

Abstract
PchB is an isochorismate-pyruvate lyase from Pseudomonas aeruginosa. A positively charged lysine residue is located in a flexible loop that behaves as a lid to the active site, and the lysine residue is required for efficient production of salicylate. A variant of PchB that lacks the lysine at residue 42 has a reduced catalytic free energy of activation of up to 4.4 kcal/mol. Construction of a lysine isosteric residue bearing a positive charge at the appropriate position leads to the recovery of 2.5–2.7 kcal/mol (about 60%) of the 4.4 kcal/mol by chemical rescue. Exogenous addition of ethylamine to the K42A variant leads to a neglible recovery of activity (0.180 kcal/mol, roughly 7% rescue), whereas addition of propylamine caused an additional modest loss in catalytic power (0.056 kcal/mol, or −2% loss). This is consistent with the view that (a) the lysine-42 residue is required in a specific conformation to stabilize the transition state and (b) the correct conformation is achieved for a lysine-mimetic sidechain at site 42 in the course of loop closure, as expected for transition-state stabilization by the side chain ammonio-function. That the positive charge is the main effector of transition state stabilization is shown by the construction of a lysine-isosteric residue capable of exerting steric effects and hydrogen bonding but not electrostatic effects, leading to a modest increase of catalytic power (0.267 – 0.505 kcal/mol of catalytic free energy, or roughly 6 – 11% rescue).
Chemical rescue experiments have been widely used in enzymology studies to obtain insight into the function of important active site amino acid sidechains (1). Non-covalent rescue experiments usually include mutation of the residue of interest to an alanine and the addition of excess compound mimicking the lost side chain (e.g. propylamine to mimic a lysine as depicted in Scheme 1A and 1B). The first chemical rescue, by Toney and Kirsch, restored activity in a lysine-to-alanine variant of aspartate aminotransferase using aliphatic amines as the rescue agents (2, 3). Since then, a variety of amino acid variants that suffered loss of activity have restored activity in the presence of exogenous compounds, including but not limited to lysine to alanine or cysteine variants rescued with aliphatic amines (4–7), arginine to alanine variants rescued with guanidinium or imidazole (8–13), histidine to glycine or alanine variants rescued with imidazole (14–17), and an aspartate to serine variant rescued with acetate (18). These experiments have sometimes been called “cavity mutants” as the exogenous rescue agent is thought to bind in the cavity generated by the mutational variation (19). Binding of the rescue agent in the vacated site has been confirmed crystallographic studies of myoglobin (14) and trypsin (18). However, the exogenous rescue agent does not always bind in the cavity, as in the case of carbonic anhydrase, where the rescue agent reestablished a hydrogen bonding network by binding in a site remote to the mutation (15). Alternatively, mutation of the residue of interest to cysteine allows for covalent chemical modification by alkylating agents (e.g. bromoethylamine to generate γ-thialysine, Scheme 1C). Covalent modification may allow for better control over the position and orientation of the rescue mimetic, with the caveat that any other reactive cysteines may be modified as well. Covalent modification has been shown to be effective in a variety of systems, including acetoacetate decarboxylase (20), ketopantoate reductase (7), fructose 1,6-bisphosphate aldoloase (21), glutamine synthase (22), arginase I (23), and aspartate aminotransferase (24, 25).
Scheme 1.
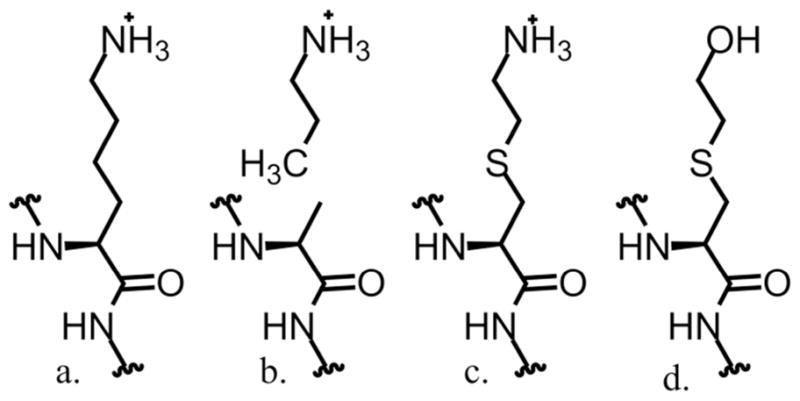
Structures of the lysine at residue 42 of WT PchB (A), a lysine mimetic assembly with propylamine (B), a lysine mimetic residue resulting from covalent modification of the cysteine variant with bromoethylamine (C), and a lysine-isosteric residue lacking electrostatic charge resulting from covalent modification of the cysteine variant with bromoethanol (D).
The enzyme under study here is the isochorismate-pyruvate lyase from Pseudomonas aeruginosa (PchB). PchB is a structural homolog of the E. coli chorismate mutase (26), and indeed has adventitious chorismate mutase activity, although the efficiency of the reaction is lower (27, 28). The chorismate mutase reaction is pericyclic, with concerted but asynchronous bond breaking and bond forming and a cyclic transition state (29). The isochorismate-pyruvate lyase activity has been shown by NMR to occur via a quantitative hydrogen transfer from C2 to C9, which led to the hypothesis that this reaction is also pericyclic with the hydrogen atom as one of the atoms of the cyclic transition state (Scheme 2) (30). The hypothesis was supported computationally (31).
Scheme 2.
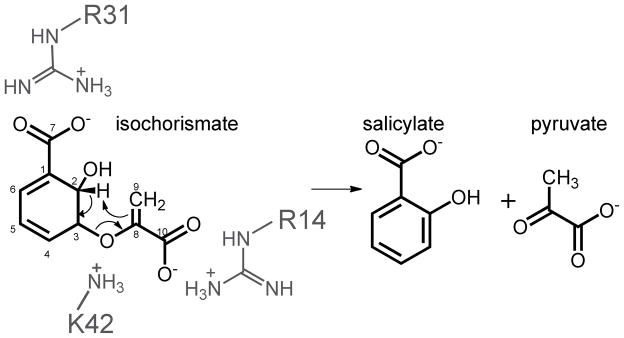
Isochorismate-pyruvate lyase activity catalyzed by PchB. The arginines that arrange the substrate carboxylates (R14 and R31) are shown in grey, as is the lysine (K42) of interest in this study, which is hypothesized to stabilize the developing negative charge of the transition state.
In our investigations of the pericyclic reactions catalyzed by the isochorismate-pyruvate lyase from Pseudomonas aeruginosa (PchB), we have provided evidence that catalytic power is derived from the organization of the substrate in the active site and from a positive charge at the 42 site of the mobile active site loop. This hypothesis has a basis in three previous experiments: 1) mutational analyses showed that variants at the K42 site did not alter the active site architecture but a change in the chemical nature of the sidechain from positively charged to neutral or negatively charged reduced or abolished activity (28); 2) pH titrations of the K42H variant of the enzyme showed that with a protonated histidine at the 42 site the enzyme had nearly full activity whereas a deprotonated histidine at the same site was an inefficient catalyst (32); and 3) thermodynamic data suggested that organization of the substrate in the active site does not account for the observed entropic change thereby implicating loop organization in catalysis (33). With these data in mind, we performed several chemical rescue experiments. It is important to note that since the reaction is pericyclic, the lysine of interest is not involved in a proton transfer (general acid-base chemistry), does not participate in a covalent intermediate, nor is it a metal ligand as frequently evaluated by chemical rescue. Instead, K42 is the residue of debate in the controversy over electrostatic transition state stabilization versus reactive substrate destabilization in the structurally homologous E. coli chorismate mutase. This lysine residue (or the comparable arginine in the B. subtilis chorismate mutase) has been hypothesized to be important either in stabilizing the developing negative charge of the ether oxygen at bond breaking (34–41) or to be unimportant in catalysis (42–46).
The first chemical rescue experiment described was to determine if exogenous rescue agents in a K42A variant were sufficient for restoration of activity, which might suggest that the aliphatic amine could bind in a preformed cavity vacated by the variant sidechain. This hypothesis seemed unlikely as the variant sidechain is found in the mobile active site loop that undergoes a disorder to order transition upon binding of ligands in the active site, as evidenced by crystallography (Figure 1) (26). In the second series of experiments, the K42C and C7A,K42C variants were generated and tested for restoration of activity after treatment with bromoethylamine, which generates a lysine mimetic, or treatment with bromoethanol, which generates an isosteric alcohol (Scheme 1D). We hypothesized that restoration of activity would be more efficient in covalent rescue experiments. The comparison of the amine versus alcohol derivatives was to consider the importance of a positive charge and/or hydrogen bond donors/acceptors at the 42 site. We report our findings here.
Figure 1. Active site of PchB in the open and closed wildtype and closed K42A-PchB structures.
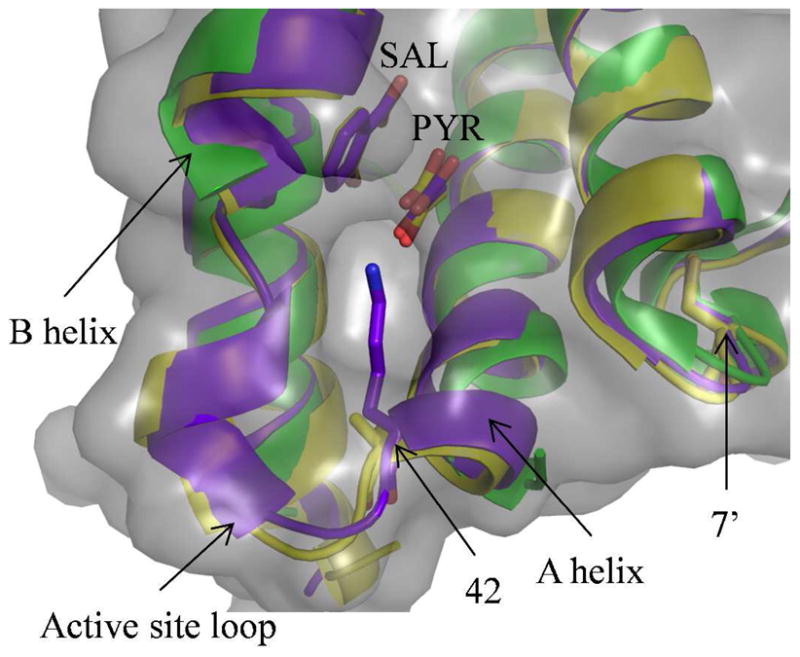
Overlay of the wildtype closed (PDB: 3REM, purple), wildtype open (PDB: 2H9C, green) and K42A –PchB (PDB: 3HGX, yellow) structures. The open structure, solved in the absence of products, has no electron density for the active site loop, which connects the A and B helices and contains a single turn of helix when ordered in the closed structures. The amino acid at the 42 position, which is the first residue of the active site loop, is shown as sticks, as is the amino acid at the 7 position from the opposing monomer (7′). The salicylate and pyruvate from the closed structures are also shown as sticks, all colored like the cartoon. The solvent accessible surface (grey) for the K42A structure shows a hole leading to the active site, highlighting the location that a chemical rescue agent must bind and be organized. Figure was generated with PyMOL (52).
EXPERIMENTAL PROCEDURES
Protein preparation
Wildtype and K42A-PchB were prepared as previously described (26). Site directed mutants of PchB were prepared with the plasmid used for wildtype PchB overexpression as the template and Quickchange II (Agilent) mutagenesis kit, using the protocol as per manufacturer’s instructions. The K42C mutation was generated using a primer of sequence 5′-GGCGTCGCGCTTCTGCGCCAGCGAGGCGG-3′ (mutated codon is underlined) and its reverse complement. The C7A mutation was generated using a primer of sequence 5′-AACTCCGAAGACGCCACCGGCCTGGCG-3′ (mutated codon is underlined) and its reverse complement. In order to prevent disulfide bond formation and aggregation of the mutants which generated a new cysteine, 20mM β mercapto-ethanol was added to the resuspension buffer. All buffers used in the purification process, including 30Q and gel filtration buffers contained 1mM TCEP (Sigma).
Preparation of isochorismate
Isochorismate was prepared as described previously (28).
Enzymatic assays
All assays were performed using a TgK Scientific Stopped Flow device operated at 20 °C, with a xenon lamp with the monochromator set at 310 nm, and a 360 nm cutoff filter. Equal 50μl volumes of enzyme and substrate were injected into a 22 μl cell and initial velocity of salicylate production was measured as an increase in fluorescence at 430 nm for 30 seconds during which the reaction was linear. Pre-injection enzyme and substrate concentrations were twice the final enzyme/substrate concentration in the cell (post mixing). Unless otherwise specified, all concentrations reported are final post-mixing concentrations. Both the substrate isochorismate and enzyme were prepared in the same buffer system to prevent buffer dilution effects post mix (50 mM Tris pH 8.0, 150 mM NaCl, 10% glycerol, 1 mM TCEP, and 500 mM rescue agent [where applicable]). Isochorismate was prepared to a desired pre-mix final concentration of 0 – 800 μM. Enzyme concentrations used were 100 nM for wildtype and C7A-PchB experiments and samples treated with bromoethylamine, and 2.5 μM for K42A-PchB, and 5 μM for PchB-K42C and C7A,K42C and samples treated with bromoethanol. Michaelis-Menten kinetic data were fit using Kaleidagraph (Synergy Software) using non-linear regression.
Covalent chemical rescue of K42C- and C7A,K42C-PchB variants with bromoethylamine and bromoethanol
PchB variants and controls were incubated for 22–24 hrs at 18° C in 2.5 mL of 1 M Tris pH 8.8 with and without 1 mM bromoacylating reagent added. Enzyme was concentrated using microcon centrifuge concentrators, and unreacted alkylating agent was removed by buffer exchange into 50 mM Tris HCl pH 8.0, 150mM NaCl, 10% Glycerol, 1mM TCEP using a PD-10 desalting column (GE Healthcare) as per manufacturer’s specifications. Protein concentration was determined by Bradford assay. Enzyme was added to 10 mL of reaction buffer without bromoacylating agent to final concentration of 200 nM – 10 μM (twice the post-mix enzyme concentration). Substrate was prepared with the same buffer components.
Circular Dichroism Spectroscopy
CD spectra of wild-type and variant PchB enzymes (~10 μM protein in 20 mM potassium phosphate, pH 7.0) with and without modification with bromoethylamine were measured with a Jasco J-815 spectrometer (Easton, MD) at 25 °C with a path length of 1 cm. Spectra were recorded three times for each sample and averaged. The step size and bandwith were 1 nm and the averaging time at each wavelength was 1 s.
Ellman’s Reagent competition assay
In order to determine whether the cysteine at position 7 was solvent accessible and thus alkylatable, 1 mL of 1 mM protein sample (wildtype, K42C-, C7A- or C7A,K42C-PchB) was incubated for 24 hours at 18 °C in the same buffer used in the alkylation process as described above, but without alkylating reagent. A volume of 100 μl of each of the samples was reacted with 1 mM 5,5′-Dithio-bis(2-nitrobenzoic acid) (DTNB or Ellman’s reagent) in 300 mM Tris pH 8.0, 25 mM sodium acetate buffer for 5 minutes at room temperature. Absorbance was measured at 412 nm and concentration of NTB−2 was determined (ε = 13600 M−1 cm−1) (47). For these experiments, protein concentration was determined by measuring the absorbance at 280 nm and using the extinction coefficient calculated by ProtParam (ε= 20,970 M−1 cm−1) (48). This concentration corresponds to a 1:1 ratio with the solvent accessible cysteines in solution. Wildtype (one cysteine at position 7) and C7A-PchB variant (no cysteines in sequence) showed no change in absorbance, indicating no cysteines are solvent accessible in these enzymes. K42C- (two cysteines at positions 7 and 42) and C7A,K42C-PchB (one cysteine at position 42) both showed a similar absorbance change corresponding to one cysteine per protein molecule being solvent accessible. Presumably, this is cysteine at position 42, since the cysteine at position 7 is not accessible in the wildtype and only one cysteine is accessible in the K42C-variant; however, it is possible that both cysteines are modified at 50% efficiency in the K42C-variant. Samples of each enzyme were then incubated as described above for the controls but with excess alkylating agent (10 mM). A DTNB competition assay was performed to determine the percent alkylation for bromoethylamine and bromoethanol using the equation
| (1) |
Wildtype and the C7A-variant showed no alkylation (0%). K42C-PchB showed 82% alkylation for two cysteines. C7A, K42C-PchB showed 98% alkylation for 1 cysteine with derivatization with bromoethylamine, and 100% alkylation for 1 cysteine with derivatization with bromoethanol. The values (% alkylation) were used to adjust the kinetic parameters found in Tables 1 and 2.
Table 1.
Catalytic constants for wildtype PchB and variants at residue 42 with or without exogenously added ethylamine or propylamine or covalent modification of the sidechain with bromoethylamine or bromoethanol.
| Samplea | kcat (×10−3 s−1) | kcatvar/kcatctrl | Km (μM) | Kmvar/Kmctrl |
|---|---|---|---|---|
| WTb | 130 ± 0 | 1.1 ± 0.1 | ||
|
| ||||
| K42A | 45 ± 1 | 1 | 30 ± 1 | 1 |
| K42A + EtA | 83 ± 1 | 1.8 | 60 ± 1 | 2 |
| K42A + PrA | 98 ± 1 | 2.2 | 48 ± 1 | 1.6 |
|
| ||||
| WTc | 178 ± 1 | 1.39 ± 0.03 | ||
|
| ||||
| K42C | 8.28 ± 0.04 | 1 | 134 ±1 | 1 |
| K42C + BrEA | 94.6 ± 0.1 | 11.4 | 23 ± 1 | 0.17 |
| K42C + BrEtOH | 18.4 ± 0.1 | 2.2 | 125 ± 1 | 0.93 |
|
| ||||
| C7A,K42C | 8.96 ± 0.01 | 1 | 123 ± 2 | 1 |
| C7A,K42C + BrEA | 105 ± 1 | 12 | 15.6 ± 0.3 | 0.13 |
| C7A,K42C + BrEtOH | 12.8 ± 0 | 1.4 | 114 ± 2 | 0.93 |
|
| ||||
| C7A | 200 ± 10 | 2.10 ± 0.02 | ||
EtA = exogenous ethylamine, PrA = exogenous propylamine, BrEA = treatment with bromoethylamine, BrEtOH = treatment with bromoethanol.
exogenous amine conditions
covalent rescue conditions
Table 2.
Effects of sidechain at residue 42 on loss or rescue of electrostatic catalysis in the lyase activity of PchB
| Samplea | kcat/Km (M−1 s−1) | Variational Effect
|
ΔΔG‡ (kcal mol−1) (−RTln [var. effect])b | % Rescue | ||
|---|---|---|---|---|---|---|
| variant/control | value | |||||
| 1. | a. WTc | 124,000 ± 4,000 | ||||
| b. K42A | 1510 ± 30 | K42A/WT | 0.01 | 2.56 ± 0.09 | ||
|
| ||||||
| 2. | a. K42A + EtA | 2,060 ± 20 | (K42A+EtA)/K42A | 1.4 | −0.180 ± 0.004 | 7% |
| b. K42A + PrA | 1,370 ± 10 | (K42A+PrA)/K42A | 0.9 | 0.056 ± 0.001 | −2% | |
|
| ||||||
| 3. | a. WTd | 128,000 ± 3,000 | ||||
| b. K42C | 62 ± 0 | K42C/WT | 0.0005 | 4.4 ± 0.1 | ||
| c. C7A | 93,000 ± 5,000 | C7A/WT | 0.7 | 0.17 ± 0.01 | ||
| d. C7A,K42C | 65 ± 1 | (C7A,K42C)/C7A | 0.0007 | 4.2 ± 0.2 | ||
|
| ||||||
| 4. | a. K42C + BrEA | 4,200 ± 100 | (K42C+BrEA)/K42C | 68 | −2.45 ± 0.06 | 55% |
| b. C7A,K42C + BrEA | 6,700 ± 100 | (C7A,K42C + BrEA)/(C7A,K42C) | 103 | −2.69 ± 0.06 | 64% | |
|
| ||||||
| 5. | a. K42C + BrEtOH | 147 ± 1 | (K42C+BrEtOH)/K42C | 2.4 | −0.505 ± 0.003 | 11% |
| b. C7A,K42C + BrEtOH | 103 ± 1 | (C7A,K42C + BrEtOH)/(C7A,K42C) | 1.6 | −0.267 ± 0.005 | 6% | |
EtA = exogenous ethylamine, PrA = exogenous propylamine, BrEA = treatment with bromoethylamine, BrEtOH = treatment with bromoethanol.
temperature is 293 K
exogenous amine conditions
covalent rescue conditions
ΔΔG‡ Calculations
To quantify chemical rescue, variant effects were calculated in terms of changes in energies of activation:
| (2) |
where R represents the gas constant (1.98×10−3 kcal K−1 M−1) and T is the experimental temperature, 293 K (20°C). Propagation of the error of kcat/Km to the ΔΔG‡ was calculated as described previously (49) using equation 3.
| (3) |
RESULTS
Kinetic measurements of isochorismate pyruvate lyase activity
The experiments presented were conducted in a new buffer system from previously reported work and the experiments were performed using a stopped-flow spectrometer. While the values are different by three-fold from those previously reported (28), the K42A-PchB variant was shown previously to have 100-fold reduction in kcat/Km relative to wildtype, which is consistent with the values presented in Table 2.
Attempted chemical rescue of K42A-PchB with exogenous alkylamines
The K42A-PchB variant was employed to further gauge the catalytic contribution of the sidechain at this position by determining the rescue efficiency in the presence of exogenous amines. Figure 2 shows the effect of exogenous amines on specific activity of wildtype and K42A-PchB. Since the molecular void volume created by the formation of the K → A variant is the same as that of propylamine (4, 7), this compound was used for an initial analysis. Panel A shows the effect of increasing propylamine concentration on lyase activity for wildtype (circles) and K42A-PchB (squares). If there were rescue, then we would expect a hyperbolic curve. Instead, a line with a slope of 0 is shown at the value of activity without exogenous amine added. Methylamine, ethylamine and butylamine were also tested at a single concentration of 0.5 M with no rescue seen for these alkylamines of smaller or large molecular volume (Figure 2B). The effective binding constants for these compounds must be higher than two-fold the highest concentration tested, giving a minimum Kd of 2 M. All of the alkylamines showed a specific activity that was within 1.3-fold the specific activity of the K42A-PchB variant without exogenous amine.
Figure 2. No chemical rescue of isochorismate-pyruvate lyase activity by exogenous amines.

A. There was no recovery of K42A-PchB activity (□) with the addition of propylamine. The value without the addition of exogenous amine is shown as a dashed line (86.2 nmol/min/μg). Propylamine similarly had no effect on the activity of wildtype enzyme (○). The value without the addition of exogenous amine is shown as a solid line (1370 nmol/min/μg). B. The amine molecular volume was unimportant in the lack of rescue, because methylamine (MeA), ethylamine (EtA), propylamine (PrA) and butylamine (BuA) were all equally ineffective as chemical rescue agents at 0.5 M. Wild-type activity without exogenous amine is shown as the upper solid lined (1370 nmol/min/μg) and with the addition of rescue agents shown as circles (○). The activity of the K42A-PchB variant without exogenous amine is shown as the lower dashed line (94.4 nmol/min/ng) and with the addition of rescue agents shown as squares (□).
Attempted chemical rescue of K42A-PchB with exogenous ethylamine or propylamine
Michaelis-Menten kinetic analyses of K42A-PchB with 0.5 M ethylamine or 0.5 M propylamine were carried out. A comparison of the kinetic parameters for wildtype and the K42A-PchB variant is found in Table 1. The K42A-PchB variant has altered values for both kcat and Km relative to wildtype. The kcat value is reduced by 2.9-fold, and the Km is increased by 27-fold, suggesting that the lysine at the 42 site is important in substrate binding. The rate of enhancement upon the addition of ethylamine or propylamine is compared relative to the variant. K42A-PchB in the presence of either ethylamine or propylamine demonstrates a 2-fold increase in kcat relative to K42A-PchB without exogenous amine. There was a comparable change in Km. The change in the Km may suggest that the lysine has a role in binding substrate, but the addition of the exogenous amine does not rescue substrate binding. A comparison of the kcat/Km values (Table 2) shows the lack of rescue more clearly. The kcat/Km values for K42A-PchA with and without exogenous amine are all approximately 100-fold less than the wildtype value. The addition of ethylamine to K42A-PchB increased the kcat/Km by 1.4-fold relative to the variant without exogenous amine, whereas propylamine reduced the value by 0.9-fold.
Successful chemical rescue of K42C-variants PchB by construction of an isosteric γ-thialysine at position 42 with bromoethylamine
A K42C-PchB variant was generated and modified by reaction with bromoethylamine to form γ-thialysine (Scheme 1C) at any solvent accessible cysteines. The K42C-PchB variant shows an increased Km relative to wildtype, presumably due to the loss of the hydrogen bond donor or positive charge at this site (Table 1). The Km values for K42C-PchB with the modification to make the lysine mimetic shows a decrease in the Km by nearly 6-fold. However, when the K42C-PchB is modified with the bromoethylamine, the kcat shows a 11.4-fold increase relative to the unmodified variant. The change in both values represent a significant rescue of activity by the lysine mimetic. Indeed, the kcat/Km increases 68-fold (Table 2).
A double mutant was generated to remove the naturally occurring cysteine from the N-terminus of the protein, thereby generating a C7A,K42C-PchB double variant. In the absence of rescue agent, the double variant showed kinetic parameters comparable to the single variant (Table 1). With the formation of the lysine mimetic, the C7A,K42C-variant kcat value was again rescued significantly (12-fold) and in this case, the Km value was decreased by ~8-fold. Rescue is even more evident in this variant with a kcat/Km value that is 103-fold higher than the variant without the modification to make the lysine mimetic.
Attempted chemical rescue of K42C- PchB and C7A,K42C- PchB by construction of an isosteric γ-thia-ω-oxa-lysine at position 42 with bromoethanol
The K42C- PchB variant and the C7A,K42C- PchB double variant were also modified with bromoethanol to generate the isosteric alcohol residue (Scheme 1D). These modified variants showed no rescue in kcat, showing values within 2.2-fold of the unmodified variant, and demonstrated Km value within a 1.1-fold of the unmodified variant (Table 1). Indeed, the kcat/Km values are ~2-fold higher than the variant without the modification (Table 2).
Secondary structure maintained as detected by circular dichroism spectroscopy
Circular dichroism spectroscopy was used to assess the secondary structure of the variant proteins with and without modification by bromoethylamine. All variant proteins were found to be comparable to wild type with strong minima at 208 and 212 nm, characteristic of helical structure (Supplement Figure 1). The variants that contained the cysteine to alanine change at the seven position may exhibit a slightly lower helical property than those with the native cysteine. Modification of the proteins with bromoethylamine had no impact of helicity (open versus closed symbols).
DISCUSSION
Recent work by Richard and colleagues on triosphosphate isomerase (50) and orotidine 5′-monophosphate decarboxylase (51) provides a framework for reporting and interpreting chemical rescue experiments. In both enzymes, a cationic sidechain (lysine12 and arginine225, respectively) is adjacent to a flexible phosphodianion gripper loop, and the sidechain forms a stabilizing ion pair with the substrate. Excision of the sidechain results in a large decrease in catalytic activity (104 to 105-fold). A substantial fraction of the catalytic activity for the variants is rescued by addition of exogenous guanidine or alkyl ammonium cations. In the work on these two enzymes, elegant calculations are used to determine the effective molarity of the sidechain and the related free energies of activation: ΔG‡rescue agent represents the portion that can be rescued by the activator, and ΔG‡S represents the advantage of having the two pieces connected, called the “connection energy.” In these previous examples,
where ΔG‡rescue agent is negative, and with the connection energy (ΔG‡S) sums to give the variational effect caused by mutation at the site in question (ΔG‡variant − ΔG‡WT). In the best case scenario, a simple model provides an estimate of the entropy advantage caused by fusing the rescue agent to the sidechain. Richard and colleagues (50) also provide enthalpic arguments, stating that the connection energy is an “empirical” transition state stabilization: there may be an enthalpic penalty due to desolvation or an enthalpic advantage due to binding interactions. The former case would be evident as a large effective molarity, since desolvation of the sidechain is already “paid” in the folding of the enzyme. An enthalpic advantage from favorable binding interactions between the rescue agent and the protein is evidenced by a small effective molarity (50).
In PchB, the site in question is the 42 position, which is located in a flexible loop as determined by crystallography (Figure 1). No electron density is observed for residues 42–48 in the wildtype structure with an open active site (26). The open structure further shows that there is no pre-formed binding site for exogenous amines in this conformation. The structure of the K42A-PchA variant shows a hole in the solvent accessible surface of this “closed” conformation where the lysine sidechain is no longer present, highlighting the site where an exogenous amine would need to bind and be organized after ordering of the loop (Figure 1). This “binding site” for exogenous amines presumably has a Kd > 2 M, since no rescue was observed at concentrations up to 1 M. Previous determination of the activation thermodynamics of the reaction suggested at the importance of loop organization for PchB lyase activity (33). Therefore, organization of the loop upon active site closure must also organize the lysine sidechain and lead to the formation of the transition state.
If Richard’s calculations are applied to the K42A-PchB protein and the rescue attempts with exogenous propylamine (Supplemental Figure S2), the effective molarity of the sidechain at lysine42 is 610 M, suggesting that if the experiments could be conducted at this concentration of propylamine, the variant would turn over at the same rate as wildtype. The effective molarity in the PchB experiments is within the range of those presented previously (50, 51), which would suggest no evidence for an enthalpic disadvantage due to desolvation. We suggest that desolvation of the rescue agent and the mobile sidechain in the flexible active site loop would be comparable. The sidechain is exposed to solvent in the ground state open conformation and becomes desolvated and buried upon active site closing, which is true regardless of whether or not the sidechain is covalently attached. Furthermore, as mentioned above, we estimate that the Kd is > 2 M, since no rescue was observed for concentrations up to 1 M. In other words, there is not tight binding of the rescue agents to PchB, since there is not a preformed cavity for binding in the ground state. A portion of the binding interactions may be required to hold the loop in the closed conformation in the transition state. Such an interaction would not be rescued by an exogenous amine, but would require a covalent connection between the amino acid sidechain and the enzyme. Indeed, the connection energy in the transition state (ΔG‡S) is calculated from the effective molarity to be 3.8 kcal/mol, which is greater than the value for the variational effect between the K42A- and WT-PchB, ΔG‡K42A − ΔG‡WT, determined to be 3.4 kcal/mol. In keeping with this result, the 1/Kd‡ for propylamine can be determined and the ΔG‡PrA calculated, giving a value of 0.4 kcal/mol (positive, not negative as previously seen). While these values are internally consistent, this analysis highlights the inability to rescue by an exogenous amine. A different analysis is desirable for PchB to account for the placement of the amino acid to be rescued in the flexible loop and for direct comparison with covalent rescue agents.
We propose that variational effects in chemical rescue experiments can be calculated more directly from the (apparent) kcat/Km in terms of differences in the free energies of activation (ΔΔG‡, Table 2). The difference in energies can be calculated using equation 2, such that the mutational effect of replacing the wildtype lysine at position 42 with an alanine (effectively removing steric and charge effects) increases the free energy of activation as compared to the wildtype lyase activity by 2.56 kcal/mol (Table 2 entry 1b). This number represents the maximal change in free energy of activation that any given exogenous chemical would be able to rescue to reach activity levels comparable to wild type enzyme. Addition of excess exogenous positive charge in the form of ethylamine does not rescue catalytic activity (−0.18 kcal/mol or 7% rescue of the mutational effect; Table 2 entry 2a). The addition of propylamine causes a further reduction in activity (0.056 kcal/mol, or −2% of the mutational effect; Table 2 entry 2b). Addition of alkylamines of different carbon chain length (methyl- or butylamine) at the same concentration did not rescue catalysis either (Figure 2).
A K→C variant was generated at the 42 position, to enable introduction of a positive charge with bromoalkylating compounds (Scheme 1C). As with the K→A mutation, the K42C variant displayed an increased energy of activation, in this case by 4.4 ± 0.1 kcal/mol (Table 2, entry 3b). This increase in ΔΔG‡ would correspond to a theoretically perfect rescue. Indeed, the covalent modification of the K42C variant with bromoethylamine (generating a γ-thialysine at this position), rescues 2.45 ± 0.06 kcal/mol, roughly 55% of the ideal rescue (Table 2 entry 4a). However, wildtype PchB treated with bromoethylamine showed a decrease in catalytic activity with a kcat/Km 2.4-fold lower (See Supplemental Table S1). This may be due to the alkylation of a natural cysteine located near the N-terminus (position 7) of the enzyme. Removal of this cysteine through the generation of a C7A variant provides better control over the effects of extraneous alkylation, and the kcat/Km value for C7A-PchB is comparable to wildtype (0.7-fold change). The K42C variant over the C7A background resulted in a 4.2 kcal/mol gain in energy of activation with respect to the C7A background variant (Table 2 entry 3d). Treatment of this double variant with bromoethylamine confirmed the results of the K42C single mutant experiments: generation of the γ-thialysine results in a significant rescue. In the case of the double mutant, a 2.69 kcal/mol rescue (64%; Table 2 entry 4b) of the possible ideal rescue of 4.2 kcal/mol is observed.
Previous work on the structural homolog Escherichia coli chorismate mutase had suggested that a positive charge at the site equivalent to PchB 42 is not important for catalysis: organization of the substrate carboxylates by arginines buried deep within the active site was hypothesized to generate a near attack conformation which was sufficient for catalysis (42–46). In order to test this hypothesis, a variant with a lysine-isosteric residue bearing a hydroxyl group in lieu of the positively charged amino group was generated using bromoethanol instead of bromoethylamine (Scheme 1D). A modest increase in activity was observed for the K42C-PchB variant (0.505 ± 0.003 kcal/mol; 11%; Table 2 entry 5a) with less observed in the C7A,K42C double variant (0.267 ± 0.005 kcal/mol; 6%; Table 2 entry 5b). Mutagenesis (28) and pH dependence (32) experiments have provided evidence that the positive charge at position 42 is important for efficient catalysis. The ability to rescue catalytic activity of the variants modified to produce a γ-thialysine at position 42, but not through the generation of the hydroxyl derivative, gives further support to the importance of electrostatic transition state stabilization in the isochorismate-pyruvate lyase reaction. Structural (26) and thermodynamic (33) experiments have led to the hypothesis that loop organization is important for catalysis. Indeed, this is supported by the ineffectiveness of the ethylamine or propylamine to serve as a non-covalent chemical rescue reagents; the positive charge must be organized with the loop for efficient catalysis. We propose that a positive charge at position 42 and correct loop organization, including organization of the lysine at position 42, lead to efficient lyase catalysis, and give further support to the idea that electrostatic transition state stabilization and loop dynamics work in concert for catalysis.
These data lead to the question of why the isosteric γ-thialysine at position 42 is less able to promote catalysis than the native lysine or the previously tested histidine variant (32) at the same site. The tested variants are all apparently properly folded and stable, and for those modified by alkylation, the chemistry of modification was either complete or the kinetics were adjusted by a small correction. The answer may be simply that the active site is better able to accommodate the positively charged sidechains lacking sulfur; however, the geometry of the lysine and γ-thialysine would place the positive charge in a more similar position than that for histidine, which is effectively one methylene group shorter (in fairness, we must note that the histidine variant has not been tested in the buffer system detailed in this report). Alternatively, the answer may be in the role that the lysine plays in catalysis. This amino acid is not involved in covalent catalysis or acid/base chemistry, but has been proposed to stabilize the developing negative charge of the transition state in this pericyclic reaction. In PchB, closing of the active site is integral to formation of the transition state from the reactant state (33), and this requires organization of the active site loop and the lysine at site 42. It is tempting to propose that the motion of the residue at the 42 site may promote development of the negative charge on the ether oxygen of bond breaking. A vibrational motion bringing the positively charge sidechain into proximity of the ether oxygen of the substrate may be more easily achieved by lysine and histidine as compared to the γ-thialysine.
Supplementary Material
Acknowledgments
This publication was made possible by funds from the NIH grants numbered R01 AI77725 and K02 AI093675 from the National Institute for Allergy and Infectious Disease (A.L.L), by the Graduate Training Program in Dynamic Aspects of Chemical Biology NIH grant number T32 GM08545 (J.O.), and by NIH grant number P20 RR016475 from the INBRE Program of the National Center for Research Resources (subaward to A.L.L.).
We are grateful to R. L. Schowen and J. M. Bollinger Jr. for insightful discussions and suggestions. We are grateful to A. S. Chilton for technical assistance.
Footnotes
The supplemental materials, which may be accessed free of charge online at http://pubs.acs.org, include: 1) Table S1 – a complete table of the catalytic constants of all controls and variants discussed in this work. 2) Figure S1 – circular dichroism spectra of the variants with and without the bromoethylamine modification. 3) Figure S2 – calculations based on the work of Richard and colleagues (50, 51) demonstrating a lack of activation of K42A-PchB variant by the addition of exogenous propylamine.
References
- 1.Peracchi A. How (and why?) to revivie a dead enzyme: the power of chemical rescue. Curr Chem Biol. 2008;2:32–49. [Google Scholar]
- 2.Toney MD, Kirsch JF. Direct Bronsted analysis of the restoration of activity to a mutant enzyme by exogenous amines. Science. 1989;243:1485–1488. doi: 10.1126/science.2538921. [DOI] [PubMed] [Google Scholar]
- 3.Toney MD, Kirsch JF. Bronsted analysis of aspartate aminotransferase via exogenous catalysis of reactions of an inactive mutant. Protein Sci. 1992;1:107–119. doi: 10.1002/pro.5560010111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arnett SO, Gerratana B, Townsend CA. Rate-limiting steps and role of active site Lys443 in the mechanism of carbapenam synthetase. Biochemistry. 2007;46:9337–9345. doi: 10.1021/bi0618464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harpel MR, Hartman FC. Chemical rescue by exogenous amines of a site-directed mutant of ribulose 1,5-bisphosphate carboxylase/oxygenase that lacks a key lysyl residue. Biochemistry. 1994;33:5553–5561. doi: 10.1021/bi00184a026. [DOI] [PubMed] [Google Scholar]
- 6.Jiang W, Locke G, Harpel MR, Copeland RA, Marcinkeviciene J. Role of lys100 in human dihydroorotate dehydrogenase: mutagenesis studies and chemical rescue by external amines. Biochemistry. 2000;39:7990–7997. doi: 10.1021/bi000630d. [DOI] [PubMed] [Google Scholar]
- 7.Zheng R, Blanchard JS. Identification of active site residues in E. coli ketopantoate reductase by mutagenesis and chemical rescue. Biochemistry. 2000;39:16244–16251. doi: 10.1021/bi002134v. [DOI] [PubMed] [Google Scholar]
- 8.Boehlein SK, Walworth ES, Richards NG, Schuster SM. Mutagenesis and chemical rescue indicate residues involved in beta-aspartyl-AMP formation by Escherichia coli asparagine synthetase B. J Biol Chem. 1997;272:12384–12392. doi: 10.1074/jbc.272.19.12384. [DOI] [PubMed] [Google Scholar]
- 9.Muratore KE, Seeliger MA, Wang Z, Fomina D, Neiswinger J, Havranek JJ, Baker D, Kuriyan J, Cole PA. Comparative analysis of mutant tyrosine kinase chemical rescue. Biochemistry. 2009;48:3378–3386. doi: 10.1021/bi900057g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Phillips MA, Hedstrom L, Rutter WJ. Guanidine derivatives restore activity to carboxypeptidase lacking arginine-127. Protein Sci. 1992;1:517–521. doi: 10.1002/pro.5560010406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qiao Y, Molina H, Pandey A, Zhang J, Cole PA. Chemical rescue of a mutant enzyme in living cells. Science. 2006;311:1293–1297. doi: 10.1126/science.1122224. [DOI] [PubMed] [Google Scholar]
- 12.Rynkiewicz MJ, Seaton BA. Chemical rescue by guanidine derivatives of an arginine-substituted site-directed mutant of Escherichia coli ornithine transcarbamylase. Biochemistry. 1996;35:16174–16179. doi: 10.1021/bi961311i. [DOI] [PubMed] [Google Scholar]
- 13.Williams DM, Wang D, Cole PA. Chemical rescue of a mutant protein-tyrosine kinase. J Biol Chem. 2000;275:38127–38130. doi: 10.1074/jbc.C000606200. [DOI] [PubMed] [Google Scholar]
- 14.Barrick D. Replacement of the proximal ligand of sperm whale myoglobin with free imidazole in the mutant His-93-->Gly. Biochemistry. 1994;33:6546–6554. doi: 10.1021/bi00187a023. [DOI] [PubMed] [Google Scholar]
- 15.Maupin CM, Castillo N, Taraphder S, Tu C, McKenna R, Silverman DN, Voth GA. Chemical rescue of enzymes: proton transfer in mutants of human carbonic anhydrase II. J Am Chem Soc. 2011;133:6223–6234. doi: 10.1021/ja1097594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Newmyer SL, de Montellano PR. Rescue of the catalytic activity of an H42A mutant of horseradish peroxidase by exogenous imidazoles. J Biol Chem. 1996;271:14891–14896. doi: 10.1074/jbc.271.25.14891. [DOI] [PubMed] [Google Scholar]
- 17.Tu CK, Silverman DN, Forsman C, Jonsson BH, Lindskog S. Role of histidine 64 in the catalytic mechanism of human carbonic anhydrase II studied with a site-specific mutant. Biochemistry. 1989;28:7913–7918. doi: 10.1021/bi00445a054. [DOI] [PubMed] [Google Scholar]
- 18.Perona JJ, Hedstrom L, Wagner RL, Rutter WJ, Craik CS, Fletterick RJ. Exogenous acetate reconstitutes the enzymatic activity of trypsin Asp189Ser. Biochemistry. 1994;33:3252–3259. doi: 10.1021/bi00177a016. [DOI] [PubMed] [Google Scholar]
- 19.Pond AE, Roach MP, Thomas MR, Boxer SG, Dawson JH. The H93G myoglobin cavity mutant as a versatile template for modeling heme proteins: ferrous, ferric, and ferryl mixed-ligand complexes with imidazole in the cavity. Inorg Chem. 2000;39:6061–6066. doi: 10.1021/ic0007198. [DOI] [PubMed] [Google Scholar]
- 20.Highbarger LA, Gerlt JA, Kenyon GL. Mechanism of the reaction catalyzed by acetoacetate decarboxylase. Importance of lysine 116 in determining the pKa of active-site lysine 115. Biochemistry. 1996;35:41–46. doi: 10.1021/bi9518306. [DOI] [PubMed] [Google Scholar]
- 21.Hopkins CE, O’Connor PB, Allen KN, Costello CE, Tolan DR. Chemical-modification rescue assessed by mass spectrometry demonstrates that gamma-thia-lysine yields the same activity as lysine in aldolase. Protein Sci. 2002;11:1591–1599. doi: 10.1110/ps.3900102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dhalla AM, Li B, Alibhai MF, Yost KJ, Hemmingsen JM, Atkins WM, Schineller J, Villafranca JJ. Regeneration of catalytic activity of glutamine synthetase mutants by chemical activation: exploration of the role of arginines 339 and 359 in activity. Protein Sci. 1994;3:476–481. doi: 10.1002/pro.5560030313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Colleluori DM, Reczkowski RS, Emig FA, Cama E, Cox JD, Scolnick LR, Compher K, Jude K, Han S, Viola RE, Christianson DW, Ash DE. Probing the role of the hyper-reactive histidine residue of arginase. Arch Biochem Biophys. 2005;444:15–26. doi: 10.1016/j.abb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 24.Kim DW, Yoshimura T, Esaki N, Satoh E, Soda K. Studies of the active-site lysyl residue of thermostable aspartate aminotransferase: combination of site-directed mutagenesis and chemical modification. J Biochem. 1994;115:93–97. doi: 10.1093/oxfordjournals.jbchem.a124311. [DOI] [PubMed] [Google Scholar]
- 25.Planas A, Kirsch JF. Reengineering the catalytic lysine of aspartate aminotransferase by chemical elaboration of a genetically introduced cysteine. Biochemistry. 1991;30:8268–8276. doi: 10.1021/bi00247a023. [DOI] [PubMed] [Google Scholar]
- 26.Zaitseva J, Lu J, Olechoski KL, Lamb AL. Two crystal structures of the isochorismate pyruvate lyase from Pseudomonas aeruginosa. J Biol Chem. 2006;281:33441–33449. doi: 10.1074/jbc.M605470200. [DOI] [PubMed] [Google Scholar]
- 27.Gaille C, Kast P, Haas D. Salicylate biosynthesis in Pseudomonas aeruginosaPurification and characterization of PchB, a novel bifunctional enzyme displaying isochorismate pyruvate-lyase and chorismate mutase activities. J Biol Chem. 2002;277:21768–21775. doi: 10.1074/jbc.M202410200. [DOI] [PubMed] [Google Scholar]
- 28.Luo Q, Olucha J, Lamb AL. Structure-function analyses of isochorismate-pyruvate lyase from Pseudomonas aeruginosa suggest differing catalytic mechanisms for the two pericyclic reactions of this bifunctional enzyme. Biochemistry. 2009;48:5239–5245. doi: 10.1021/bi900456e. [DOI] [PubMed] [Google Scholar]
- 29.Gustin DJ, Mattei P, Kast P, Wiest O, Lee L, Cleland WW, Hilvert D. Heavy Atom Isotope Effects Reveal a Highly Polarized Transition State for Chorismate Mutase. J Am Chem Soc. 1999;121:1756–1757. [Google Scholar]
- 30.DeClue MS, Baldridge KK, Kunzler DE, Kast P, Hilvert D. Isochorismate Pyruvate Lyase: A Pericyclic Reaction Mechanism? J Am Chem Soc. 2005;127:15002–15003. doi: 10.1021/ja055871t. [DOI] [PubMed] [Google Scholar]
- 31.Marti S, Andres J, Moliner V, Silla E, Tunon I, Bertran J. Mechanism and plasticity of isochorismate pyruvate lyase: a computational study. J Am Chem Soc. 2009;131:16156–16161. doi: 10.1021/ja905271g. [DOI] [PubMed] [Google Scholar]
- 32.Olucha J, Ouellette AN, Luo Q, Lamb AL. pH dependence of catalysis by Pseudomonas aeruginosa isochorismate-pyruvate lyase: Implications for transition state stabilization and the role of lysine 42. Biochemistry. 2011;50:7198–7207. doi: 10.1021/bi200599j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo Q, Meneely KM, Lamb AL. Entropic and enthalpic components of catalysis in the mutase and lyase activities of Pseudomonas aeruginosa PchB. J Am Chem Soc. 2011;133:7229–7233. doi: 10.1021/ja202091a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chook YM, Gray JV, Ke H, Lipscomb WN. The monofunctional chorismate mutase from Bacillus subtilis Structure determination of chorismate mutase and its complexes with a transition state analog and prephenate, and implications for the mechanism of the enzymatic reaction. J Mol Biol. 1994;240:476–500. doi: 10.1006/jmbi.1994.1462. [DOI] [PubMed] [Google Scholar]
- 35.Chook YM, Ke H, Lipscomb WN. Crystal structures of the monofunctional chorismate mutase from Bacillus subtilis and its complex with a transition state analog. Proc Natl Acad Sci USA. 1993;90:8600–8603. doi: 10.1073/pnas.90.18.8600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kast P, Asif-Ullah M, Jiang N, Hilvert D. Exploring the active site of chorismate mutase by combinatorial mutagenesis and selection: the importance of electrostatic catalysis. Proc Natl Acad Sci USA. 1996;93:5043–5048. doi: 10.1073/pnas.93.10.5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kast P, Grisostomi C, Chen IA, Li S, Krengel U, Xue Y, Hilvert D. A strategically positioned cation is crucial for efficient catalysis by chorismate mutase. J Biol Chem. 2000;275:36832–36838. doi: 10.1074/jbc.M006351200. [DOI] [PubMed] [Google Scholar]
- 38.Lee AY, Karplus PA, Ganem B, Clardy J. Atomic structure of the buried catalytic pocket of Escherichia coli chorismate mutase. J Am Chem Soc. 1995;117:3627–3628. [Google Scholar]
- 39.Lee AY, Stewart JD, Clardy J, Ganem B. New insight into the catalytic mechanism of chorismate mutases from structural studies. Chem Biol. 1995;2:195–203. doi: 10.1016/1074-5521(95)90269-4. [DOI] [PubMed] [Google Scholar]
- 40.Liu DR, Cload ST, Pastor RM, Schultz PG. Analysis of Active Site Residues in Escherichia coli Chorismate Mutase by Site-Directed Mutagenesis. J Am Chem Soc. 1996;118:1789–1790. [Google Scholar]
- 41.Zhang S, Kongsaeree P, Clardy J, Wilson DB, Ganem B. Site-directed mutagenesis of monofunctional chorismate mutase engineered from the E. coli P-protein. Bioorg Med Chem. 1996;4:1015–1020. doi: 10.1016/0968-0896(96)00099-5. [DOI] [PubMed] [Google Scholar]
- 42.Hur S, Bruice TC. The mechanism of catalysis of the chorismate to prephenate reaction by the Escherichia coli mutase enzyme. Proc Natl Acad Sci USA. 2002;99:1176–1181. doi: 10.1073/pnas.022628599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hur S, Bruice TC. The near attack conformation approach to the study of the chorismate to prephenate reaction. Proc Natl Acad Sci USA. 2003;100:12015–12020. doi: 10.1073/pnas.1534873100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hur S, Bruice TC. Just a near attack conformer for catalysis (chorismate to prephenate rearrangements in water, antibody, enzymes, and their mutants) J Am Chem Soc. 2003;125:10540–10542. doi: 10.1021/ja0357846. [DOI] [PubMed] [Google Scholar]
- 45.Hur S, Bruice TC. Comparison of formation of reactive conformers (NACs) for the Claisen rearrangement of chorismate to prephenate in water and in the E. coli mutase: the efficiency of the enzyme catalysis. J Am Chem Soc. 2003;125:5964–5972. doi: 10.1021/ja0210648. [DOI] [PubMed] [Google Scholar]
- 46.Zhang X, Zhang X, Bruice TC. A definitive mechanism for chorismate mutase. Biochemistry. 2005;44:10443–10448. doi: 10.1021/bi050886p. [DOI] [PubMed] [Google Scholar]
- 47.Sedlak J, Lindsay RH. Estimation of total, protein-bound, and nonprotein sulfhydryl groups in tissue with Ellman’s reagent. Anal Biochem. 1968;25:192–205. doi: 10.1016/0003-2697(68)90092-4. [DOI] [PubMed] [Google Scholar]
- 48.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, WIlkinds MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy Server. In: Walker JM, editor. The Proteomics Protocols Handook. Humana Press; Totowa, NJ: 2005. pp. 571–607. [Google Scholar]
- 49.Taylor RA. An Introduction to Error Analysis. University Science Books; Mill Valley, CA: 1982. [Google Scholar]
- 50.Go MK, Amyes TL, Richard JP. Rescue of K12G triosephosphate isomerase by ammonium cations: the reaction of an enzyme in pieces. J Am Chem Soc. 2010;132:13525–13532. doi: 10.1021/ja106104h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barnett SA, Amyes TL, Wood BM, Gerlt JA, Richard JP. Activation of R235A mutant orotidine 5′-monophosphate decarboxylase by the guanidinium cation: effective molarity of the cationic side chain of Arg-235. Biochemistry. 2010;49:824–826. doi: 10.1021/bi902174q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.DeLano W. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA: 2002. www.pymol.org. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.