

Recently, enormous strides have been made in our understanding of the crosstalk among innate immunity, adaptive immunity, inflammation, and tissue damage. T cells play a key role in immunostimulation and immunoregulation [1–3]. Upon activation by various physiological and pathophysiological cues, CD4+ T cells can differentiate into different lineages of T helper (Th) cells with distinct biological functions (Figure 1). Th1 cells are characterized by the transcription factor T-bet and signal transducer and activator of transcription (STAT) 4, and the production of IFN-γ, IL-2 and TNF-β. These cells are involved in the cellular immunity and inflammation (Figure 1). On the other hand, transcription factors like GATA- 3 and STAT6 drive the generation of Th2 cells that produce ant-iinflammatory cytokines (IL-4, IL-10 and IL-13) and mediate humoral immunity. It is known that autoimmune diseases are mainly mediated by Th1 response and that switching of Th1 to a Th2 phenotype is a way to ameliorate autoimmune diseases [3–6]. However, this hypothesis is being challenged as after the discovery of IL-23, one new T helper cell (Th17) has been identified (Figure 1) that expresses signature transcription factor RORγT and secretes IL-17 and IL-21 [7]. Although Th17 cells are involved in the host defense against bacteria, fungi, and viruses, these cells are nowadays considered to play a more active role than Th1 cells in the disease process of multiple sclerosis (MS) and other autoimmune disorders [2,3,7].
Figure 1. Differentiation of CD4+ T cells into Th1, Th2, Th9, and Th17 cells.
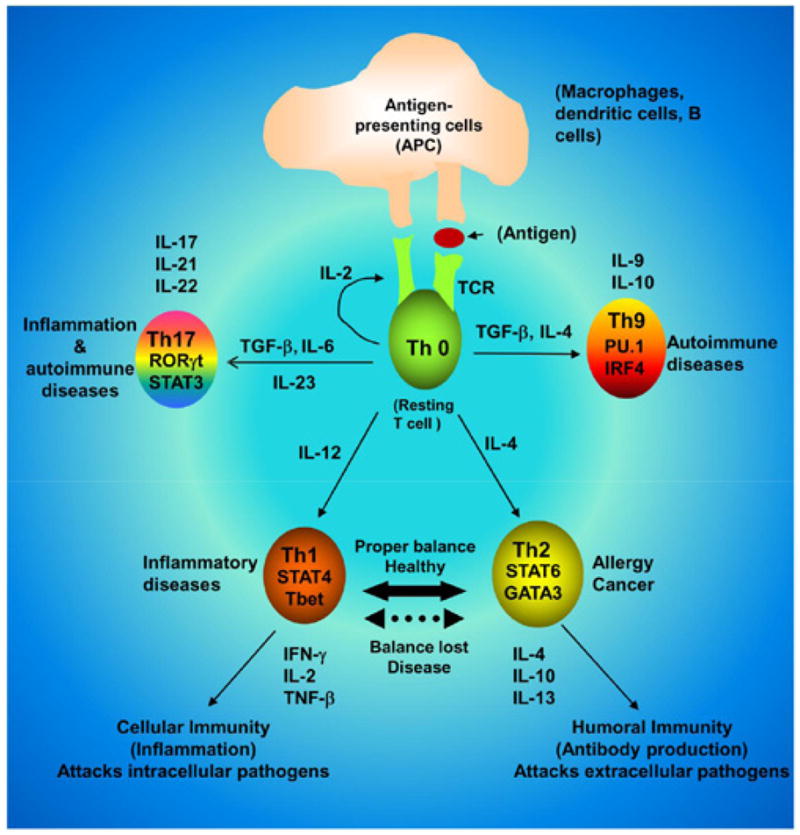
Distinct set of cytokines drive the differentiation of a particular Th cells. While Th1, Th9 and Th17 cells participate in autoimmune disorders, Th2 cells play a key role in cancer and allergy.
This is the era of T helper cells because in addition to Th1, Th2 and Th17 cells, another subset of Th cell is recently making headlines as a possible player in autoimmune disorders. This is Th9 cells, which are driven by the collaborative efforts of TGF-β and IL-4 (Figure 1). Here we must remember while TGF-β and IL-6 are required for Th17 cells, TGF-β and IL-4 are required for the generation of Th9 cells [8]. Recent studies have shown the involvement of transcription factors like PU.1 and IRF4 in the expression of IL-9 [8,9]. As evident from the name, Th9 cells produce large amounts of IL-9 and some IL-10. It is puzzling because IL-9 together with TGF-β can contribute to the differentiation of Th17 cells, and alternatively Th17 cells themselves can also produce some IL-9 [2,8,9]. Therefore, a better understanding of IL-9 will have important consequences in the field of immune regulation and autoimmunity.
Although the differentiation of naive CD4+ T cells into Th1, Th2, Th17, and Th9 is initiated by the engagement of their T cell receptor (TCR) and costimulatory molecules in the presence of specific cytokines produced by the innate immune cells, during any immune challenge, a gaseous lipophilic molecule called nitric oxide (NO) is often generated [10–12]. Being a double edged sword depending on its concentration in the microenvironment, NO is involved in both physiological and pathological processes of many organ systems including immune organs. NO is enzymatically formed from L-arginine by the enzyme nitric oxide synthase (NOS). The NOS are basically divided into two forms. The constitutive form, present in neurons (nNOS) and endothelial cells (eNOS), is a calcium-dependent enzyme. The inducible form (iNOS), expressed in various cell types including innate immune cells in response to wide variety of immunological cues [10,11], is however, regulated mainly at the transcriptional level and does not require calcium for its activity.
Recent discoveries have identified an important crosstalk between NO and Th cells. We have demonstrated that NO is a key regulator of T-bet and GATA3 in neuroantigen-primed T cells [13]. While iNOS:macrophage co-localization is directly proportional to T-bet:T cell co-localization, GATA3:T cell co-localization inversely correlates to iNOS:macrophage in spleen [13]. Gemfibrozil, an FDA-approved lipid-lowering drug, suppresses the production of NO and the expression of iNOS in different cells [14–16]. This drug also suppresses T-bet and stimulates GATA3 in MBP-primed T cells [13]. However, gemfibrozil is unable to suppress T-bet and stimulate GATA3 in MBP-primed splenocytes when NO is added during antigen priming [13]. Furthermore, the differential effect of gemfibrozil on T-bet and GATA3 could be replicated by adding a NO scavenger alone during antigen priming (Figure 2). Increase in NO production in MBP-primed lymph node cells (LNC) as compared to normal LNC (Figure 2A) suggests that MBP-priming induces the production of NO. Expectedly, PTIO, a NO scavenger, reduced the level of NO in MBPprimed LNC (Figure 2A). As evident from Figure 2, MBP-priming induced the mRNA expression of T-bet and IFN-γ (Figure 2B) and the production of IFN-γ protein (Figure 2C) in LNC. Interestingly, removal of NO by PTIO during MBP-priming inhibited the mRNA expression of T-bet and IFN-γ (Figure 2B) and the production of IFN-γ protein (Figure 2C) in LNC. On the other hand, treatment of MBP-primed LNC with a NO donor (DETA-NONOate) stimulated the mRNA expression of T-bet and IFN-γ (Figure 2B) and the production of IFN-γ protein (Figure 2C). In contrast, MBP-priming inhibited the mRNA expression of GATA3 and IL-10 (Figure 2D) and the protein level of IL-10 (Figure 2E) in splenocytes. Interestingly, this inhibition was also abolished by NO scavenger PTIO and magnified by NO donor DETA-NONOate (Figures 2D and 2E). These novel results highlight differential regulation of T-bet/IFN-γ/Th1 and GATA3/IL-10/Th2 cells by NO (Figure 3).
Figure 2. Differential regulation of T-bet/IFN-γ and GATA3/IL-10 by NO.
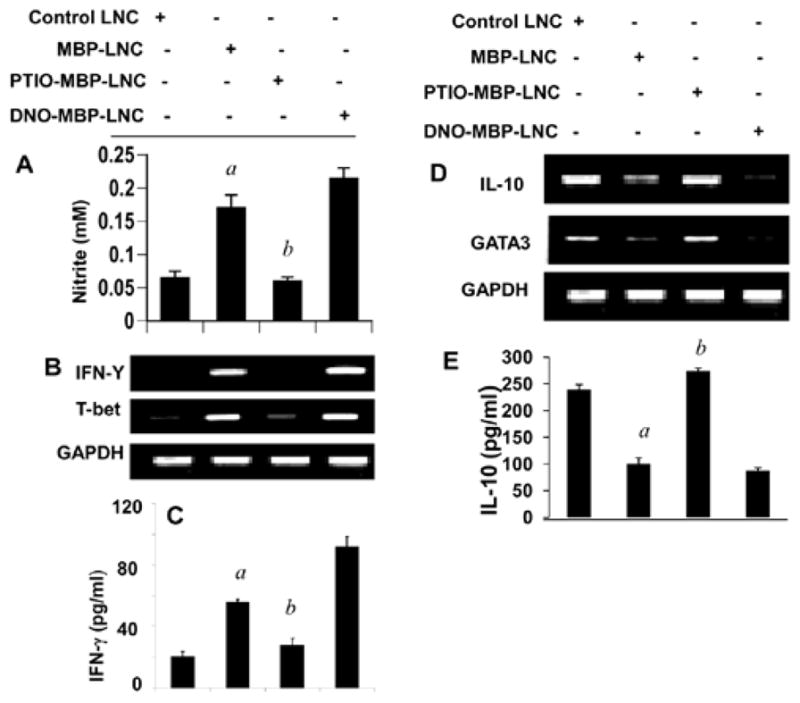
Lymph node cells (LNC) isolated from MBP-immunized female SJL/J mice were stimulated with 50 μg/ml MBP in the presence or absence of PTIO (50 μM) and DETA-NONOate (DNO; 100 μM), respectively. After 24 h of stimulation, the concentration of nitrite was measured in supernatants using ‘Griess’ reagent (A) and the mRNA expression of T-bet and IFN-γ (B) and GATA3 and IL-10 (D) was monitored in cells by semi-quantitative RT-PCR. After 72 h of stimulation, supernatants were assayed for either IFN-γ (C) or IL-10 (E) by ELISA. Results are mean + SD of three different experiments. ap<0.001 vs control; bp<0.001 vs MBP.
Figure 3. Complex regulation of Th1, Th2, Th9, and Th17 cells by NO.
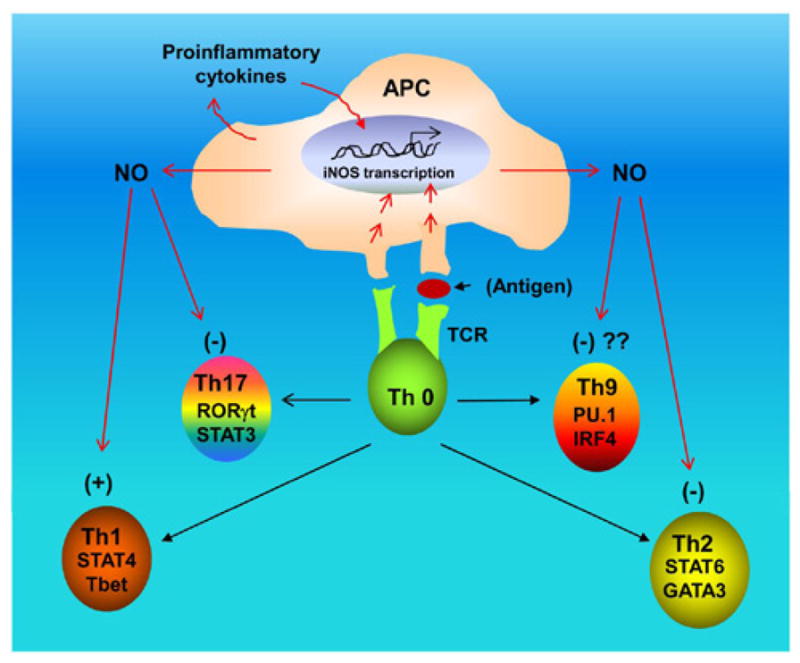
While NO suppresses the differentiation of Th2 and Th17 cells, the differentiation of Th1 cells is favored by NO. Although it is yet to be determined, NO may also suppress the differentiation of Th9 cells.
Although both Th1 and Th17 cells are autoimmune in nature, NO does not favor the differentiation of Th17 cells. According to Niedbala et al. [17], NO can suppress the proliferation and function of polarized murine and human Th17 cells. Development of Th17 cells can be enhanced by the activation of aryl hydrocarbon receptor (AHR). It has been shown that NO inhibits AHR expression in Th17 cells and the downstream events of AHR activation, including IL-22 and IL-23 receptor [17]. Accordingly, mice lacking iNOS developed more severe experimental autoimmune encephalomyelitis (EAE), an animal model of MS, than WT mice, with elevated AHR expression, increased IL-17A, and IL-22 synthesis [17]. According to Nath et al. [18], S-nitrosoglutathione (GSNO), a NO donor, attenuates the disease process of EAE by reducing the production of IL-17 (from Th0 or Th17 cells) and the infiltration of CD4+ T cells into the CNS. STAT3 and RORγt are positive regulators of Th17 signaling (Figure 1). They have also demonstrated that GSNO suppresses the phosphorylation of STAT3 and attenuates the expression of RORγt in splenocytes [18]. These evidences point towards a negative regulation of Th17 cells by NO (Figure 3).
On the other hand, until now it is not known whether the differentiation of Th9 cells depends on NO. However, careful analysis of differentiation and regulation of Th9 cells also indicates inverse relationship between NO and Th9 cells. For example, the differentiation of Th9 cells depends on TGF-β and IL-4, cytokines that are known to antagonize the expression of iNOS and the production of NO [19]. IFN-γ is a potent inducer of iNOS in many cell types including macrophages where it is known to induce the transcription of iNOS gene and the production of NO via STAT1α [10]. It has been shown that IFN-γ suppresses the generation of Th9 cells [20]. Therefore, it is possible that TGF-β and IL-4 favor Th9 via the suppression of NO production and that IFN-γ antagonizes Th9 via the induction of NO production. Such possibilities are currently being investigated in our lab.
Different immunomodulatory molecules secreted by Th1, Th2, Th9, and Th17 cells interact with the target organ to determine the outcome of an immunomodulatory process, leading to either tissue damage or repair. Recently, we have demonstrated that NO is a key player in controlling the encephalitogenicity of T cells, in which scavenging of NO reduces the EAE-inducing activity of T cells [21]. We have also described that NO is a critical molecule for the development of self tolerance in male as NO alone is sufficient to break the tolerance of male myelin-specific T cells [21]. Therefore, differential regulation of Th1, Th2, Th9, and Th17 cells by NO (Figure 3) is important as it would help in understanding the complex regulation of different inflammatory autoimmune disorders by NO and may assist in designing therapeutic avenues for numerous immunological disorders.
Acknowledgments
This study was supported by a grant from NIH (AT6681).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Joller N, Peters A, Anderson AC, Kuchroo VK. Immune checkpoints in central nervous system autoimmunity. Immunol Rev. 2012;248:122–139. doi: 10.1111/j.1600-065X.2012.01136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zepp J, Wu L, Li X. IL-17 receptor signaling and T helper 17-mediated autoimmune demyelinating disease. Trends Immunol. 2011;32:232–239. doi: 10.1016/j.it.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pahan K. Neuroimmune pharmacological control of EAE. J Neuroimmune Pharmacol. 2010;5:165–167. doi: 10.1007/s11481-010-9219-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steinman L. Multiple sclerosis: a coordinated immunological attack against myelin in the central nervous system. Cell. 1996;85:299–302. doi: 10.1016/s0092-8674(00)81107-1. [DOI] [PubMed] [Google Scholar]
- 5.Pahan K. Immunomodulation of experimental allergic encephalomyelitis by cinnamon metabolite sodium benzoate. Immunopharmacol Immunotoxicol. 2011;33:586–593. doi: 10.3109/08923973.2011.561861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Racke MK, Bonomo A, Scott DE, Cannella B, Levine A, et al. Cytokine-induced immune deviation as a therapy for inflammatory autoimmune disease. J Exp Med. 1994;180:1961–1966. doi: 10.1084/jem.180.5.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.El-behi M, Rostami A, Ciric B. Current views on the roles of Th1 and Th17 cells in experimental autoimmune encephalomyelitis. J Neuroimmune Pharmacol. 2010;5:189–197. doi: 10.1007/s11481-009-9188-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li H, Rostami A. IL-9: basic biology, signaling pathways in CD4+ T cells and implications for autoimmunity. J Neuroimmune Pharmacol. 2010;5:198–209. doi: 10.1007/s11481-009-9186-y. [DOI] [PubMed] [Google Scholar]
- 9.Ramming A, Druzd D, Leipe J, Schulze-Koops H, Skapenko A. Maturation-related histone modifications in the PU.1 promoter regulate Th9-cell development. Blood. 2012;119:4665–4674. doi: 10.1182/blood-2011-11-392589. [DOI] [PubMed] [Google Scholar]
- 10.Saha RN, Pahan K. Regulation of inducible nitric oxide synthase gene in glial cells. Antioxid Redox Signal. 2006;8:929–947. doi: 10.1089/ars.2006.8.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saha RN, Pahan K. Signals for the induction of nitric oxide synthase in astrocytes. Neurochem Int. 2006;49:154–163. doi: 10.1016/j.neuint.2006.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ignarro LJ, Kadowitz PJ. The pharmacological and physiological role of cyclic GMP in vascular smooth muscle relaxation. Annu Rev Pharmacol Toxicol. 1985;25:171–191. doi: 10.1146/annurev.pa.25.040185.001131. [DOI] [PubMed] [Google Scholar]
- 13.Dasgupta S, Roy A, Jana M, Hartley DM, Pahan K. Gemfibrozil ameliorates relapsing-remitting experimental autoimmune encephalomyelitis independent of peroxisome proliferator-activated receptor-alpha. Mol Pharmacol. 2007;72:934–946. doi: 10.1124/mol.106.033787. [DOI] [PubMed] [Google Scholar]
- 14.Xu J, Chavis JA, Racke MK, Drew PD. Peroxisome proliferator-activated receptor-alpha and retinoid X receptor agonists inhibit inflammatory responses of astrocytes. J Neuroimmunol. 2006;176:95–105. doi: 10.1016/j.jneuroim.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 15.Pahan K, Jana M, Liu X, Taylor BS, Wood C, et al. Gemfibrozil, a lipid-lowering drug, inhibits the induction of nitric-oxide synthase in human astrocytes. J Biol Chem. 2002;277:45984–45991. doi: 10.1074/jbc.M200250200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jana M, Jana A, Liu X, Ghosh S, Pahan K. Involvement of phosphatidylinositol 3-kinase-mediated up-regulation of I kappa B alpha in anti-inflammatory effect of gemfibrozil in microglia. J Immunol. 2007;179:4142–4152. doi: 10.4049/jimmunol.179.6.4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Niedbala W, Alves-Filho JC, Fukada SY, Vieira SM, Mitani A, et al. Regulation of type 17 helper T-cell function by nitric oxide during inflammation. Proc Natl Acad Sci USA. 2011;108:9220–9225. doi: 10.1073/pnas.1100667108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nath N, Morinaga O, Singh I. S-nitrosoglutathione a physiologic nitric oxide carrier attenuates experimental autoimmune encephalomyelitis. J Neuroimmune Pharmacol. 2010;5:240–251. doi: 10.1007/s11481-009-9187-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oswald IP, Gazzinelli RT, Sher A, James SL. IL-10 synergizes with IL-4 and transforming growth factor-beta to inhibit macrophage cytotoxic activity. J Immunol. 1992;148:3578–3582. [PubMed] [Google Scholar]
- 20.Murugaiyan G, Beynon V, Pires Da Cunha A, Joller N, Weiner HL. IFN-γ Limits Th9-Mediated Autoimmune Inflammation through Dendritic Cell Modulation of IL-27. J Immunol. 2012;189:5277–5283. doi: 10.4049/jimmunol.1200808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mondal S, Brahmachari S, Pahan K. Regulation of Encephalitogenicity of Neuroantigen-Primed T Cells by Nitric Oxide: Implications for Multiple Sclerosis. J Clin Cell Immunol. 2012;3:124. doi: 10.4172/2165-8048.1000124. [DOI] [PMC free article] [PubMed] [Google Scholar]