-CAN-04-3913. [DOI] [PubMed] [Google Scholar]
- Huang X, Gao L, Wang S, McManaman JL, Thor AD, Yang X, et al. Heterodimerization of the growth factor receptors erbB2, erbB3, and insulin-like growth factor-I receptor in breast cancer cells resistant to Trastuzumab. Cancer Res. 2010;70:1204–1214. doi: 10.1158/0008-5472.CAN-09-3321. [DOI] [PubMed] [Google Scholar]
- Zhang S, Huang WC, Li P, Guo H, Poh SB, Brady SW, et al. Combating Trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nature Med. 2001;17:461–470. doi: 10.1038/nm.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Lee DA, Li S. IL-12 and IL-27 sequential gene therapy via intramuscular electroporation delivery for eliminating distal aggressive tumors. J Immunol. 2010;184:2348–2354. doi: 10.4049/jimmunol.0902371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez CG, Ma CX, Crowder RJ, Guintoli T, Phommaly C, Gao F, et al. Preclinical modeling of combined phosphatidylinositol-3-kinase inhibition with endocrine therapy for estrogen receptor-positive breast cancer. Breast Cancer Res. 2011;13:R21. doi: 10.1186/bcr2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.