

Background: MqsR, an endoribonuclease, and MqsA, a transcriptional regulator, form a unique toxin-antitoxin (TA) pair.
Results: The high affinity, stable MqsR-MqsA complex is unable to bind DNA.
Conclusion: MqsR is the only toxin shown to disrupt the antitoxin-DNA complex, which would promote transcription.
Significance: The MqsR toxin may promote multidrug tolerance in E. coli by disrupting MqsA-mediated transcriptional repression of several genes related to persistence.
Keywords: Bacterial Toxins, Bacterial Transcription, Biophysics, DNA-binding Protein, DNA-Protein Interaction, Microbiology, Ribonuclease, Endoribonuclease, Persister Cells, Toxin-Antitoxin
Abstract
Bacterial biofilms are complex communities of cells containing an increased prevalence of dormant cells known as persisters, which are characterized by an up-regulation of genes known as toxin-antitoxin (TA) modules. The association of toxins with their cognate antitoxins neutralizes toxin activity, allowing for normal cell growth. Additionally, protein antitoxins bind their own promoters and repress transcription, whereas the toxins serve as co-repressors. Recently, TA pairs have been shown to regulate their own transcription through a phenomenon known as conditional cooperativity, where the TA complexes bind operator DNA and repress transcription only when present in the proper stoichiometric amounts. The most differentially up-regulated gene in persister cells is mqsR, a gene that, with the antitoxin mqsA, constitutes a TA module. Here, we reveal that, unlike other TA systems, MqsR is not a transcription co-repressor but instead functions to destabilize the MqsA-DNA complex. We further show that DNA binding is not regulated by conditional cooperativity. Finally, using biophysical studies, we show that complex formation between MqsR and MqsA results in an exceptionally stable interaction, resulting in a subnanomolar dissociation constant that is similar to that observed between MqsA and DNA. In combination with crystallographic studies, this work reveals that MqsA binding to DNA and MqsR is mutually exclusive. To our knowledge, this is the first TA system in which the toxin does not function as a transcriptional co-repressor, but instead functions to destabilize the antitoxin-operator complex under all conditions, and thus defines another unique feature of the mqsRA TA module.
Introduction
Bacterial biofilms are communities of bacterial cells that adhere to an inert surface and are enclosed in a self-produced extracellular polymeric matrix (1). Notably, biofilms are present in at least 65% of all bacterial infections (2). Their presence is particularly challenging because biofilms are extremely recalcitrant to antibiotic treatment (3). The cellular population of biofilms differs from that of normal planktonic cultures in that they display an increased prevalence of a subpopulation of cells known as persister cells (4). First identified in 1944 (5), persister cells are phenotypic variants of wild type cells that exhibit multidrug tolerance (6–8). Although much remains unknown about the persister state and how it is regulated in vivo, it is becoming widely evident that a class of genes known as toxin-antitoxin (TA)2 modules play a significant role in the persister phenotype (7, 9). For instance, deletion of certain TA operons has been shown to decrease the levels of persistence (10, 11). More importantly, it has also been shown that many TA genes, especially toxins, are preferentially up-regulated in persister versus nonpersister cells (9), and it is the overexpression of toxins that leads to increased antibiotic tolerance (12, 13).
There are currently five known types of TA systems (14–16). Whereas the toxin genes always code for proteins, the antitoxins are either protein (types II, IV, and V) (15–17), or RNA (types I and III) (18, 19). Type II TA systems are the most well characterized and are comprised of a two-gene operon that encodes a labile protein antitoxin and a stable toxin (17). Under normal conditions, the protein products associate to form a nontoxic complex. However, under conditions of environmental stress, such as nutrient starvation, oxidative stress, or antibiotic challenge, the antitoxins are rapidly degraded by cellular proteases, mainly Lon or ClpXP (20–23), and the toxins are free to exert their cellular effects.
Previously, gene expression profiling revealed that the gene most highly up-regulated in Escherichia coli persister cells is the toxin mqsR (9). The mqsR gene encodes a small 98-amino acid protein that functions as a sequence-specific mRNA endoribonuclease belonging to the RelE family of bacterial toxins (24–26). Its cognate antitoxin mqsA, which is located immediately downstream of mqsR in the same operon, encodes a 131-amino acid protein that functions to neutralize MqsR toxicity. There is a growing body of evidence that the mqsRA TA system plays a significant role in multiple cellular processes in E. coli. For instance, MqsR has been shown to facilitate biofilm formation by affecting cellular motility as a result of autoinducer-2 signaling (27). Also, deletion of mqsR and the mqsRA operon leads to decreased persister cell formation, the only type II TA system known to do so, whereas overexpression of MqsR increases persister cell formation (10). Finally, the antitoxin MqsA has been shown to serve as a transcriptional regulator not only of the mqsRA operon, but also of several other E. coli genes, including mcbR, cspD, and the general stress response regulator rpoS (21, 24, 28).
We showed previously that the antitoxin MqsA is unique among all antitoxins studied to date. First, MqsA is completely structured throughout its entire sequence both in the free and DNA- or toxin-bound states (24, 29), whereas other antitoxins are either partially or completely unstructured in the free state (30–34). Second, antitoxins typically bind DNA and their cognate toxins via their N- and C-terminal domains, respectively (31, 32, 35, 36). Conversely, MqsA binds its toxin, MqsR, via its N-terminal domain and DNA via its C-terminal domain. Third, MqsA is the only antitoxin that binds metal, zinc, at its N-terminal toxin-binding domain for structural stability (24).
However, although the unique features of the MqsA antitoxin are well established, it is only now becoming evident that the MqsR toxin is also atypical compared with other E. coli toxins. Instead of binding MqsA and enhancing transcriptional repression, as has been observed in other TA systems (37–41), here we show that MqsR serves solely to destabilize the complex formed between MqsA and DNA. This occurs because both the MqsA-DNA and MqsR-MqsA complexes have comparably strong affinities and because the binding sites of DNA and MqsR on MqsA partially overlap. This makes it impossible for MqsA to bind DNA and MqsR simultaneously without conformational changes in either the MqsA antitoxin, the MqsR toxin, or both. Thus, these data provide further evidence that both the MqsA antitoxin and MqsR toxin have multiple unique characteristics that are not observed in canonical TA systems, and thus mqsRA is the founding member of a unique family of TA pairs.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Free MqsR was obtained by co-expressing pET30a-MqsR, which included an N-terminal hexahistidine (His6) tag, with untagged pCA21a-MqsA-N I44A (MqsA-N is the N-terminal domain of MqsA and includes residues 1–76) at 18 °C in BL21(DE3) E. coli competent cells. After lysis, the His6-MqsR-MqsA-N complex was bound to nickel metal affinity resin and denatured with 6 m guanidine hydrochloride. His6-MqsR (hereafter referred to as MqsR) was eluted with 500 mm imidazole and refolded by stepwise dialysis in buffers containing decreasing amounts of guanidine hydrochloride followed by a final preparative gel filtration step (Superdex 75 16/60; GE Healthcare). The untagged MqsR-MqsA co-expressed and co-purified complex was prepared as described previously (24). The His6-MqsR-MqsA co-expressed and co-purified complex (CoExp-MqsR-MqsA) was formed by co-expressing pET30a-His6-MqsR with untagged full-length pCA21a-MqsA (MqsA-F, residues 1–131) at 18 °C in BL21(DE3) E. coli competent cells. The complex was co-purified using nickel affinity chromatography and eluted with 50 mm Tris, pH 8.0, 500 mm NaCl, 500 mm imidazole. The pooled protein was concentrated and purified by a final preparative gel filtration step (Superdex 75 26/60 or 16/60; GE Healthcare). The His6-MqsR-MqsA reconstituted complex (Rec-MqsR-MqsA) was prepared by incubating equimolar amounts of purified His6-MqsR and MqsA for 30 min on ice and purified by a final preparative gel filtration step (Superdex 75 16/60). Mutagenesis of mqsA was carried out using the QuikChange Mutagenesis kit (Agilent Technologies) using the manufacturer's protocols; all constructs were verified by sequencing. Finally, full-length MqsA constructs (WT and each mutant) were expressed and purified as described previously (24). All protein samples were stored in 10 mm Tris, pH 7.5, 100 mm NaCl, 0.5 mm TCEP.
Circular Dichroism
All circular dichroism (CD) measurements were performed with a Jasco J-815 CD spectropolarimeter. Dilute samples in 10 mm Tris, pH 7.5, 100 mm NaCl, 0.5 mm TCEP were placed in a 0.2-cm glass cuvette. Wavelength scans were performed in the far-UV region from 260 to 195 nm at 25 °C at a speed of 20 nm/min. Thermal denaturation scans were performed at 208 nm from 25 to 100 °C for MqsA (2.5 μm) and MqsR (5 μm) or 25 to 110 °C for MqsR-MqsA, MqsR-MqsA R61A, and MqsR-MqsA R61D (2.5 and 5.0 μm) at a scan speed of 1 °C/min. Raw data were processed using the denatured protein analysis routine implemented in the SepctraAnalysis software and converted to mean residue molar ellipticity using the following equation: [θ] = θ/(10×C×l×n), where θ is ellipticity, C is the molar concentration, l is the cell path length in centimeters, and n is the number of residues. Plots are the average of three replicate scans.
Isothermal Titration Calorimetry
Isothermal titration calorimetry (ITC) experiments were performed using a VP-ITC (GE Healthcare) at 25 °C. All proteins were exchanged into the same protein buffer using size exclusion chromatography (Superdex 75 16/60) and then concentrated immediately prior to the ITC experiments. MqsA-F (30 μm; syringe) was titrated into MqsR (2.8 μm; sample cell) with constant stirring. Due to the extremely tight binding between MqsR and MqsA, the injection volume was decreased from 10 to 5 μl in the middle of the titrations (injections 8–27 of 38 total injections) and then increased back to their initial value. This allowed more data points to be obtained at the isotherm transition which, in turn, allowed the binding transition to be fit with higher accuracy. Binding isotherms were fit using a one-site binding model and association constant (Ka) values calculated based on the known binding stoichiometry (n) of one (24) using Origin.
mRNA Cleavage Assay
DNA containing a T7 RNA polymerase promoter sequence was obtained using the primers shown in supplemental Table S1 and purified using the PureLink PCR purification kit (Invitrogen). The PCR products were used as templates for the in vitro RNA synthesis reaction with the AmpliScribe T7-Flash transcription kit (Epicentre). The MqsR cleavage assay contained 2 μg of RNA and 15 ng of protein. For the RNA-only control reaction, protein was replaced by buffer (10 mm Tris, pH 7.5, 100 mm NaCl, 0.5 mm TCEP). The reactions were incubated at 37 °C for 15 min and quenched by the addition of an equal volume of 2× Novex TBE-urea sample loading buffer (Invitrogen). The reaction products were resolved by electrophoresis with RNA denaturing gels (15% polyacrylamide with 7 m urea; Invitrogen) and stained with 0.5 μg/ml ethidium bromide.
Electrophoretic Mobility Shift Assay
The mqsRA operon promoter (PmqsRA, Table 1) was PCR-amplified using primers shown in supplemental Table S2 from BW25113 WT genomic DNA as described previously (24). The DNA was then labeled using the biotin 3′ End DNA Labeling Kit (Pierce). Palindromes 1 and 2 (each 26 bp, Table 1) were obtained by annealing. Specifically, following synthesis of the individual oligonucleotides (IDT Technologies, each oligonucleotide included a 3′ biotin label; supplemental Table S2), the complementary oligonucleotides were combined, heated to 95 °C, and then cooled by 1 °C/min to a final temperature of 25 °C. For titration experiments, increasing concentrations (0.2, 1, 2, 5, or 10 pmol) of MqsA-F, MqsA R61A, MqsA R61D, MqsR, or the MqsR-MqsA-F complex were added to a constant amount (100 fmol) of labeled DNA, and all reactions were carried out in binding buffer (10 mm Tris, pH 7.5, 50 mm KCl, 1 mm DTT) in the presence of poly(dI-dC) DNA probe (50 ng/μl) to prevent nonspecific binding. The binding reactions were incubated at room temperature for 20 min. Samples were then loaded onto a 6% DNA retardation gel (Invitrogen) and subjected to electrophoresis at 4 °C for either 75 min (Pal1 and Pal2) or 90 min (PmqsRA) at 100 V in 0.5× TBE buffer (45 mm Tris, pH 8.3, 45 mm boric acid, 1 mm EDTA). The DNA was transferred to a nylon membrane at 390 mA for 30 min and subsequently UV cross-linked at 302 nm by placing the membrane face-down on a UV illuminator for 15 min. Chemiluminescence was performed with the LightShift Chemiluminescent EMSA Kit (Pierce), and the samples were detected using a CCD imager (Typhoon 9410 Imager).
TABLE 1.
DNA constructs used in EMSA experiments
| DNA construct | Base pairs | Palindrome 1 | Palindrome 2 |
|---|---|---|---|
| PmqsRA | 234 | X | X |
| Pal1 | 26 | X | |
| Pal2 | 26 | X |
For experiments altering the relative amount of MqsR, a constant amount of MqsA-F (400 fmol) was incubated with increasing amounts of MqsR (either 200, 300, 400, or 800 fmol for experiment 1 or 50, 100, 150, 200, or 400 fmol for experiment 2). The MqsR-MqsA-F complex was allowed to incubate at room temperature for 10 min. Next, 50 fmol of labeled PmqsRA DNA was added to the preformed MqsR-MqsA-F complex and the ternary binding reaction allowed to incubate an additional 20 min at room temperature. For the second experiment, the reverse reaction was also prepared in which a constant amount of MqsA was incubated with a constant amount of labeled PmqsRA DNA followed by addition of increasing amounts of MqsR. The electrophoresis, transfer, and chemiluminescent detection were carried out as described above.
RESULTS
Free MqsR Readily Degrades Its Own mRNA
Free MqsR, an endoribonuclease toxin, was isolated by purifying and denaturing the MqsR-MqsA-N complex (MqsA-N is the N-terminal domain of MqsA, residues 1–76) and then isolating and refolding MqsR by stepwise dialysis followed by size exclusion chromatography (Fig. 1A). Circular dichroism (CD) polarimetry (Fig. 1B) was used to verify that MqsR was folded. Refolded MqsR also formed a complex with full-length MqsA (reconstituted MqsR-MqsA, or Rec-MqsR-MqsA) that was indistinguishable from co-expressed and co-purified MqsR-MqsA (CoExp-MqsR-MqsA), as determined by size exclusion chromatography experiments (Fig. 1A). We used electrophoretic mobility shift assays (EMSAs) to demonstrate that, in contrast to MqsA, MqsR does not bind the full mqsRA promoter (Fig. 1C; the specificity of MqsA for PmqsRA is shown in supplemental Fig. S1 and in Ref. 29 for an individual mqsRA palindrome). To verify that the refolded MqsR was active, we performed mRNA cleavage assays with conditions similar to those reported previously (26) and using mRNA transcripts of mqsR and mqsA as substrates. MqsR was previously shown to specifically cleave mRNA at GCU and, to a lesser extent, GCA sequences (24–26); mqsR and mqsA transcripts contain one and three GCU motifs, respectively. As can be seen in Fig. 1D, incubation of refolded MqsR with in vitro transcribed mqsR and mqsA mRNA (amplified using primers in supplemental Table S1) resulted in the cleavage of both mRNAs. Significantly more cleavage products were observed for mqsA than mqsR mRNA, consistent with the presence of three versus one GCU cleavage sites in the mqsA versus mqsR transcript, respectively. These data also showed that MqsR activity is potently inhibited when bound to MqsA, independent of whether the MqsR-MqsA complex was co-expressed and co-purified or the MqsA and MqsR proteins were purified individually and the complex reconstituted in vitro. Collectively, these data show that refolded MqsR behaves identically to WT MqsR and that it is one of only a few examples in which a bacterial toxin has been shown to cleave its own mRNA transcript, an activity that may serve as a mechanism of auto-regulation to alleviate the persister state (42).
FIGURE 1.

MqsR readily cleaves mqsRA mRNA. A, size exclusion chromatogram of free MqsR, the MqsR-MqsA complex which was co-expressed and co-purified (CoExp-MqsR-MqsA), and the MqsR-MqsA complex which was produced by incubating free MqsA with refolded MqsR prior to SEC (Rec-MqsR-MqsA). B, CD wavelength scan of refolded MqsR. C, EMSA with increasing amounts of either MqsA alone (lanes 2 and 3) or MqsR alone (lanes 5 and 6) with biotin-labeled PmqsRA. D, MqsR toxin readily degrades both mqsA and mqsR transcripts (lanes 4 and 9). MqsR activity was inhibited when bound to MqsA, independent of whether the MqsR-MqsA complex was formed in vitro (lanes 2 and 7) or co-expressed and co-purified (lanes 3 and 8). Full-length mqsA mRNA (415 nucleotides, lane 1) and full-length mqsR mRNA (316 nucleotides, lane 6) were incubated with buffer under the same reaction conditions as a negative control. Lane 5 contains a single-stranded RNA ladder.
Unlike Most Antitoxins, MqsA Binds MqsR and Its Operator DNA with Similar Subnanomolar Affinities
Several toxins associate with their cognate antitoxins with extremely high affinities, likely to allow for proper cell growth under nonstressful environmental conditions (43–45). To determine the affinity of the MqsR-MqsA complex, we used ITC. ITC measurements using MqsA and MqsR reported a dissociation constant (KD) of 0.08 ± 0.07 nm (a representative of three replicates is shown in Fig. 2A). This KD is similar to those reported for other TA systems, including CcdA-CcdB (0.03 nm) (43), RelB-RelE (0.33 nm) (44), and PezA-PezT (<1 nm) (45).
FIGURE 2.
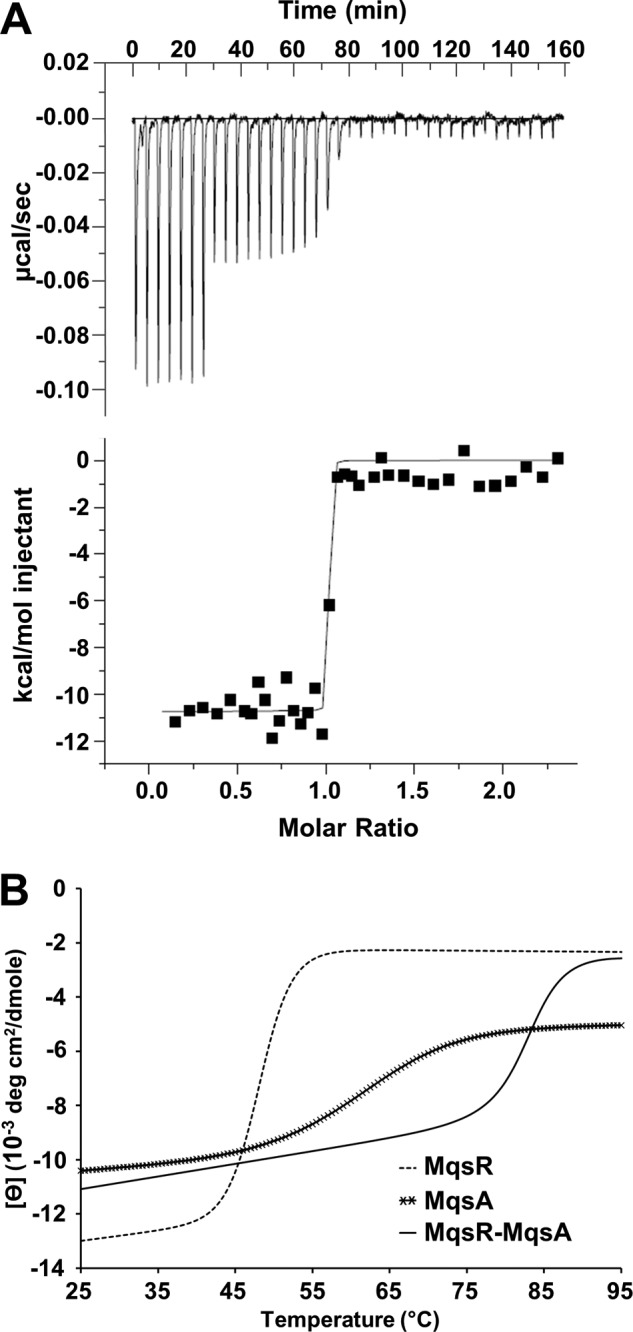
MqsR forms an extremely stable complex with MqsA. A, raw isothermal titration calorimetry data (upper) and derived binding isotherm plotted versus the molar ratio of titrant which was fit using a one-site model (lower) for MqsA (titrant) into MqsR (sample). Due to the high affinity, the volume of injections 8–27 (of 38 total) was decreased from 10 to 5 μl to better monitor the binding transition. The solid line in the lower panel is the best fit to the data using the nonlinear least squares regression algorithm (ORIGIN). B, thermal denaturation at 208 nm of MqsR-MqsA (2.5 μm, solid line). The complex unfolds with a single-state transition and a Tm of 83.4 ± 0.3 °C. The Tm for MqsR (5 μm, dashed line) and MqsA (2.5 μm, crossed line) is 48.1 ± 0.3 °C and 61.1 ± 3.3 °C, respectively.
However, the KD of MqsA for MqsR is nearly identical to that between MqsA and its DNA operator sequence (0.8 nm), which we measured previously using quantitative EMSA (29). This similarity in affinities of an antitoxin for DNA and for its cognate toxin is not typical of most TA systems. Instead, in other cases, the binding affinity between the antitoxin and DNA is much lower than that between the antitoxin and toxin. For example, RelB binds its DNA operator with a KD of 200–300 μm, which is nearly 1000-fold higher (weaker affinity) than that between RelB and RelE (34). Similarly, the KD of CcdA and its DNA operator is 2–90 μm, or 100–1000-fold higher (weaker affinity) than that between CcdA and CcdB (31).
The MqsR-MqsA Complex Is Exceptionally Stable
To compare the stabilities of free MqsR, free MqsA, and the MqsR-MqsA complex, we monitored protein unfolding with increasing temperature using CD polarimetry. Both MqsA and MqsR exhibited a two-state melting curve, with melting temperatures (Tm) of 61.1 ± 3.3 °C and 48.1 ± 0.3 °C, respectively (Fig. 2B, Table 2). The MqsR-MqsA complex also exhibited a two-state melting curve, demonstrating that the complex unfolds cooperatively and behaves like a single folded protein. However, the Tm of the MqsR-MqsA complex was 83.4 ± 0.3 °C (Fig. 2B), more than 22 °C and 35 °C higher than for free MqsA and free MqsR, respectively. This shows that complex formation significantly enhances the stability of both proteins, resulting in a complex that is exceptionally stable.
TABLE 2.
Thermal melting transition temperatures determined by CD (n (≥2))
| Protein | Tm |
|---|---|
| °C | |
| MqsR | 48.1 ± 0.3 |
| MqsA-F | 61.1 ± 3.3 |
| MqsR-MqsA-F | 83.4 ± 0.3 |
| MqsR-MqsA-F R61A | 75.2 ± 0.4 |
| MqsR-MqsA-F R61D | 70.5 ± 1.0 |
MqsR Destabilizes the MqsA-DNA Transcriptional Repressor Complex
In most type II TA systems, both the antitoxin and the TA complex are able to bind DNA and regulate the transcription of the TA operon. Moreover, in many cases, the purified TA complex binds DNA with higher affinity. The mqsRA promoter contains two palindromic MqsA binding sites (Fig. 3A and Table 1) (26, 29). Using EMSAs and x-ray crystallography, we have shown previously that MqsA binds robustly and with high affinity to both palindromic binding sites (29, 46). However, detailed analysis of the structures of the MqsA-DNA (29) and MqsR-MqsA-N complexes (24) also revealed that, if the MqsR-MqsA complex was able to bind DNA, either the MqsA-DNA or the MqsA-MqsR interface would have to change for both DNA and MqsR to bind MqsA simultaneously. This suggested that the MqsR-MqsA complex might interact with DNA in a completely different manner than MqsA alone.
FIGURE 3.

The MqsR-MqsA complex does not bind to the mqsRA promoter in vitro. A, sequence of the mqsRA promoter. DNA constructs used in this study are illustrated. Palindromes 1 and 2 are boxed (nucleotides that interact specifically with MqsA, as determined from the MqsA-DNA crystal structure, are in blue). The PmqsRA DNA is highlighted in green, and the primers used to amplify it are underlined. B, EMSA with increasing amounts of either MqsA alone (lanes 2–6) or the untagged MqsR-MqsA complex (lanes 8–12) incubated with biotin-labeled palindrome 1 (Pal1) of PmqsRA. C, same as B, except proteins were incubated with biotin-labeled Palindrome 2 (Pal2). D, same as B, except proteins were incubated with biotin-labeled PmqsRA promoter DNA. The two shifted bands represent protein bound to either one (middle arrow; 1× MqsA-PmqsRA) or both palindromes (top arrow; 2× MqsA-PmqsRA). DNA binding in the presence of the MqsR-MqsA complex is due to trace amounts of free MqsA, as the observed migration positions are identical to that seen with MqsA alone. For all gels, the negative control (100 fmol of labeled DNA lacking protein) is shown in lanes indicated as 0.
To test this, we examined the ability of the MqsR-MqsA complex to bind either the first (Fig. 3B) or second palindrome (Fig. 3C) in PmqsRA using EMSAs; free MqsA was tested in parallel for comparison. As expected, free MqsA robustly binds to both DNA palindromes at all concentrations tested (10–500 nm; Fig. 3, B and C, lanes 2–6). Unexpectedly, however, at the same concentrations, the MqsR-MqsA complex shows practically no DNA binding to either palindrome, even up to 500 nm (Fig. 3, B and C, lanes 8–12). Notably, these protein concentrations represent a 2-, 10-, 20-, 50-, and 100-fold excess of protein versus DNA. The inability of the MqsR-MqsA complex to bind DNA is contrary to other TA systems, in which the toxin serves as a transcriptional co-repressor and enhances the effective DNA binding affinity of their cognate antitoxins. This result is also in contrast to previous reports, which concluded that the MqsR-MqsA complex does bind the mqsRA promoter (24, 26). To investigate this difference further, we combined increasing amounts of both the MqsA antitoxin and the MqsR-MqsA complex with the full PmqsRA promoter DNA (this DNA contains both palindromes) and monitored binding. Critically, in these EMSA experiments, both free MqsA and the MqsR-MqsA complex reactions were run on the same gel to allow, for the first time, a side-by-side comparison of the observed shift with MqsA and the MqsR-MqsA complex. As can be seen in Fig. 3D, our data show that the shift in DNA migration that occurs in the presence of the MqsR-MqsA complex (54.2 kDa) is identical to that which occurs in the presence of MqsA alone (29.8 kDa). Thus, the slower migration (higher position on the gel) expected with the MqsR-MqsA complex due to its nearly 2-fold higher molecular mass is not observed. This result shows that at the high molar excess of protein to DNA tested here, the MqsR-MqsA and MqsA-DNA interactions, which have similar KD values, result in a fraction of MqsA dissociating from MqsR and binding the DNA. Thus, because previous EMSA experiments with MqsA and MqsR-MqsA were never performed on the same gel, the observed shift for the MqsR-MqsA complex was attributed to the complex. However, these results show unequivocally that the shift is not due to the MqsR-MqsA complex binding DNA, but instead due to a small fraction of free MqsA that binds DNA. Critically, this binding is observed only when the concentration of MqsR-MqsA significantly exceeds that of the DNA. Collectively, these data show for the first time that, unlike most other TA systems, the MqsR-MqsA complex does not bind DNA in vitro.
mqsRA Is Also Unique in That It Does Not Exhibit Conditional Cooperativity
An emerging feature of type II TA systems is their ability to autoregulate transcription by a mechanism known as conditional cooperativity (47–50). In this mechanism, the relative ratio of toxin and antitoxin present in the cell dictates the repression state of the operon. For canonical TA systems, when the amount of antitoxin is in excess or equal to that of the toxin, transcription is repressed, and the toxin serves as a co-repressor by increasing DNA binding affinity. However, when this ratio is reversed and the amount of toxin exceeds that of the antitoxin, the antitoxin dissociates from the DNA, allowing transcription to proceed.
We have already shown, using the co-purified complex, that when MqsR and MqsA are present in equimolar ratios, the complex does not bind DNA (Fig. 3). To determine whether the mqsRA TA system exhibits conditional cooperativity, we tested the ability of MqsA to bind DNA in the presence of subequimolar ratios of MqsR (Fig. 4A). In the absence of MqsR, 20 nm MqsA is able to simultaneously bind both palindromic sites and completely shift the PmqsRA DNA (Fig. 4A, lane 2). However, upon the addition of MqsR toxin, DNA binding is immediately destabilized, as can be seen by a decrease in intensity of the band corresponding to MqsA bound to both palindromes and a gradual increase in intensity of the band corresponding MqsA bound to only a single palindrome (Fig. 4A, lanes 3–4). At a 1:1 ratio of MqsR to MqsA, which is the same ratio of the complex in solution, practically all DNA binding has been abolished. Similar results were obtained regardless of whether MqsR was added to the preformed MqsA-DNA complex or whether DNA was added to the preformed MqsR-MqsA complex (Fig. 4B). Thus, these experiments show that at all concentrations, MqsR serves simply to dissociate the MqsA-DNA complex in vitro and never functions to enhance binding of MqsA to the operon promoter.
FIGURE 4.

MqsR destabilizes the interaction of MqsA with DNA. A, EMSA of MqsA binding to PmqsRA DNA in the presence of increasing amounts of MqsR. The two shifted bands represent protein bound to either one palindrome (middle arrow; 1× MqsA-PmqsRA) or both palindromes (top arrow; 2× MqsA-PmqsRA) simultaneously. Lanes 2–6 contain a constant amount of MqsA (20 nm) whereas lanes 3–6 contain 10, 15, 20, or 40 nm MqsR. Biotin-labeled PmqsRA in the absence of protein (50 fmol, lane 1) was used as a negative control. B, preformed MqsR-MqsA samples. MqsA was incubated with MqsR at the indicated ratios for 10 min at room temperature. Biotin-labeled PmqsRA DNA was then added to the MqsR-MqsA complex and the entire reaction incubated at room temperature for an additional 20 min followed by electrophoresis. Preformed MqsA-PmqsRA samples: a constant amount of MqsA (400 fmol) was first incubated with PmqsRA DNA (50 fmol; 10 min, room temperature) then increasing amounts of MqsR added at the indicated ratios. MqsR amounts were 0, 50, 100, 150, 200, or 400 fmol. The control reaction (50 fmol of biotin-labeled PmqsRA only) is shown in the first lane.
DISCUSSION
The central finding of our work is that, unlike other type II toxins, MqsR does not enhance the binding of MqsA to the mqsRA promoter. Instead, under all circumstances, we show that MqsR functions solely to disrupt the MqsA-DNA interaction (Figs. 3 and 4), a function due in part to the tight binding (KD of 0.08 ± 0.07 nm; Fig. 2A) and exceptional stability of the MqsR-MqsA complex (Fig. 2B). This behavior is in stark contrast to most other TA systems, in which the toxin functions as a transcriptional co-repressor until a certain threshold is reached, above which it then functions as a de-repressor resulting in robust transcription (known as conditional cooperativity) (48–50).
There are two proposed mechanisms that underlie conditional cooperativity which rely on interactions made either by the toxin monomer (mechanism 1) or the toxin dimer (mechanism 2). In the first mechanism, shown for the PhD-Doc (48) TA system, the antitoxin interacts with the toxin at two separate interfaces present on the antitoxin, a high affinity site and a low affinity site. At low levels of toxin, both sites are populated, and the toxin molecules serve to bridge antitoxin dimers onto multiple DNA binding sites, which leads to increased affinity for DNA (48). However, when the toxin levels increase to exceed that of the antitoxin, only the high affinity binding sites on the antitoxin are populated. This alters the toxin-antitoxin complex stoichiometry, which in turn destabilizes DNA binding. In the second mechanism, which was recently reported for the VapB-VapC system (50), the VapC toxin dimerizes and these toxin dimers serve to bridge antitoxin molecules bound to multiple DNA sites to increase antitoxin avidity for DNA. However, when there is excess toxin, the toxin dimer interactions are broken which subsequently destabilizes antitoxin DNA binding.
We believe that MqsR cannot utilize either mechanism to serve as a transcriptional co-repressor for two reasons. First, there is no evidence for multiple binding interfaces between MqsR and MqsA. Our analysis of the MqsR-MqsA complex in solution has not revealed the presence of any oligomeric species larger than the MqsR-MqsA2-MqsR heterotetramer, as determined using size exclusion chromatography (Fig. 1A). Second, MqsR exists as a monomer in solution and has not been demonstrated to dimerize, which we also showed using size exclusion chromatography (Fig. 1A). Therefore, MqsR cannot serve as a co-repressor either alone (mechanism 1) or as a dimer (mechanism 2). Instead, and in contrast to all other characterized TA systems, even subequimolar quantities of MqsR toxin serve to destabilize the MqsA-DNA interaction (Fig. 4). In fact, at a 1:1 ratio of MqsR toxin to MqsA antitoxin, DNA binding by MqsA is essentially ablated. This behavior is unique to mqsRA because in all other TA systems, transcriptional derepression occurs only when toxin concentrations exceed their normal stoichiometric levels.
A detailed analysis of the x-ray crystal structures of the various states of MqsR and MqsA provides insight as to why MqsR destabilizes the interaction between MqsA and the mqsRA promoter. A model of the hypothetical MqsR-MqsA-DNA ternary complex based on our crystal structures of MqsA bound to DNA (Protein Data Bank (PDB) ID code 3O9X) and MqsR bound to the N-terminal domain of MqsA (PDB ID code 3HI2) revealed the potential for clashes between the DNA and MqsR when both are bound to MqsA (Fig. 5A). Moreover, the binding sites on MqsA for both MqsR and DNA partially overlap as MqsA Arg-61 forms interactions with both binding partners in the respective crystal structures (Fig. 5, B and C). In support of the role of Arg-61 in DNA and MqsR binding, mutation of this residue to either alanine (R61A) or aspartic acid (R16D) leads to both a decrease in DNA binding, as determined using EMSAs (Fig. 5D), and a decrease in the thermal stability of the MqsR-MqsAmut complexes, as determined using CD thermal denaturation (Table 2 and supplemental Fig. S2). These observations suggested two possibilities: (i) that a conformational rearrangement in one or more of the macromolecules is necessary for simultaneous binding, or (ii) that all three macromolecules are not capable of forming a single complex.
FIGURE 5.

MqsR and DNA have overlapping binding sites on MqsA. A, model of the hypothetical MqsR-MqsA-PmqsRA ternary complex. The model was generated by superimposing the MqsR-MqsA-N crystal structure (PDB 3HI2; colored blue/brown, respectively) with the MqsA-DNA crystal structure (PDB 3O9X; colored orange/gray, respectively) using the respective MqsA N-terminal domains; for clarity, only one monomer of MqsA from the MqsA-DNA crystal structure is shown. As modeled, there are structural clashes between MqsR and the DNA. The MqsA binding sites for MqsR and the PmqsRA DNA partially overlap as MqsA residue Arg-61 provides electrostatic interactions in both the toxin- and DNA-bound states; Arg-61 from both structures shown as sticks. Asp-33 (MqsR, blue) and the phosphate backbone from nucleotides thymine 14 and 15 (pink) are also shown; nitrogen atoms in dark blue, oxygen atoms in red. B, zoom-in view of overlay in A of just the MqsR-MqsA-N structure. MqsA residue Arg-61 forms a salt bridge with MqsR Asp-33. C, zoom-in view of overlay in A of just the MqsA-DNA structure. MqsA residue Arg-61 forms a salt bridge with the phosphate backbone of nucleotides thymine 14 and 15 from one strand. Electrostatic interactions in B and C are indicated by black dashed lines. D, EMSA with increasing amounts of WT MqsA (lanes 1–4), MqsA R61A (lanes 5–8), or MqsA R61D (lanes 9–12) incubated with biotin-labeled palindrome 1 (Pal1) of PmqsRA.
Here, we show that the MqsR-MqsA-DNA ternary complex does not exist in vitro. Instead, MqsR, which binds the MqsA N-terminal domain with a 0.08 nm KD, prevents this domain from interacting with the DNA due to steric hindrance. Although, in isolation, the MqsA N-terminal domains do not bind DNA, the interactions they provide within the context of full-length MqsA are important, as we previously showed that the binding affinity of MqsA for DNA decreases ∼50-fold in the absence of the N-terminal domain interactions, from a KD of 0.8 nm for full-length MqsA to 41.5 nm for the MqsA C-terminal domain (29). Consequently, MqsR binding results in a decrease in the effective binding affinity of MqsA for DNA and, at equimolar concentrations of MqsR, results in the disruption of the MqsA-DNA complex and the formation only of the MqsR-MqsA complex. Thus, unlike most toxins, MqsR does not act as a transcriptional co-repressor but instead a transcriptional derepressor. Critically, this newly identified function of MqsR may also explain previous results which showed that overexpression of MqsR led to an increase in the transcription of genes known to be regulated by MqsA, including cspD, rpoS, and mqsA itself (28). Therefore, both in vitro (these studies) and in vivo experiments (28) show that MqsR also functions as a transcriptional derepressor.
We previously determined that the mqsRA toxin-antitoxin module is unique among E. coli TA modules based upon the novel features of the MqsA antitoxin. However, it is now becoming evident that the MqsR toxin also displays unusual toxin characteristics which, together with the unique features of the MqsA antitoxin, may also lead to differences in transcriptional regulation of the mqsRA operon. We report that the MqsR-MqsA complex does not bind its own operon promoter, which would lead to transcriptional repression, as is common for several other TA systems. Instead, in all instances, the MqsR toxin serves solely to disrupt the MqsA-DNA complex in vitro and thus results in transcriptional activation. In addition, we show that MqsR is capable of cleaving mRNA corresponding not only to mqsA, but to mqsR as well. This suggests that MqsR may autoregulate its own activity, which could allow for rapid exit from the persister state upon removal of environmental stress. Expression of MqsR was previously shown to play a role in persistence as well as biofilm formation through positively regulating the expression of various genes including the toxin cspD and the two-component motility regulatory system qseBC (27, 28). Because MqsA regulates multiple genes that play a vital role in E. coli physiology, including activation of the general stress response (21), the function of MqsR in alleviating MqsA-mediated repression would further elucidate the mechanism by which MqsR exerts some of its cellular effects. Thus, MqsR may be a central regulator of bacterial persistence not only through its function as an endoribonuclease but also through its role in indirectly activating transcription of other genes that lead to cell dormancy.
Supplementary Material
Acknowledgment
We thank Dr. Thomas K. Wood (Pennsylvania State University, University Park) for stimulating discussions.
This work was supported by National Science Foundation-CAREER Award MCB-0952550 (to R. P.), National Institutes of Health-NINDS Predoctoral Fellowship Award F31NS062630 (to B. L. B.), and a National Science Foundation-Experimental Program to Stimulate Competitive Research (EPSCoR) graduate fellowship (to D. M. L.). This research is based in part upon work conducted using the Rhode Island National Science Foundation-EPSCoR Proteomics Shared Resource Facility, which is supported in part by the National Science Foundation-EPSCoR Grant 1004057, National Institutes of Health Grant 1S10RR020923, a Rhode Island Science and Technology Advisory Council grant, and the Division of Biology and Medicine, Brown University. This research is based in part upon work conducted in the Center for Genomics and Proteomics Core Facility with partial support from the National Institutes of Health (NCRR Grants P30RR031153, P20RR018728, and S10RR02763), National Science Foundation-EPSCoR Grant 0554548, Lifespan Rhode Island Hospital, and the Division of Biology and Medicine, Brown University.

This article contains supplemental Tables S1 and S2 and Figs. S1 and S2.
- TA
- toxin-antitoxin
- ITC
- isothermal titration calorimetry
- TCEP
- tris(2-carboxyethyl)phosphine.
REFERENCES
- 1. López D., Vlamakis H., Kolter R. (2010) Biofilms. Cold Spring Harb. Perspect Biol. 2, a000398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cvitkovitch D. G., Li Y. H., Ellen R. P. (2003) Quorum sensing and biofilm formation in streptococcal infections. J. Clin. Invest. 112, 1626–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Costerton J. W., Stewart P. S., Greenberg E. P. (1999) Bacterial biofilms: a common cause of persistent infections. Science 284, 1318–1322 [DOI] [PubMed] [Google Scholar]
- 4. Spoering A. L., Lewis K. (2001) Biofilms and planktonic cells of Pseudomonas aeruginosa have similar resistance to killing by antimicrobials. J. Bacteriol. 183, 6746–6751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bigger J. W. (1944) Treatment of staphylococcal infections with penicillin. Lancet 497–500 [Google Scholar]
- 6. Balaban N. Q., Merrin J., Chait R., Kowalik L., Leibler S. (2004) Bacterial persistence as a phenotypic switch. Science 305, 1622–1625 [DOI] [PubMed] [Google Scholar]
- 7. Keren I., Shah D., Spoering A., Kaldalu N., Lewis K. (2004) Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J. Bacteriol. 186, 8172–8180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Keren I., Kaldalu N., Spoering A., Wang Y., Lewis K. (2004) Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 230, 13–18 [DOI] [PubMed] [Google Scholar]
- 9. Shah D., Zhang Z., Khodursky A., Kaldalu N., Kurg K., Lewis K. (2006) Persisters: a distinct physiological state of E. coli. BMC Microbiol. 6, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim Y., Wood T. K. (2010) Toxins Hha and CspD and small RNA regulator Hfq are involved in persister cell formation through MqsR in Escherichia coli. Biochem. Biophys. Res. Commun. 391, 209–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dörr T., Vulić M., Lewis K. (2010) Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol. 8, e1000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Falla T. J., Chopra I. (1998) Joint tolerance to β-lactam and fluoroquinolone antibiotics in Escherichia coli results from overexpression of hipA. Antimicrob. Agents Chemother. 42, 3282–3284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Correia F. F., D'Onofrio A., Rejtar T., Li L., Karger B. L., Makarova K., Koonin E. V., Lewis K. (2006) Kinase activity of overexpressed HipA is required for growth arrest and multidrug tolerance in Escherichia coli. J. Bacteriol. 188, 8360–8367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Blower T. R., Salmond G. P., Luisi B. F. (2011) Balancing at survival's edge: the structure and adaptive benefits of prokaryotic toxin-antitoxin partners. Curr. Opin. Struct. Biol. 21, 109–118 [DOI] [PubMed] [Google Scholar]
- 15. Masuda H., Tan Q., Awano N., Wu K. P., Inouye M. (2012) YeeU enhances the bundling of cytoskeletal polymers of MreB and FtsZ, antagonizing the CbtA (YeeV) toxicity in Escherichia coli. Mol. Microbiol. 84, 979–989 [DOI] [PubMed] [Google Scholar]
- 16. Wang X., Lord D. M., Cheng H. Y., Osbourne D. O., Hong S. H., Sanchez-Torres V., Quiroga C., Zheng K., Herrmann T., Peti W., Benedik M. J., Page R., Wood T. K. (2012) A new type V toxin-antitoxin system where mRNA for toxin GhoT is cleaved by antitoxin GhoS. Nat. Chem. Biol. 8, 855–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gerdes K., Christensen S. K., Løbner-Olesen A. (2005) Prokaryotic toxin-antitoxin stress response loci. Nat. Rev. Microbiol. 3, 371–382 [DOI] [PubMed] [Google Scholar]
- 18. Fozo E. M., Hemm M. R., Storz G. (2008) Small toxic proteins and the antisense RNAs that repress them. Microbiol. Mol. Biol. Rev. 72, 579–589, Table of Contents [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Blower T. R., Fineran P. C., Johnson M. J., Toth I. K., Humphreys D. P., Salmond G. P. (2009) Mutagenesis and functional characterization of the RNA and protein components of the toxin abortive infection and toxin-antitoxin locus of Erwinia. J. Bacteriol. 191, 6029–6039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Christensen S. K., Maenhaut-Michel G., Mine N., Gottesman S., Gerdes K., Van Melderen L. (2004) Overproduction of the Lon protease triggers inhibition of translation in Escherichia coli: involvement of the yefM-yoeB toxin-antitoxin system. Mol. Microbiol. 51, 1705–1717 [DOI] [PubMed] [Google Scholar]
- 21. Wang X., Kim Y., Hong S. H., Ma Q., Brown B. L., Pu M., Tarone A. M., Benedik M. J., Peti W., Page R., Wood T. K. (2011) Antitoxin MqsA helps mediate the bacterial general stress response. Nat. Chem. Biol. 7, 359–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Van Melderen L., Bernard P., Couturier M. (1994) Lon-dependent proteolysis of CcdA is the key control for activation of CcdB in plasmid-free segregant bacteria. Mol. Microbiol. 11, 1151–1157 [DOI] [PubMed] [Google Scholar]
- 23. Aizenman E., Engelberg-Kulka H., Glaser G. (1996) An Escherichia coli chromosomal “addiction module” regulated by guanosine [corrected] 3′,5′-bispyrophosphate: a model for programmed bacterial cell death. Proc. Natl. Acad. Sci. U.S.A. 93, 6059–6063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brown B. L., Grigoriu S., Kim Y., Arruda J. M., Davenport A., Wood T. K., Peti W., Page R. (2009) Three dimensional structure of the MqsR:MqsA complex: a novel TA pair comprised of a toxin homologous to RelE and an antitoxin with unique properties. PLoS Pathog. 5, e1000706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Christensen-Dalsgaard M., Jørgensen M. G., Gerdes K. (2010) Three new RelE-homologous mRNA interferases of Escherichia coli differentially induced by environmental stresses. Mol. Microbiol. 75, 333–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yamaguchi Y., Park J. H., Inouye M. (2009) MqsR, a crucial regulator for quorum sensing and biofilm formation, is a GCU-specific mRNA interferase in Escherichia coli. J. Biol. Chem. 284, 28746–28753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. González Barrios A. F., Zuo R., Hashimoto Y., Yang L., Bentley W. E., Wood T. K. (2006) Autoinducer 2 controls biofilm formation in Escherichia coli through a novel motility quorum-sensing regulator (MqsR, B3022). J. Bacteriol. 188, 305–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim Y., Wang X., Zhang X. S., Grigoriu S., Page R., Peti W., Wood T. K. (2010) Escherichia coli toxin/antitoxin pair MqsR/MqsA regulate toxin CspD. Environ. Microbiol. 12, 1105–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brown B. L., Wood T. K., Peti W., Page R. (2011) Structure of the Escherichia coli antitoxin MqsA (YgiT/b3021) bound to its gene promoter reveals extensive domain rearrangements and the specificity of transcriptional regulation. J. Biol. Chem. 286, 2285–2296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Loris R., Marianovsky I., Lah J., Laeremans T., Engelberg-Kulka H., Glaser G., Muyldermans S., Wyns L. (2003) Crystal structure of the intrinsically flexible addiction antidote MazE. J. Biol. Chem. 278, 28252–28257 [DOI] [PubMed] [Google Scholar]
- 31. Madl T., Van Melderen L., Mine N., Respondek M., Oberer M., Keller W., Khatai L., Zangger K. (2006) Structural basis for nucleic acid and toxin recognition of the bacterial antitoxin CcdA. J. Mol. Biol. 364, 170–185 [DOI] [PubMed] [Google Scholar]
- 32. Oberer M., Zangger K., Gruber K., Keller W. (2007) The solution structure of ParD, the antidote of the ParDE toxin antitoxin module, provides the structural basis for DNA and toxin binding. Protein Sci. 16, 1676–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cherny I., Gazit E. (2004) The YefM antitoxin defines a family of natively unfolded proteins: implications as a novel antibacterial target. J. Biol. Chem. 279, 8252–8261 [DOI] [PubMed] [Google Scholar]
- 34. Li G. Y., Zhang Y., Inouye M., Ikura M. (2008) Structural mechanism of transcriptional autorepression of the Escherichia coli RelB/RelE antitoxin/toxin module. J. Mol. Biol. 380, 107–119 [DOI] [PubMed] [Google Scholar]
- 35. Mattison K., Wilbur J. S., So M., Brennan R. G. (2006) Structure of FitAB from Neisseria gonorrhoeae bound to DNA reveals a tetramer of toxin-antitoxin heterodimers containing pin domains and ribbon-helix-helix motifs. J. Biol. Chem. 281, 37942–37951 [DOI] [PubMed] [Google Scholar]
- 36. Kamada K., Hanaoka F., Burley S. K. (2003) Crystal structure of the MazE/MazF complex: molecular bases of antidote-toxin recognition. Mol. Cell 11, 875–884 [DOI] [PubMed] [Google Scholar]
- 37. Magnuson R., Yarmolinsky M. B. (1998) Corepression of the P1 addiction operon by Phd and Doc. J. Bacteriol. 180, 6342–6351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gotfredsen M., Gerdes K. (1998) The Escherichia coli relBE genes belong to a new toxin-antitoxin gene family. Mol. Microbiol. 29, 1065–1076 [DOI] [PubMed] [Google Scholar]
- 39. Marianovsky I., Aizenman E., Engelberg-Kulka H., Glaser G. (2001) The regulation of the Escherichia coli mazEF promoter involves an unusual alternating palindrome. J. Biol. Chem. 276, 5975–5984 [DOI] [PubMed] [Google Scholar]
- 40. Tam J. E., Kline B. C. (1989) The F plasmid ccd autorepressor is a complex of CcdA and CcdB proteins. Mol. Gen. Genet. 219, 26–32 [DOI] [PubMed] [Google Scholar]
- 41. Wilbur J. S., Chivers P. T., Mattison K., Potter L., Brennan R. G., So M. (2005) Neisseria gonorrhoeae FitA interacts with FitB to bind DNA through its ribbon-helix-helix motif. Biochemistry 44, 12515–12524 [DOI] [PubMed] [Google Scholar]
- 42. Zhang Y., Zhang J., Hoeflich K. P., Ikura M., Qing G., Inouye M. (2003) MazF cleaves cellular mRNAs specifically at ACA to block protein synthesis in Escherichia coli. Mol. Cell 12, 913–923 [DOI] [PubMed] [Google Scholar]
- 43. Dao-Thi M. H., Van Melderen L., De Genst E., Afif H., Buts L., Wyns L., Loris R. (2005) Molecular basis of gyrase poisoning by the addiction toxin CcdB. J. Mol. Biol. 348, 1091–1102 [DOI] [PubMed] [Google Scholar]
- 44. Overgaard M., Borch J., Gerdes K. (2009) RelB and RelE of Escherichia coli form a tight complex that represses transcription via the ribbon-helix-helix motif in RelB. J. Mol. Biol. 394, 183–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Khoo S. K., Loll B., Chan W. T., Shoeman R. L., Ngoo L., Yeo C. C., Meinhart A. (2007) Molecular and structural characterization of the PezAT chromosomal toxin-antitoxin system of the human pathogen Streptococcus pneumoniae. J. Biol. Chem. 282, 19606–19618 [DOI] [PubMed] [Google Scholar]
- 46. Brown B. L., Page R. (2010) Preliminary crystallographic analysis of the Escherichia coli antitoxin MqsA (YgiT/b3021) in complex with mqsRA promoter DNA. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66, 1060–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Afif H., Allali N., Couturier M., Van Melderen L. (2001) The ratio between CcdA and CcdB modulates the transcriptional repression of the ccd poison-antidote system. Mol. Microbiol. 41, 73–82 [DOI] [PubMed] [Google Scholar]
- 48. Garcia-Pino A., Balasubramanian S., Wyns L., Gazit E., De Greve H., Magnuson R. D., Charlier D., van Nuland N. A., Loris R. (2010) Allostery and intrinsic disorder mediate transcription regulation by conditional cooperativity. Cell 142, 101–111 [DOI] [PubMed] [Google Scholar]
- 49. Overgaard M., Borch J., Jørgensen M. G., Gerdes K. (2008) Messenger RNA interferase RelE controls relBE transcription by conditional cooperativity. Mol. Microbiol. 69, 841–857 [DOI] [PubMed] [Google Scholar]
- 50. Winther K. S., Gerdes K. (2012) Regulation of enteric vapBC transcription: induction by VapC toxin dimer-breaking. Nucleic Acids Res. 40, 4347–4357 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.