

Abstract
Background
The cuticle is an important adaptive structure whose origin played a crucial role in the transition of plants from aqueous to terrestrial conditions. HvABCG31/Eibi1 is an ABCG transporter gene, involved in cuticle formation that was recently identified in wild barley (Hordeum vulgare ssp. spontaneum). To study the genetic variation of HvABCG31 in different habitats, its 2 kb promoter region was sequenced from 112 wild barley accessions collected from five natural populations from southern and northern Israel. The sites included three mesic and two xeric habitats, and differed in annual rainfall, soil type, and soil water capacity.
Results
Phylogenetic analysis of the aligned HvABCG31 promoter sequences clustered the majority of accessions (69 out of 71) from the three northern mesic populations into one cluster, while all 21 accessions from the Dead Sea area, a xeric southern population, and two isolated accessions (one from a xeric population at Mitzpe Ramon and one from the xeric ‘African Slope’ of “Evolution Canyon”) formed the second cluster. The southern arid populations included six haplotypes, but they differed from the consensus sequence at a large number of positions, while the northern mesic populations included 15 haplotypes that were, on average, more similar to the consensus sequence. Most of the haplotypes (20 of 22) were unique to a population. Interestingly, higher genetic variation occurred within populations (54.2%) than among populations (45.8%). Analysis of the promoter region detected a large number of transcription factor binding sites: 121–128 and 121–134 sites in the two southern arid populations, and 123–128,125–128, and 123–125 sites in the three northern mesic populations. Three types of TFBSs were significantly enriched: those related to GA (gibberellin), Dof (DNA binding with one finger), and light.
Conclusions
Drought stress and adaptive natural selection may have been important determinants in the observed sequence variation of HvABCG31 promoter. Abiotic stresses may be involved in the HvABCG31 gene transcription regulations, generating more protective cuticles in plants under stresses.
Keywords: Wild barley, HvABCG31, Promoter, Phylogenetic, TFBSs
Background
Over their long evolutionary history, plants have evolved morphological, physiological, biochemical, and genetic traits that enable them to respond to abiotic and biotic stresses [1]. Reports on adaptive evolution in plants are numerous, but in general, specific phenotypes associated with putative adaptive events are not well understood [2,3]. One such event, the origin of the cuticle covering their outermost surfaces, was a key adaptation in the transition of plants from aqueous to terrestrial conditions. The cuticle plays an important role in protecting tissue from environmental stresses [4-6] and is especially important for plants growing in drought-prone environments [7]. One of the most important roles of the cuticle is to provide a diffusion barrier against the uncontrolled loss or uptake of water and gases, although it has many other functions such as protection against UV irradiation, mechanical damage, and phytopathogens and herbivorous insects [8]. Among the many genes involved in the process of cuticle formation, Eibi1 is the only full ABC transporter gene for releasing cuticle compounds reported in cereals. It was identified in wild barley, (Hordeum spontaneum), as “HvABCG31” by analogy with the rice gene [9,10]. The HvABCG31 gene was cloned from a spontaneous mutant of wild barley from Israel. This mutant had a drought-hypersensitive phenotype, with a 50% reduction in the major cutin monomers and an incomplete cuticle surface. HvABCG31 gene was mapped and sequenced and its major function was related to cuticle secretion [10-12].
The distribution centre of Hordeum vulgare ssp. spontaneum, the progenitor of cultivated barley, lies in the “Fertile Crescent”, starting from Israel and Jordan to southern Turkey, Iraq, Kurdistan, and south-western Iran [13,14], and eastward into Tibet [14]. Israel is located at the junction of three continents and harbours remarkable ecosystemic diversity, such as sharp climatic divisions between northern mesic Mediterranean and the southern xeric desert, topographical diversity from the Dead Sea (the lowest altitude on our planet) to the Golan Heights and Mount Hermon. Environmental heterogeneity is a direct driving force for plant evolution in nature, and wide genetic variation is needed for populations to adapt and survive in highly variable and stressful environments. Domestication and modern plant breeding practices have narrowed the genetic diversity in cultivated plants [15-19]. Extraordinary genetic diversity of wild barley from Israel has been recorded [13,19-21]. Wild barley provides a rich source of potential variation for barley improvement because there are no biological barriers to crossing and meiotic recombination between wild and cultivated barley [13,22,23]. The basic characteristics of barley, such as a short life cycle, few chromosomes (2n = 14), predominant self-pollination, along with a high-density genetic map and high genetic synteny with other Triticeae species, facilitate the exploitation of its potential value for crop improvement.
The coding regions of HvABCG31 are conserved in monocot and dicot plants [10]. Increasing evidence indicates that the flanking gene regions are highly dynamic [24-26]. Gene sequence variations reflect the genetic and evolutionary history of organisms [27]. Transcription factor binding sites (TFBSs) located at promoter regions are important for regulating gene expression [28].
In this study, the genetic diversity, phylogenetic relationships, and distribution of TFBSs in the HvABCG31 promoter region were analyzed using 112 wild barley accessions from five natural populations from different habitats in Israel, ranging from the southern xeric Negev desert to the northern mesic Golan Heights. The phylogeny of the HvABCG31 promoter sequence suggested that adaptation to ecogeographic environmental stresses, including aridity, was an important explanatory factor for the observed variation and history of HvABCG31. The analysis of TFBSs in the HvABCG31 promoter region indicated that gibberellin (GA), light, and abiotic stresses may be involved in the HvABCG31 transcriptional regulation.
Results
Genetic analysis of the HvABCG31 promoter sequence from five H. vulgare ssp. spontaneum populations in Israel
Sequences of the HvABCG31 promoter were compared among 112 wild barley accessions from five natural populations: Mitzpe Ramon (P1; southern Israel), Dead Sea (P2; southern), "Evolution Canyon" (P3; northern), Arbel (P4; northern), and Yehudiyya (P5; northern) (Figure 1; Table 1). The total length of the aligned HvABCG31 promoter sequences was 2148 bp. We identified 22 haplotypes; all haplotype sequences were deposited in the DDBJ database ( http://sakura.ddbj.nig.ac.jp/top-e.html) with the accession numbers AB709909–AB709930. Over 90% of the haplotypes were unique to a population: 20 haplotypes were population-specific, while only two were shared between populations in this study (Figure 1; Table 2).
Figure 1.
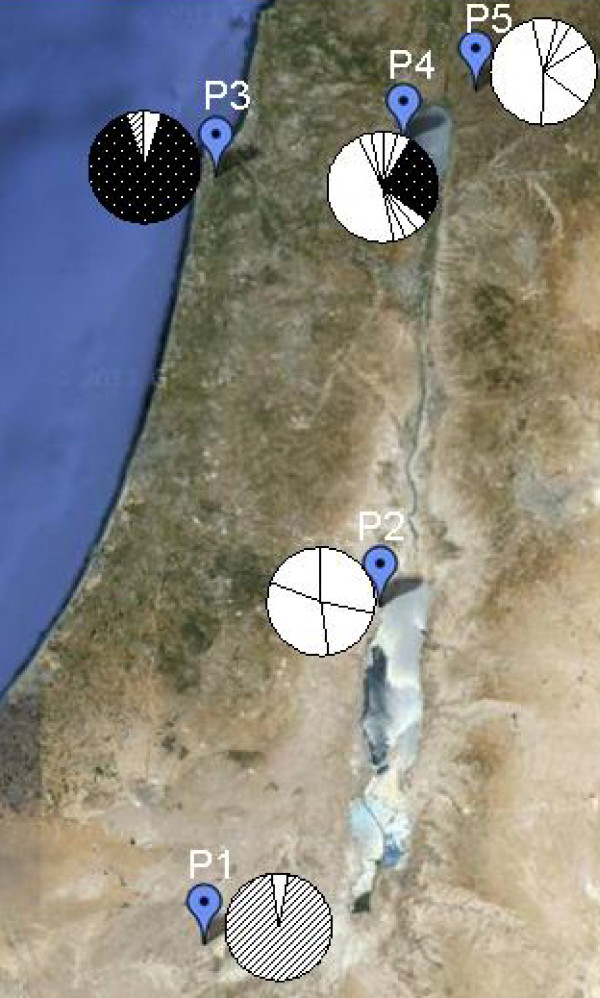
Haplotype distribution in five natural populations of Hordeum vulgare ssp. s (Spontaneum) in Israel. The white sectors in the pies indicate the frequencies of haplotypes unique to that population, while patterned sectors denote the frequencies of haplotypes shared between populations (for frequency details, see Table 2). P1, Mitzpe Ramon; P2, Dead Sea; P3, "Evolution Canyon"; P4, Arbel; P5, Yehudiyya.
Table 1.
Description of the natural wild barley (Hordeum vulgare ssp. spontaneum) populations sampled in this study
| Population | Number of accessions | Location | Latitude (N) Longitude(E) | Altitude(m) | Mean annual rainfall(mm) | Mean January temperature(°C) | Minimal January temperature(°C) | Annual evaporation(cm) | Soil type |
|---|---|---|---|---|---|---|---|---|---|
| P1 |
20 |
Mitzpe Ramon |
30°37'26.6" 34°50'22.4" |
519 |
69.4 |
9.8 |
2.3 |
240 |
Desert alluvium |
| P2 |
21 |
Dead Sea |
31°43'15.8" 35°27'05.3" |
−390 |
97.7 |
14 |
4.8 |
260 |
Desert alluvium |
| P3 |
19 |
“Evolution Canyon” |
32°42'48" 34°58'34" |
61 |
621.8 |
11.8 |
2.5 |
180 |
Terra rossa |
| P4 |
26 |
Arbel |
32°49'19.0" 35°29'43.2" |
133 |
448.7 |
10.5 |
2.2 |
230 |
Terra rossa |
| P5 | 26 | Yehudiyya | 32°56'07.6" 35°41'50.3" | 172 | 521.1 | 9.9 | 2.1 | 230 | Basaltic |
Table 2.
Haplotype frequencies in five Israeli natural population of wild barley (Hordeum vulgare ssp. spontaneum)
| P1 (20) | P2 (21) | P3 (19) | P4 (26) | P5 (26) | |
|---|---|---|---|---|---|
| Hap 1 |
0.95 (19) |
0 |
0.0526(ES) (1) |
0 |
0 |
| Hap 2 |
0.05 (1) |
0 |
0 |
0 |
0 |
| Hap 3 |
0 |
0.286 (6) |
0 |
0 |
0 |
| Hap 4 |
0 |
0.19 (4) |
0 |
0 |
0 |
| Hap 5 |
0 |
0.333 (7) |
0 |
0 |
0 |
| Hap 6 |
0 |
0.19 (4) |
0 |
0 |
0 |
| Hap 7 |
0 |
0 |
0.895(ES + AS) (17) |
0.269 (7) |
0 |
| Hap 8 |
0 |
0 |
0.0526(AS) (1) |
0 |
0 |
| Hap 9 |
0 |
0 |
0 |
0.0385 (1) |
0 |
| Hap 10 |
0 |
0 |
0 |
0.0385 (1) |
0 |
| Hap 11 |
0 |
0 |
0 |
0.0385 (1) |
0 |
| Hap 12 |
0 |
0 |
0 |
0.462 (12) |
0 |
| Hap 13 |
0 |
0 |
0 |
0.0385 (1) |
0 |
| Hap 14 |
0 |
0 |
0 |
0.0385 (1) |
0 |
| Hap 15 |
0 |
0 |
0 |
0.0385 (1) |
0 |
| Hap 16 |
0 |
0 |
0 |
0.0385 (1) |
0 |
| Hap 17 |
0 |
0 |
0 |
0 |
0.0769 (2) |
| Hap 18 |
0 |
0 |
0 |
0 |
0.192 (5) |
| Hap 19 |
0 |
0 |
0 |
0 |
0.154 (4) |
| Hap 20 |
0 |
0 |
0 |
0 |
0.462 (12) |
| Hap 21 |
0 |
0 |
0 |
0 |
0.0769 (2) |
| Hap 22 | 0 | 0 | 0 | 0 | 0.0385 (1) |
Note: ES refers to ‘European Slope’ and AS refers to ‘African Slope’ of “Evolution Canyon”, Lower Nahal Oren, Mount Carmel [31]; The number in the bracket refers to the number of accessions in each population.
Based on the major environmental factors of average rainfall and soil type (Table 1), the five populations could be divided into the southern xeric group (P1 and P2) and the northern mesic group (P3, P4, and P5). The northern mesic group included 17 haplotypes, most with low frequencies; whereas six haplotypes were found in the southern xeric group. Four of five populations harboured a population-specific high-frequency haplotype; the exception, P2, comprised several haplotypes in similar proportions (Figure 1). In population P1, the majority of accessions (19 of 20) had one haplotype (Hap 1), and in population P3, 17 of 19 accessions shared one haplotype (Hap 7). Interestingly, no haplotypes were shared between P1 and P2 (southern) or between P4 and P5 (northern). Two haplotypes were shared between populations: Hap 7 occurred in most accessions (17 of 19) of P3 and in some accessions in P4, which has the same soil type (terra rossa); Hap 1 was shared in P3 and P1, although the P3 accession was an isolated individual from the ‘European slope’ of “Evolution Canyon”.
The genetic variation within and between populations was calculated by AMOVA. The genetic divergences were remarkably high (Table 3). Interestingly, more genetic variation occurred within populations than among populations (54.2% and 45.8%, respectively); the estimated Fst was.
Table 3.
Analysis of molecular variance (AMOVA)
| Source of variation | d.f. | Sum of squares | Variance components | Percentage of variation |
|---|---|---|---|---|
| Among groups |
1 |
5.913 |
0.022 Va |
4.4 |
| Among populations within groups |
3 |
14.729 |
0.207 Vb |
41.4 |
| Within populations |
107 |
28.929 |
0.270 Vc |
54.2 |
| Total | 111 | 49.571 | 0.499 |
The genetic diversity analysis and the neutrality test results in different populations are summarized in Table 4. There were more haplotypes, and greater haplotype diversity (Hd) in two northern populations (P4 and P5), but the highest Hd and nucleotide diversity (π) were observed in P2 (Dead Sea). Two neutrality tests (Tajima, and Fu & Li tests) showed that different populations experienced different selection pressures. No significant evidence for selection was identified in P4 and P5, which together harboured 15 haplotypes. Significant positive values were obtained for P2 using both the Tajima and Fu & Li tests, suggesting balancing selection on the HvABCG31 promoter region in this population. In contrast, significant negative values for both tests were obtained for P1 and P3, which could have been caused by purifying selection, demographic effects (i.e., population expansion), or low frequencies of harmful mutations [2,29,30]. However, despite predominant haplotypes, low-frequency haplotypes were found in both P1 and P3 (Figure 1; Table 2). One accession from P1 had Hap 2, and two accessions from P3 had Hap 1 and Hap 8.
Table 4.
Genetic diversity and neutrality test
| P1 | P2 | P3 | P4 | P5 | Total | |
|---|---|---|---|---|---|---|
| Number of accessions |
20 |
21 |
19 |
26 |
26 |
112 |
| Number of haplotypes, h |
2 |
4 |
3 |
9 |
6 |
22 |
| Haplotype diversity, Hd |
0.100 |
0.771 |
0.205 |
0.732 |
0.742 |
0.893 |
| Nucleotide diversity, π |
0.001 |
0.009 |
0.002 |
0.0004 |
0.001 |
0.006 |
| Tajima's D test |
−2.452 *** |
2.244 * |
−2.278 ** |
−1.062 |
−1.119 |
−0.553 |
| Fu and Li's F* test | −3.917 ** | 2.179 ** | −3.068 ** | −2.176 | 0.567 | −0.133 |
Statistical significance in Tajima's D test: * P < 0.05, ** P < 0.01, *** P < 0.001; Statistical significance in Fu & Li's F* test: **P < 0.02.
Phylogenetic analysis of H. vulgare ssp. spontaneum HvABCG31 promoter sequence
To investigate the relationships among the different populations, a phylogenetic tree based on SNP haplotypes was constructed using maximum likelihood as implemented in MEGA5 software (Figure 2). The tree was rooted with the promoter sequence of a putative HvABCG31 homologue from the wheat (Triticum aestivum var. Chinese Spring) 3B chromosome. Most accessions (69/71 = 97.2%) from the three northern mesic populations (from 89.5% of P3, 100% of P4, and 100% of P5) formed one cluster (Cluster 1). This cluster was further divided into two major sub-clusters with high support values: Cluster 1-a from P3 and P4 and Cluster 1-b from P5. Interestingly, the structure of the sub-clusters was in accordance with the soil types of their collection sites (terra rossa in Cluster 1-a; basalt in Cluster 1-b). The second cluster (Cluster 2) included all 21 accessions from P2, one accession from the xeric population of P1, and one accession from the xeric ‘African Slope’ of P3. One shared haplotype (Hap 1) from both P1 and P3 proved very distant from both clusters and represented most of the accessions from P1 (19/20 = 95%) and one accession from the ‘European Slope’ of P3.
Figure 2.
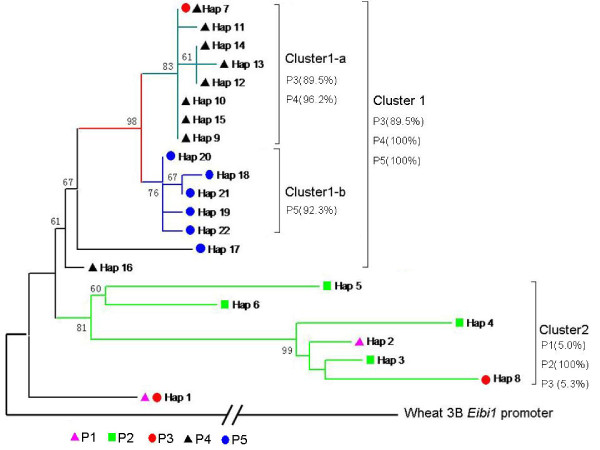
Maximum likelihood bootstrap consensus phylogeny of 2 kb sequence of the HvABCG31 promoter. The phylogenetic consensus tree was constructed using the method of maximum likelihood in MEGA5. Values at the nodes are bootstrap percentage > 60%. Percentages below the cluster names refer to the accession proportions in specific populations. P1, Mitzpe Ramon; P2, Dead Sea; P3, "Evolution Canyon"; P4, Arbel; P5, Yehudiyya.
The distances among the haplotypes in Cluster 1 (including Clusters 1-a and 1-b) were closer than those in Cluster 2 (see Figures 2, 3). Figure 3 depicts this pattern in more detail. The total number of polymorphic sites (90) was determined from the alignment of all 22 haplotypes (see Additional file 1: Table S1). The number of differences between each of the haplotypes and the consensus sequence varied from 2–30. There were 15 haplotypes in Cluster 1 and six in Cluster 2; one haplotype (Hap 1) was separated from all others. The majority of haplotypes (13 of 15) in Cluster 1 differed by 2–5 bases from the consensus sequence, with the exceptions of Hap 16 (8 differences) and Hap 17 (13 differences). In Cluster 2, haplotypes differed from the consensus sequence by 16–30 bases, and the isolated Hap 1 had 16 differences. A total of 21 rare polymorphisms were scattered among different haplotypes represented by one or two accessions. Rare sites were found in about half of the haplotypes (5/9 = 55.6%) from P4, in one haplotype from each P1 and P3, and in two haplotypes from P5. Most of the rare sites occurred in a few haplotypes, like Hap 17 from P5 (with 6 rare sites) and Hap 8 from P3 (with 7 rare sites). Most of the rare polymorphic sites (19 of 21) appeared in three northern mesic populations (P3, P4, and P5). Southern arid populations had fewer haplotypes, but they differed from the consensus sequence at a large number of positions, while northern mesic populations had more haplotypes that were, on average, closer to the consensus sequence.
Figure 3.
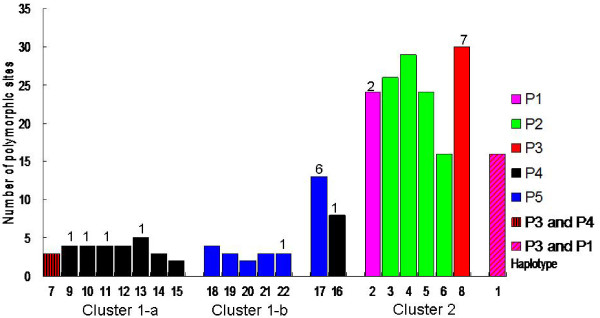
Differences between haplotypes and the consensus sequence of HvABCG31 promoter in Hordeum vulgare ssp. spontaneum. The number of rare polymorphic sites for each haplotype are above the bars. P1, Mitzpe Ramon; P2, Dead Sea; P3, "Evolution Canyon"; P4, Arbel; P5, Yehudiyya.
Transcription factor binding sites (TFBSs) in the HvABCG31 promoter
The total number of TFBSs was 148 with a range of 121–134 among the haplotypes. There were 121–128 (P1) and 121–134 (P2) TFBSs in the two southern arid populations, and 123–128 (P3), 125–128 (P4), and 123–125 (P5) sites in the three northern mesic populations. The TFBSs could be classified into 8 categories according to their basic functions (see Additional files 2 and 3: Table S2 and Table S3). Because the TFBSs were rather short (4–12 bases) and could have occurred randomly, their presence in the promoter sequences were statistically analyzed using a permutation test (10,000 runs). A few TFBSs related to GA, Dof, and light were significant (Figure 4; red). Many binding sequences related to various functions occurred at low frequency and were not significant. Most were singletons or duplets, but some more common ones for GA (9–10 copies) and light (5 copies) also proved to be non-significant. One singleton with a long binding sequence (12 bp) proved statistically significant (see Figure 4).
Figure 4.
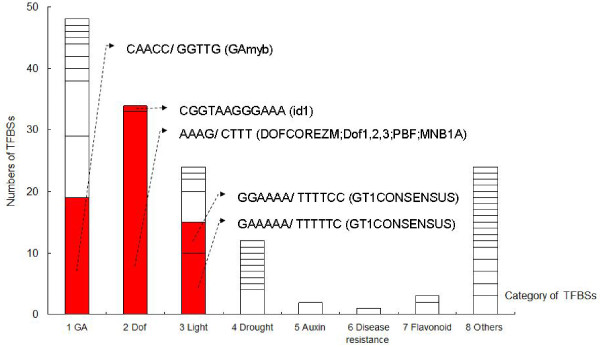
Numbers and categories of transcription factor binding sites (TFBSs) in the HvABCG31 promoter of Hordeum vulgare ssp. spontaneum. Different grids in the histogram refer to specific transcription factor binding sites; Sites indicated in red occurred at significantly higher frequencies than random, based on a permutation test with 10,000 runs; their sequences are given and related transcription factors are listed in brackets.
Of the 148 TFBSs, about 70% were located in the conserved regions of the 22 haplotypes, whereas the rest appeared in the non-conserved regions (Figure 5, upper pie chart). Interestingly, the conserved regions included a high proportion of TFBSs related to GA, Dof, and light (Figure 5, bottom left). In contrast, in the non-conserved regions, the GA TFBSs signals were predominant (44%) (Figure 5, bottom right). For example: among the 45 TFBSs in non-conserved regions, 20 were related to GA. Figure 6 shows the distribution of non-conserved TFBSs in all 22 haplotypes: of the 45 non-conserved TFBSs, 21 were absent in the minority haplotypes (Figure 6, upper dash area), while 19 appeared in some minor haplotypes (Figure 6, bottom dash area). A high proportion of non-conserved TFBSs (40/45 = 88.9%) affected minor haplotypes. Most of these minor haplotypes (bold and italic numbers in Additional file 2: Table S2) were related to haplotypes 1–6, 8 in 43 accessions; most of these accessions (41/43 = 95.3%) originated from southern xeric populations (P1 and P2); the other two accessions came from the two slopes (‘African Slope’ and ‘European Slope’) of P3.
Figure 5.
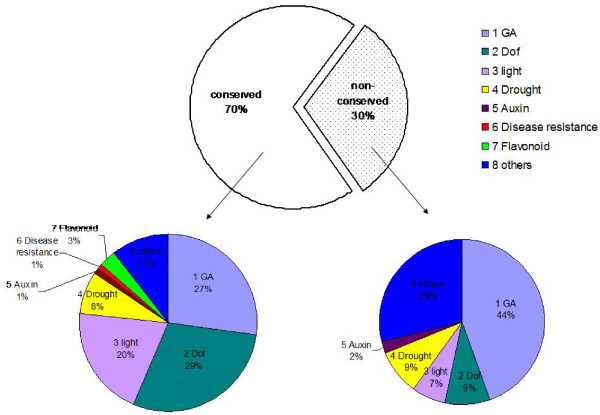
Proportions of conserved and non-conserved TFBSs in the Hordeum vulgare ssp. spontaneum HvABCG31 promoter sequence. The TFBSs were analysed using PlantPan database ( http://plantpan.mbc.nctu.edu.tw/index.php). The upper pie chart showed the over all proportions of conserved and non-conserved TFBSs, and the two lower pie charts showed the proportions of different categories of TFBSs within each group.
Figure 6.
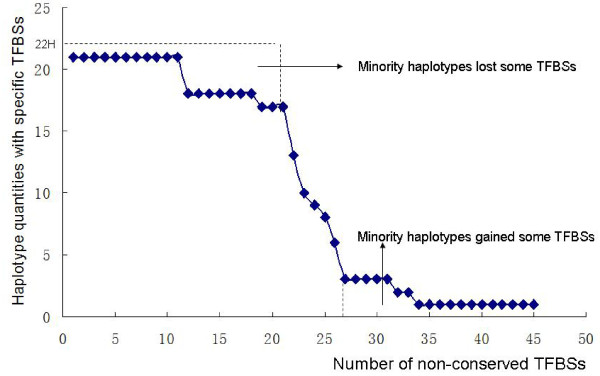
Distribution of non-conserved TFBSs in all 22 haplotypes of the HvABCG31 promoter. The upper dashed area refer to TFBSs that occurred in a majority of haplotypes, and the lower dashed area refer to those occurring in a minority of haplotypes.
Discussion
Genetic differentiation of HvABCG31 promoter sequences
High genetic diversity was found in the HvABCG31 promoter region (Hd = 0.893; π = 0.006) (Table 4) in the present study. Similar results for the Dehydrin 1 promoter region (Hd = 0.946; π = 0.0054) were reported in a study of wild barley populations from Israel [31]. Numerous studies at global, regional, and local scales have demonstrated that genetic diversity is associated with stressful environments. In wild barley, this was shown using allozymes and DNA markers (RAPDs, SSRs, AFLPs, and rDNA) [32-36]. Drought is a major evolutionary driving force affecting plant genetic diversity and population genetic structure [37,38]. The sharp climatic division between the northern mesic Mediterranean region and southern xeric Negev Desert in Israel generates a gradient of increasing aridity. Our phylogenetic analysis of HvABCG31 promoter sequences grouped most accessions (69/71 = 97.2%) of the northern populations (P3, P4, and P5) into one cluster, whereas all accessions from the Dead Sea (P2), one accession from Mitzpe Ramon (P1), and one accession from the ‘African Slope’ of “Evolution Canyon” (P3) formed a second cluster. Interestingly, there were many indel polymorphisms in our five natural populations but the basic phylogenetic structure was similar whether or not indels were considered (results not shown). The major ecological factors differentiating the northern (P3, P4, and P5) and southern (P1 and P2) populations, excluding geographic distance, were average annual rainfall and soil type. Water availability is the most important environmental factor for plant growth [22,39]. Drought stress is influenced by several factors including soil type, rainfall, temperature, and rate of evaporation. Interestingly, the sub-clusters in Cluster 1 were characterized by different soil types, terra rossa in Cluster 1-a and basalt in Cluster 1-b. Therefore, drought stress may explain this separation [40-42]; however, we cannot exclude the influence of other factors, such as isolation by distance.
In the current study significant evidence against neutrality (based on Tajima's D test and Fu & Li's F* test) was found in the two southern populations (P1 and P2) and in one northern populations (P3), while no significant deviations from neutrality were found in the other two northern populations (P4 and P5). Significant positive values of the test statistics indicate balancing selection, while significant negative values may suggest purifying selection and demographic effects (population expansion) [2,29,30,43-45]. Balancing selection detected in population P2 may relate to environmental heterogeneity. Our Mitzpe Ramon (P1) population is located in stressful habitats in the Negev Desert, and P2 is located in the Dead Sea area, with highly heterogeneous and stressful conditions including severe drought and salinity stresses. “Evolution Canyon” (P3) consist of two opposite slopes: the warm-dry ‘African Slope’ is drastically divergent from the cool and humid ‘European Slope’, although they are separated by an average distance of only 200 meters. Much research on “Evolution Canyon” has proved that the interslope divergent selection is due to microclimate [46,47] (see the publication list: http://evolution.haifa.ac.il/index.php/component/content/article/29).Different selection pressures in different regions are important in adaptation to local habitats [48,49]. Our results allowed us to assume that different selection pressures in our five populations affected adaptation to different environments. Water availability may be an important selection pressure [38]. However, more extensive sampling is needed to support the hypothesis that polymorphisms in the HvABCG31 promoter are maintained by variable drought and salinity.
HvABCG31 expression may be regulated by transcription factors related to abiotic stress
Although HvABCG31 is involved in cuticle formation [10], only three TFBSs related to flavonoid biosynthesis were found in its promoter region. TFBSs copy numbers are considered to be important for transcriptional regulation [50]. The TFBSs related to three factors (GA, Dof, and light) were significantly enriched in the HvABCG31 promoter. Previous studies showed that cuticle formation is affected by drought and light [51-53]. The Dof proteins are a family of plant-specific transcription factors involved in the control of multiple functions in plant growth and development including stress, light, and plant hormone responses [54]. GA is an important plant hormone that controls diverse aspects of growth and development [55]. In the non-conserved regions of the HvABCG31 promoter, most base changes (44%) added or deleted GA-binding sites. Of note, a recent microarray analysis of drought-stressed wild emmer wheat roots indicated that GA may be involved in drought resistance [56]. In addition, GA plays a crucial role in barley protection under drought and other stresses [57]. GA, light, and abiotic stresses may affect HvABCG31 transcription, which may indirectly indicate that HvABCG31 affects abiotic stress tolerance, although other processes may influence HvABCG31.
Base variation can dramatically change TFBSs and thus affect gene regulation [28]. We found 45 non-conserved TFBSs, some of which were lost or gained in the HvABCG31 promoters of minor haplotypes, mainly in the southern (xeric) populations. An early paper reported that TFBSs can appear or disappear among closely related species and even within populations [26]. Nevertheless, poor correlation exists between the divergence of TFBS sequences and gene expression, suggesting that gene expression might be regulated by compensatory mechanisms [58]. Loss of binding sites may be buffered by the presence of other binding sites for additional factors that are involved in the same process or that bind cooperatively with the factor whose binding site has diverged [59]. It remains unknown whether such compensatory mechanisms have evolved in the control of barley HvABCG31; more expression experiments are required to further understand its regulation.
Conclusions
Our phylogenetic analysis of eco-geographical variation in the 2kb promoter region of the HvABCG31 gene in five natural populations of H. vulgare ssp. spontaneum in Israel suggests that aridity may be an important factor affecting the observed sequence variation. The analysis of TFBSs in the HvABCG31 promoter region indicated that GA, light, and abiotic stresses may be involved in the HvABCG31 transcription regulation and that HvABCG31 may be involved in abiotic stress responses, both locally and regionally, thereby generating adaptive structures in response to environmental stresses.
Methods
Plant materials
Seeds of 112 accessions of H. vulgare ssp. spontaneum were collected from five natural populations in Israel during April 2007. The seeds were collected randomly from individuals situated at least 1–2 m apart [31]. We analyzed the genotypes of 19 to 26 accessions per population. The five collection sites were characterized by distinct environmental factors (Table 1). The two sites in southern Israel were (P1) Mitzpe Ramon, located in the Negev Desert and (P2) the Dead Sea area, known for its highly saline and arid environment. In northern Israel, “Evolution Canyon” (P3), at Lower Nahal Oren, Mount Carmel, near the Mediterranean coast, is a designated research microsite harbouring two distinct habitats (cool-mesic and warm-xeric) with an average distance of 200 m [32,46,47,60]. Plants were collected from similar altitudes but different slopes: 11 from the xeric south facing slope designated as ‘African Slope’ and 8 from the mesic northern facing slope, designated as ‘European Slope’; The final two populations in northern Israel were (P4) Arbel mountain, close to the Sea of Galilee; and (P5) Yehudiyya, located in the Golan Heights. The latitude, longitude, and altitude data indicated in Table 1 were recorded by a GPS (Global Positioning System) receiver, and soil types were recorded when sampled in the fields. The climatic data (average rainfall, temperatures in the barley growth season, and evaporation rates) were obtained from the BioGIS-Israel biodiversity web site ( http://www.biogis.huji.ac.il/Map.aspx). All the data were checked using a relevant reference [61] and the records of the nearest meteorological stations from 1980–2009 (provided by the Israeli Meteorological Service: http://ims.gov.il/IMS/CLIMATE).
DNA extraction, amplification, and sequencing
Seeds were germinated in Petri dishes on filter paper with purified water, treated with cold (4°C) for 7 d then held at 25°C for 3 d. Seedlings were then transplanted into pots (16 cm high, 18 cm diameter) with a cycle of 16 h, 22°C day and 8 h, 18°C night. Genomic DNA was extracted from young leaves using a small-scale isolation method [62]. Based on our previous studies of HvABCG31[11,12], one pair of primers was designed to amplify the gene promoter region: EIBI1antiR13678: TGAGCAAAGGAGCAAGGA and ABCcontig7F1550: CGGGGAGCAAAGAAAATGTA. Four single primers were used to sequence the promoter: ABCcontig7F1767: ACTACGGGCGACCTGAGCA; EIBI1F11844: TCTTTGATCGTTGGGGTTTT; EIBI1F12436: GTGCCCCGTATTGTTCTCAT and EIBI1antiF13068: AGATTTTCCACCATGCCTGT. All primers were designed using Primer3 ( http://frodo.wi.mit.edu/primer3/).
The PCR amplifications were carried out in a C1000TM thermal cycler (Bio-Rad, Hercules, CA, USA). Each 50 μL PCR reaction contained 20 ng template DNA, 5 μL 10× PCR buffer, 1.25 U ExTaq DNA polymerase (Takara, Beijing, China), 5 μL MgCl2 (25 mM), 4 μL dNTP (2.5 uM), 1.5 μL each primer (10 uM), 3 μL DMSO (Dimethyl Sulfoxide, Tianjin, China) and water. The PCR procedure consisted of an initial denaturing step (94°C, 5 min); followed by 30 cycles of 94°C for 1 min, 60°C for 1 min, and 72°C for 2 min, and completed by an incubation at 72°C for 15 min. Amplified DNA products were electrophoresed on 1–2% w/v agarose gels and sequenced on an ABI 3100 DNA sequencer (Applied Biosystems, Foster City, CA, USA).
DNA sequences analysis
Overlapping DNA sequences were assembled using DNAMAN6.0 ( http://en.bio-soft.net/format/DNAMAN.html), and ambiguous sites were checked manually in accordance with the DNA peak files in Chromas2.01 ( http://www.technelysium.com.au/chromas_lite.html). Sequences were aligned with the MUSCLE feature in MEGA5 ( http://www.megasoftware.net/) [63,64]. Genetic diversity, haplotype identification, and neutrality tests (Tajima tests and Fu & Li tests) were conducted in DnaSP5.10 ( http://www.ub.edu/dnasp/). Gaps were considered a fifth state [65]. Haplotypes occurring in a single population were considered to be unique, and in more than one population were considered shared. An analysis of molecular variance (AMOVA) was performed in Arlequin 3.1 ( http://cmpg.unibe.ch/software/arlequin3/) based on the haplotype frequencies [66]. Genetic differentiation was evaluated by the F statistics, Fst [67,68]. Polymorphic sites were determined manually based on an alignment of all haplotypes, and each haplotype was characterized by the number of sites by which it differed from the consensus sequence [69,70]. An allele was considered to be rare in fewer than 2% (1–2 accessions) in this study. Tandem gaps were treated as a single mutant event (insertion or deletion) and were therefore counted as single polymorphic sites. Each polymorphic (polymorphic) site was considered an independent unit of variation in the analysis [71].
A phylogenetic tree of haplotypes sequences was constructed using the maximum likelihood (ML) algorithm in MEGA5, with 1000 bootstrap replicates to gauge support, and a 60% cut off was used in the analysis. The phylogenetic tree was rooted using 2 kb of the putative homologue from wheat which was obtained from a BLAST search of the 3B chromosome sequences (kindly provided by Frédéric Choulet, INRA; Clermont Ferrand, France). All haplotypes of the H. vulgare ssp. spontaneum HvABCG31 promoter were scanned for TFBSs using the PlantPan database ( http://plantpan.mbc.nctu.edu.tw/index.php), which combines several plant TFBS databases, such as PLACE, TRANSFAC, JASPER, and AGRIS [72]. The search was conducted using the monocots transcription factor libraries (wheat, barley, maize, and rice) because distinct differences in the evolution of an upstream region exist between monocots and dicots [73]. A special script was developed to test the significance of highly abundant binding signals of specific TFs. Using this program, the original DNA sequences were reshuffled to produce randomized sequences, which were scored for the occurrence of specific TFBS signals that proved abundant in our original sequences. Using 10,000 such permutation runs, we evaluated the statistical significance of these original binding signals.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XM, GJ, and GC had the main responsibility for the study and participated in all aspects, including developing the main idea, hypotheses, setting up the study, collecting some of the data, analyzing the data, and preparing and revising the manuscript. CL, AW, MP, SS, and TK carried out the molecular genetic studies, including DNA extraction, sequencing and data analysis. HS, DW, TK, XM, EN, and AK interpreted data and helped to revise the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Table S1. Detailed information for the polymorphic sites sample.
Table S2. Detailed information for 22 haplotype in wild barley (Hordeum vulgare ssp. spontaneum).
Table S3. Detailed information for TFBSs classification.
Contributor Information
Xiaoying Ma, Email: mxy1203@126.com.
Hanan Sela, Email: dooshra@gmail.com.
Genlin Jiao, Email: jiaogenlin@szum.gov.cn.
Chao Li, Email: chaoli@lzb.ac.cn.
Aidong Wang, Email: cabin.wang@gmail.com.
Mohammad Pourkheirandish, Email: mkheiran@affrc.go.jp.
Dmitry Weiner, Email: dmitry.weiner@gmail.com.
Shun Sakuma, Email: ssakuma@affrc.go.jp.
Tamar Krugman, Email: krugman@research.haifa.ac.il.
Eviatar Nevo, Email: nevo@research.haifa.ac.il.
Takao Komatsuda, Email: takao@affrc.go.jp.
Abraham Korol, Email: korol@research.haifa.ac.il.
Guoxiong Chen, Email: guoxiong@lzb.ac.cn.
Acknowledgements
We thank Frédéric Choulet for kindly providing the wheat 3B HvABCG31 promoter sequence, and Avigdor Beiles for constructive discussions. This work was supported by Grant O827751002 and 29Y127E71 from “One Hundred Talents” Project of The Chinese Academy of Sciences, grants 30970449 and 31170369 from the National Natural Science Foundation of China, and grant from the Ancell Tericher Research Foundation of Genetics and Molecular Evolution.
References
- Nevo E. Evolution under environmental stress at macro-and microscales. Genome Biol Evol. 2011;2:1039–1052. doi: 10.1093/gbe/evr052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SI, Gaut BS. Molecular population genetics and the search for adaptive evolution in plants. Mol Biol Evol. 2005;22:506–519. doi: 10.1093/molbev/msi035. [DOI] [PubMed] [Google Scholar]
- Leino MW, Hagenblad J. Nineteenth century seeds reveal the population genetics of landrace barley (Hordeum vulgare) Mol Biol Evol. 2010;27:964–973. doi: 10.1093/molbev/msp308. [DOI] [PubMed] [Google Scholar]
- Samuels L, Kunst L, Jetter R. Sealing plant surfaces: cuticular wax formation by epidermal cells. Plant Biol. 2008;59:683–707. doi: 10.1146/annurev.arplant.59.103006.093219. [DOI] [PubMed] [Google Scholar]
- Raven JA, Edwards D. In: The evolution of plant physiology: from whole plants to ecosystems. Alan R, editor. Amsterdam: Elsevier; 2004. Physiological evolution of lower embryophytes: adaptations to the terrestrial environment; pp. 17–41. [Google Scholar]
- Nawrath C. Unraveling the complex network of cuticular structure and function. Curr Opin Plant Biol. 2006;9:281–287. doi: 10.1016/j.pbi.2006.03.001. [DOI] [PubMed] [Google Scholar]
- Riederer M, Schreiber L. Protecting against water loss: analysis of the barrier properties of plant cuticles. J Exp Bot. 2001;52:2023–2032. doi: 10.1093/jexbot/52.363.2023. [DOI] [PubMed] [Google Scholar]
- Kunst L, Jetter R, Samuels AL. Biosynthesis and transport of plant cuticular waxes. Annual Plant Reviews Volume 23: Biology of the Plant Cuticle. 2006. pp. 182–215.
- Li C, Wang AD, Ma XY, Nero E, Chen GX. Consensus maps of cloned plant cuticle genes. Sci Cold Arid Regi. 2010;2:465–476. [Google Scholar]
- Chen G, Komatsuda T, Ma JF, Nawrath C, Pourkheirandish M, Tagiri A, Hu YG, Sameri M, Li X, Zhao X. An ATP-binding cassette subfamily G full transporter is essential for the retention of leaf water in both wild barley and rice. Proc Natl Acad Sci U S A. 2011;108:12354–12359. doi: 10.1073/pnas.1108444108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Sagi M, Weining S, Krugman T, Fahima T, Korol AB, Nevo E. Wild barley eibi1 mutation identifies a gene essential for leaf water conservation. Planta. 2004;219:684–693. doi: 10.1007/s00425-004-1277-7. [DOI] [PubMed] [Google Scholar]
- Chen GX, Komatsudu T, Pourkheirandish M, Sameri M, Sato K, Krugman T, Fahima T, Korol AB, Nevo E. Mapping of the eibi1 gene responsible for the drought hypersensitive cuticle in wild barley (Hordeum spontaneum) Breed Sci. 2009;59:21–26. doi: 10.1270/jsbbs.59.21. [DOI] [Google Scholar]
- Nevo E. Origin, evolution, population genetics and resources for breeding of wild barley, Hordeum spontaneum, in the Fertile Crescent. Barley: Genetics, Molecular Biology and Biotechnology, Shewry, P (ed) CAB International. 1992. pp. 19–43.
- Zohary D, Hopf M, Weiss E. Domestication of Plants in the Old World: The Origin and Spread of Domesticated Plants in Southwest Asia, Europe, and the Mediterranean Basin. New York: Oxford University Press; 2012. [Google Scholar]
- Doebley JF, Gaut BS, Smith BD. The molecular genetics of crop domestication. Cell. 2006;127:1309–1321. doi: 10.1016/j.cell.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Ross-Ibarra J, Morrell PL, Gaut BS. Plant domestication, a unique opportunity to identify the genetic basis of adaptation. Proc Natl Acad Sci U S A. 2007;104:8641–8648. doi: 10.1073/pnas.0700643104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger JC, Chapman MA, Burke JM. Molecular insights into the evolution of crop plants. Am J Bot. 2008;95:113–122. doi: 10.3732/ajb.95.2.113. [DOI] [PubMed] [Google Scholar]
- Buckler ES, Thornsberry JM, Kresovich S. Molecular diversity, structure and domestication of grasses. Genet Res. 2001;77:213–218. doi: 10.1017/s0016672301005158. [DOI] [PubMed] [Google Scholar]
- Nevo E. Genome evolution of wild cereal diversity and prospects for crop improvement. Plant Genet Resour. 2006;4:36–46. doi: 10.1079/PGR2006108. [DOI] [Google Scholar]
- Nevo E, Beiles A, Zohary D. Genetic resources of wild barley in the Near East: structure, evolution and application in breeding. Biol J Linn Soc. 1986;27:355–380. doi: 10.1111/j.1095-8312.1986.tb01742.x. [DOI] [Google Scholar]
- Vanhala T, Van Rijn C, Buntjer J, Stam P, Nevo E, Poorter H, Van Eeuwijk F. Environmental, phenotypic and genetic variation of wild barley (Hordeum spontaneum) from Israel. Euphytica. 2004;137:297–309. [Google Scholar]
- Nevo E, Chen G. Drought and salt tolerances in wild relatives for wheat and barley improvement. Plant Cell Environ. 2010;33:670–685. doi: 10.1111/j.1365-3040.2009.02107.x. [DOI] [PubMed] [Google Scholar]
- Nevo E. In: Advance in Barley Sciences. 15-20 April 2012; Hangzhou, China. Li C, Zhang G, Liu X, Eglinton J, editor. Hangzhou: Zhejiang University Press- Springer; 2012. Evolution of wild barley and barley improvement; pp. 1–16. (Proceedings of 11th Int. Barley Genetics Symposium). Volume. [Google Scholar]
- Wunderlich Z, Mirny LA. Different gene regulation strategies revealed by analysis of binding motifs. Trends Genet. 2009;25:434–440. doi: 10.1016/j.tig.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens S, Vingron M. Studying the evolution of promoter sequences: a waiting time problem. J Comput Biol. 2010;17:1591–1606. doi: 10.1089/cmb.2010.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JR, Wray GA. Rapid evolution of cis-regulatory sequences via local point mutations. Mol Biol Evol. 2001;18:1764–1770. doi: 10.1093/oxfordjournals.molbev.a003964. [DOI] [PubMed] [Google Scholar]
- Maeso I, Roy SW, Irimia M. Widespread recurrent evolution of genomic features. Genome Biol Evol. 2012;4:486–500. doi: 10.1093/gbe/evs022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TM, Park PJ. Advances in analysis of transcriptional regulatory networks. Wiley Interdiscip Rev Syst Biol Med. 2011;3:21–35. doi: 10.1002/wsbm.105. [DOI] [PubMed] [Google Scholar]
- Fu YX. New statistical tests of neutrality for DNA samples from a population. Genetics. 1996;143:557–570. doi: 10.1093/genetics/143.1.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. The amount of DNA polymorphism maintained in a finite population when the neutral mutation rate varies among sites. Genetics. 1996;143:1457–1465. doi: 10.1093/genetics/143.3.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Zhang T, Bolshoy A, Beharav A, Nevo E. Adaptive microclimatic structural and expressional dehydrin 1 evolution in wild barley, Hordeum spontaneum, at 'Evolution Canyon', Mount Carmel, Israel. Mol Ecol. 2009;18:2063–2075. doi: 10.1111/j.1365-294X.2009.04140.x. [DOI] [PubMed] [Google Scholar]
- Nevo E. Evolution of genome–phenome diversity under environmental stress. Proc Natl Acad Sci U S A. 2001;98:6233–6240. doi: 10.1073/pnas.101109298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronin JK, Bundock PC, Henry RJ, Nevo E. Adaptive climatic molecular evolution in wild barley at the Isa defense locus. Proc Natl Acad Sci U S A. 2007;104:2773–2778. doi: 10.1073/pnas.0611226104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevo E. Molecular evolution and ecological stress at global, regional and local scales: The Israeli perspective. J Exp Zool. 1998;282:95–119. doi: 10.1002/(SICI)1097-010X(199809/10)282:1/2<95::AID-JEZ12>3.0.CO;2-F. [DOI] [Google Scholar]
- Owuor E, Beharav A, Fahima T, Kirzhner V, Korol A, Nevo E. Microscale ecological stress causes RAPD molecular selection in wild barley, Neve Yaar microsite, Israel. Genet Resour Crop Ev. 2003;50:213–224. doi: 10.1023/A:1022995214885. [DOI] [Google Scholar]
- Zongyun F, Lili Z, Yizheng Z, Hongqing L. Further molecular evidence for the Hordeum vulgare ssp. spontaneum in Tibet as ultimate progenitor of Chinese cultivated barley. High Technology Letters. 2005;11:20–324. [Google Scholar]
- Nevo E. In: Cereal Genomics. Gupta PK, Varshney RK, editor. Dordrecht: Kluwer Academic Publishers; 2004. Population genetic structure of wild barley and wheat in the Near East Fertile Crescent: regional and local adaptive patterns; pp. 135–163. [Google Scholar]
- Lee CR, Mitchell-olds T. Quantifying effects of environmental and geographical factors on patterns of genetic differentiation. Mol Ecol. 2011;20:4631–4642. doi: 10.1111/j.1365-294X.2011.05310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Krugman T, Fahima T, Chen K, Hu Y, Röder M, Nevo E, Korol A. Chromosomal regions controlling seedling drought resistance in Israeli wild barley, Hordeum spontaneum. Genet Resour Crop Ev. 2010;57:85–99. doi: 10.1007/s10722-009-9453-z. [DOI] [Google Scholar]
- Owuor ED, Fahima T, Beharav A, Korol A, Nevo E. RAPD divergence caused by microsite edaphic selection in wild barley. Genetica. 1999;105:177–192. doi: 10.1023/A:1003781711908. [DOI] [PubMed] [Google Scholar]
- Nosil P, Funk DJ, O-B D. Divergent selection and heterogeneous genomic divergence. Mol Ecol. 2009;18:375–402. doi: 10.1111/j.1365-294X.2008.03946.x. [DOI] [PubMed] [Google Scholar]
- Ivandic V, Hackett C, Zhang Z, Staub J, Nevo E, Thomas W, Forster B. Phenotypic responses of wild barley to experimentally imposed water stress. J Exp Bot. 2000;51:2021–2029. doi: 10.1093/jexbot/51.353.2021. [DOI] [PubMed] [Google Scholar]
- Tajima F. Evolutionary relationship of DNA sequences in finite populations. Genetics. 1983;105:437. doi: 10.1093/genetics/105.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–925. doi: 10.1093/genetics/147.2.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevo E. "Evolution Canyon", a potential microscale monitor of global warming across life. Proc Natl Acad Sci U S A. 2012;109:2960–2965. doi: 10.1073/pnas.1120633109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevo E. Asian, African and European biota meet at "Evolution Canyon" Israel: local tests of global biodiversity and genetic diversity patterns. Proc R Soc Lond B Biol Sci. 1995;262:149–155. doi: 10.1098/rspb.1995.0189. [DOI] [Google Scholar]
- Hofinger BJ, Russell JR, Bass CG, Baldwin T, Dos Reis M, Hedley PE, Li Y, Macaulay M, Waugh R, Hammond-Kosack KIME. An exceptionally high nucleotide and haplotype diversity and a signature of positive selection for the eIF4E resistance gene in barley are revealed by allele mining and phylogenetic analyses of natural populations. Mol Ecol. 2011;20:3653–3668. doi: 10.1111/j.1365-294X.2011.05201.x. [DOI] [PubMed] [Google Scholar]
- Raz S, Retzkin S, Pavlicek T, Hoffman A, Kimchi H, Zehavi D, Beiles A, Nevo E. Scorpion Biodiversity and Interslope Divergence at "Evolution Canyon", Lower Nahal Oren Microsite, Mt. Carmel, Israel. PLoS One. 2009;4:e5214. doi: 10.1371/journal.pone.0005214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou C, Sun K, Mackaluso JD, Seddon AE, Jin R, Thomashow MF, Shiu SH. Cis-regulatory code of stress-responsive transcription in Arabidopsis thaliana. Proc Natl Acad Sci U S A. 2011;108:14992–14997. doi: 10.1073/pnas.1103202108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosma DK, Jenks MA. In: Advances in Molecular Breeding Toward Drought and Salt Tolerant Crops. Paul M, Hasegawa , Matthew A, Jenks S, Mohan Jain , editor. Dordrecht: Springer; 2007. Eco-physiological and molecular-genetic determinants of plant cuticle function in drought and salt stress tolerance; pp. 91–120. [Google Scholar]
- Luo B, Xue XY, Hu WL, Wang LJ, Chen XY. An ABC transporter gene of Arabidopsis thaliana, AtWBC11, is involved in cuticle development and prevention of organ fusion. Plant Cell Physiol. 2007;48:1790–1802. doi: 10.1093/pcp/pcm152. [DOI] [PubMed] [Google Scholar]
- Kunst L, Samuels AL. Biosynthesis and secretion of plant cuticular wax. Prog Lipid Res. 2003;42:51–80. doi: 10.1016/S0163-7827(02)00045-0. [DOI] [PubMed] [Google Scholar]
- Yanagisawa S. The Dof family of plant transcription factors. Trends Plant Sci. 2002;7:555–560. doi: 10.1016/S1360-1385(02)02362-2. [DOI] [PubMed] [Google Scholar]
- Yamaguchi S. Gibberellin metabolism and its regulation. Annu Rev Plant Biol. 2008;59:225–251. doi: 10.1146/annurev.arplant.59.032607.092804. [DOI] [PubMed] [Google Scholar]
- Krugman T, Peleg Z, Quansah L, Chagué V, Korol AB, Nevo E, Saranga Y, Fait A, Chalhoub B, Fahima T. Alteration in expression of hormone-related genes in wild emmer wheat roots associated with drought adaptation mechanisms. Funct Integr Genomics. 2011;11:565–583. doi: 10.1007/s10142-011-0231-6. [DOI] [PubMed] [Google Scholar]
- Vettakkorumakankav NN, Falk D, Saxena P, Fletcher RA. A crucial role for gibberellins in stress protection of plants. Plant Cell Physiol. 1999;40:542. doi: 10.1093/oxfordjournals.pcp.a029575. [DOI] [Google Scholar]
- Huang W, Nevins JR, Ohler U. Phylogenetic simulation of promoter evolution: estimation and modeling of binding site turnover events and assessment of their impact on alignment tools. Genome Biol. 2007;8:R225. doi: 10.1186/gb-2007-8-10-r225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Weinberger A, Bezalel D, Kaganovich M, Barkai N. On the relation between promoter divergence and gene expression evolution. Mol Syst Biol. 2008;4:159–170. doi: 10.1038/msb4100198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevo E. " Evolution Canyon": a microcosm of life's evolution focusing on adaptation and speciation. Israel Journal of Ecology and Evolution. 2006;52:485–506. [Google Scholar]
- Goldreich Y. The climate of Israel: observation, research, and application. Us: Springer; 2003. [Google Scholar]
- Komatsuda T, Nakamura I, Takaiwa F, Oka S. Development of STS markers closely linked to the vrs1 locus in barley, Hordeum vulgare. Genome. 1998;41:680–685. [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- Excoffier LL, Schneider S. Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evol Bioinform Online. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- Weir B, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.2307/2408641. [DOI] [PubMed] [Google Scholar]
- Del Cerro I, Marmi J, Ferrando A, Chashchin P, Taberlet P, Bosch M. Nuclear and mitochondrial phylogenies provide evidence for four species of Eurasian badgers (Carnivora) Zool Scr. 2010;39:415–425. doi: 10.1111/j.1463-6409.2010.00436.x. [DOI] [Google Scholar]
- Malysheva-Otto LV, Ganal MW, der MS R. Analysis of molecular diversity, population structure and linkage disequilibrium in a worldwide survey of cultivated barley germplasm(Hordeum vulgare L.) BMC Genet. 2006;7:6. doi: 10.1186/1471-2156-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JZ, Brown AHD, Clegg MT. Heterogeneous geographic patterns of nucleotide sequence diversity between two alcohol dehydrogenase genes in wild barley (Hordeum vulgare subspecies spontaneum) Proc Natl Acad Sci U S A. 2001;98:531–536. doi: 10.1073/pnas.98.2.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob SS, Martinez-Meyer E, Blattner FR. Phylogeographic analyses and paleodistribution modeling indicate Pleistocene in situ survival of Hordeum species (Poaceae) in southern Patagonia without genetic or spatial restriction. Mol Biol Evol. 2009;26:907–923. doi: 10.1093/molbev/msp012. [DOI] [PubMed] [Google Scholar]
- Chang WC, Lee TY, Huang HD, Huang HY, Pan RL. PlantPAN: Plant promoter analysis navigator, for identifying combinatorial cis-regulatory elements with distance constraint in plant gene groups. BMC Genomics. 2008;9:561–575. doi: 10.1186/1471-2164-9-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reineke AR, Bornberg-Bauer E, Gu J. Evolutionary divergence and limits of conserved non-coding sequence detection in plant genomes. Nucleic Acids Res. 2011;39:6029–6043. doi: 10.1093/nar/gkr179. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Detailed information for the polymorphic sites sample.
Table S2. Detailed information for 22 haplotype in wild barley (Hordeum vulgare ssp. spontaneum).
Table S3. Detailed information for TFBSs classification.