

Abstract
During the past 5 years an increasing number of patients were diagnosed with congenital myasthenic syndromes (CMS) and a number of novel syndromes were recognized and investigated. This presentation focuses on the CMS caused by defects in choline acetyltransferase, novel fast-channel syndromes that hinder isomerization of the acetylcholine receptor from the closed to the open state, the consequences of deleterious mutations in the intermediate filament linker plectin, altered neuromuscular transmission in a centronuclear myopathy, and two recently identified CMS caused by congenital defects in glycosylation.
Keywords: congenital myasthenic syndromes, acetylcholine receptor, fast-channel syndromes, choline acetyltransferase, plectin, centronuclear myopathy, GFPT1, DPAGT1
Introduction
The molecular era of the congenital myasthenic syndromes (CMS) came of age in 1995 with discovery that a highly disabling slow-channel CMS was caused by a gain-of-function point mutation in the second transmembrane domain of the AChR ε subunit. Since then, no fewer than 14 disease genes have been identified and the tools of molecular genetics empowered the candidate gene approach. Linkage analysis and more recently exome sequencing studies also helped pinpoint disease genes and mutations. Table 1 shows a classification of 350 CMS kinships investigated at the Mayo Clinic to date. The table is still incomplete because in half as many kinships the molecular basis of the CMS remains undefined. This presentation focuses on selected CMS recently investigated in our laboratory.
Table 1.
Classification of the Mayo Cohort of CMS Patientsa
| Index cases | |
|---|---|
| Presynaptic defects (6%) | |
| Choline acetyltransferase deficiencyb | 18 |
| Paucity of synaptic vesicles and reduced quantal release | 1 |
| Lambert-Eaton syndrome like | 1 |
| Synaptic basal lamina-associated defects (13%) | |
| Endplate AChE deficiencyb | 45 |
| β2-laminin deficiencyb | 1 |
| Postsynaptic defects (67%) | |
| Primary kinetic abnormality of AChR(slow- and fast-channel syndromes)b | 62 |
| Primary AChR deficiencyb | 118 |
| Rapsyn deficiencyb | 51 |
| Plectin deficiencyb | 2 |
| Na-channel myastheniab | 1 |
| Defects in endplate development and maintenance (14%) | |
| Dok-7 myastheniab | 35 |
| GFPT1 deficiencyb | 11 |
| DAPGT1 deficiencyb | 2 |
| Agrin deficiencyb | 1 |
| CMS with centronuclear myopathyc | 1 |
| Total (100%) | 350 |
One-hundred and twenty-six patients also underwent intercostal muscle biopsies..
Choline acetyltransferase (ChAT) deficiency
We have investigated 18 unrelated patients with this CMS. The first 5 were seen before 20011 and 13 others in the past decade; 11 of 13 patients harboring one nonsense and 12 missense mutation were further investigated2 (Fig. 1A). Abrupt episodes of apnea or severe dyspnea against a variable background of myasthenic symptoms remains a hallmark of the disease. Five patients presented with apnea and with or without other myasthenic symptoms at birth and 5 had similar presentation during the first 5 months of life. Three patients never breathed spontaneously; one died and 2 others are ventilator dependent at 4 and 10 years of age. One patient was born with multiple joint contractures. Eight affected siblings of 4 patients died in infancy. Pyridostigmine treatment improved but did not remove the symptoms in 9 patients and had no effect in 4 severely affected patients.
Figure 1.

Recently identified mutations in ChAT. (A) Human ChAT showing the identified mutations and the catalytic His442 at the active site. Asterisk indicates the active-site tunnel. (B) Positions of mutated residues in the active site tunnel and kinetic landscapes of wild-type and mutant enzymes. The p.M202R mutant is essentially inactive. T553N and A557T mutants fail to saturate within the indicated range of substrate concentrations. (C) Position of S572 and kinetic landscape of the S572W-ChAT. S572 is close to the AcCoA and choline binding sites. This mutation markedly reduces kcat and Km for both substrates. (PDB 2FY2). (Reprinted from Reference 2, by permission.)
To better understand the phenotypic variability of the disease, we determined expression of the recombinant mutants in heterologous cells, analyzed their kinetic properties and thermal stability, and interpreted their functional effects in the context of the atomic structural model of human ChAT at 2.2 Å resolution.3,4 We examined expression of missense mutants at the protein level by genetically engineering mutant and wild-type CHAT cDNAs into Bosc 23 cells and analyzed immunoblots of cell lysates. Eight mutants expressed at significantly lower levels than wild-type, and five of these (W421S, S498P, T553N, p.A557T, p.S572W) expressed at <50% of wild-type.
To evaluate the kinetic parameters and thermal stability of wild-type and mutant ChATs, we transformed E. coli with histidine-tagged CHAT cDNAs and purified the enzymes recovered from the bacterial cell lysates on a Ni-NTA column followed by fast protein liquid chromatography. Ten mutations alter one or more rate constants of ChAT activation and hence the catalytic efficiency of the enzyme. Two mutations that introduce a Pro residue into an alpha helix (S498P and S704P) have little effect on enzyme kinetics but compromise the thermal stability of the mutant protein. The most severely affected patients harbored at least one mutation near the active site tunnel (M202R, T553N, and A557T) (Fig. 1B) or the agonist binding site (S572W) (Fig. 1C) of the enzyme and some of these also curtailed enzyme expression.
Novel fast-channel mutations in AChR
Fast-channel CMS are caused by recessive loss-of-function mutations of the AChR. The mutations become pathogenic when accompanied by a low-expressor or null mutation in the second allele or if they occur at homozygosity. The mutations exert their effect by decreasing agonist affinity or by impeding isomerization of the receptor from the closed to the open state.
Homozygous εW55R mutation at the α/ε binding site interface
This mutation was observed in an 8-year-old boy born to consanguineous parents with severe myasthenic symptoms that responded poorly to pyridostigmine.5 Three similarly affected siblings died in infancy. The mutated Trp55 is one of several negatively charged aromatic residues at the α/ε binding site required to stabilize cationic ACh by electrostatic forces (Fig. 2A B, and C). Thus replacement of the electron-rich Trp by a cationic Arg was expected to hinder binding of ACh to the α/ε binding site. Single-channel recordings from εW55R-AChR expressed in HEK cells at low ACh concentration (50 nM) revealed that the length of the dominant component of channel opening bursts was reduced to 10% of wild type (Fig. 2E). Analysis of channel openings over a range of ACh concentrations demonstrated that the εW55R mutation reduces apparent agonist affinity at the α/ε binding site by 30-fold and decreases apparent gating efficiency by 75-fold. The mutation also curtails the opening probability of the receptor (Popen) over a wide range of ACh concentration (Fig. 2D). In the presence of 1 mM ACh, which corresponds to the estimated peak ACh concentration in the synaptic space after quantal release, the Popen of the mutant receptor is only 10% of wild-type, which explains the patient’s refractoriness to clinically attainable doses of pyridostigmine.
Figure 2.

The εW55R mutation at the AChR α/ε binding site. (A): Structural model of extracellular domains of human AChR viewed from the synaptic space indicating positions of Trp residues at the α/δ and α/ε binding sites. (PDB 1 9B) (B) Side-view of the α and ε subunits showing position of loops E, D, G, and F in the ε subunit, and loops A, B, and C in the α subunit. (C) Stereo view of the binding site showing positions of aromatic residues shrouding the binding pocket. In each panel, the mutated εTrp55 at the α/ε binding site is highlighted in red. (Based on the crystal structure of the ACh binding protein (PDB 19B) and lysine scanning mutagenesis delineating the structure of the human AChR binding domain. (D). Channel open probability (Popen) of ε W55R-AChR as function of ACh concentration. Symbols and vertical lines indicate means and standard deviations. Smooth curves are Popen predicted by the fitted rate constants. (E) Single-channel currents elicited from HEK cells transfected with wild-type- and εW55R-AChR. Left: Representative channel openings elicited by 50 nM ACh. Right: Logarithmically binned burst duration histograms fitted to the sum of exponentials. Arrows indicate mean duration of burst components. (Reprinted from Reference 5, by permission.)
αV188M mutation in the AChR C-loop hinders initiation of channel gating
According to current knowledge, the C-loop alters its conformation during agonist binding. Without agonist, it adopts a range of open conformations that allow the agonist to enter the binding pocket. When the agonist binds, the C-loop moves inward, traps the agonist, and thereby initiates the chain of events that culminate in opening of the ion channel.6–9
An αV188M mutation in the C-loop (Fig. 3A) was observed in a 42-year-old woman with severe myasthenic symptoms since birth.10 She also carries a heteroallelic low-expressor αG74C mutation in the main immunogenic region; hence αV188M determines the phenotype. Light and electron microscopy studies of intercostal muscle endplates (EPs) demonstrated no structural abnormality.
Figure 3.

The α1AChR subunit binding site and mutant cycle analysis of potentially interacting residues. (A) Stereo view of the binding site at 1.9 AǺ resolution (PDB 2QC1) shows spatial disposition of the potentially interacting residues. (B) Cubic mutant cycle analysis of energetic interactions betweenαV188, αY190 and αK145. (C) Cubic mutant cycle analysis of energetic interactions between αV188, αD200 and αK145. In (B) and (C), numbers on arrows show difference in free energy change (ΔΔG) between 2 different AChRs in kcal/mol. The indicated diagonal lines in planes show first-order coupling free energy (ΔΔGint) for a given pair of mutants except for the front planes of B and C, where it is ΔΔGint=−0.78 kcal/mol, and for the back planes of B and C where it is ΔΔGint=0.13 and 0.78 kcal/mol, respectively.( Reprinted from Reference 10, by permission.)
When wild-type and mutant receptors were expressed in HEK cells and their openings monitored by patch-clamp, the length of the predominant burst duration of the mutant receptor was only 20% of the wild-type. Detailed kinetic analysis of single channel recordings obtained over a range of ACh concentrations revealed that the most significant effect of αV188M is a ~70-fold decrease of the apparent channel gating efficiency θ, defined by the ratio of the channel opening rate α to the channel closing rate β. The derived rate constants of activation allowed calculation of Popen as a function of ACh concentration. A plot of Popen over 3 orders of magnitude of ACh concentrations revealed a marked right-shift of Popen of the mutant compared to the wild-type receptor. Substitution for Val188 with residues of larger or smaller side-chain volume again reduced the channel opening rate.
To elucidate how αV188M hinders channel opening, we employed thermodynamic mutant cycle analysis11 to test whether αV188 is energetically coupled to nearby αY190, αD200, and αK145, and whether α these residues contribute jointly to closure of the C-loop after agonist binding (Fig. 3A).12 The principle of mutant cycle analysis is that the free energy change caused by mutation of residue A depends on other residues in the protein. If mutation of a second residue B alters the free energy change caused by mutation of A, then A and B are energetically coupled. If mutation of a third residue C alters the interactive free energy of the coupling residues A and B, then C also contributes to the interaction between A and B.
We cast the results of the mutant cycle experiments into two 3-dimensional cubic mutant cycles. This analysis revealed that αV188 is strongly coupled with αY190 (Figure 3B, top face) and also with αD200 (Figure 3C, top face), but not with αK145 (front faces in in Fig. 3B and 3C). We also found that αK145 contributes to energetic coupling between αV188 and αY190 (Figure 6B, top and bottom faces) and between αV188 and αD200 (top and bottom faces in Fig. 3C). Further studies revealed that energetic coupling between αK145/αY190 also depends on αV188 (left and right faces in Fig. 3B). Moreover, αV188 also contributes to the energetic coupling between αK145 and αD200 (left and right faces in Figure 3C). Thus αV188 emerges as a key residue that orchestrates rearrangement of the C-loop during initial coupling of binding to gating.
CMS caused by plectin deficiency
Plectin, encoded by PLEC, is a 499–533 kDa dumbbell-shaped intermediate filament cytolinker with multiple isoforms due to exon 1 splice variants. Muscle isoforms link the intermediate filament cytoskeleton to myofibrils, sarcolemma, mitochondria, and nuclear envelope proteins. A skin isoform links epidermal keratin filaments to dermal integrin filaments. The predictable consequences of plectin deficiency in muscle are dislocation of myofiber organelles, gaps in the sarcolemma, and degenerating junctional folds. In skin, plectin deficiency results in epidermolysis bullosa simplex (EBS) due to dermoepidermal disjunction.
The two patients observed by us respectively carry a Q2057X and p.R2319X and a shared c.12043dupG mutation13,14 Both patients had EBS since infancy and later developed a progressive myopathy and myasthenic syndrome refractory to pyridostigmine. Both had a decremental EMG response on repetitive nerve stimulation, and half-normal amplitude miniature EP potentials (MEPPs). One patient became motionless at age 37 years and died at age 42. The second patient is severely disabled in her thirties. Morphologic studies of both patients revealed dislocated and degenerating muscle fiber organelles (Fig. 4A–D), and plasma membrane defects (Fig. 4E) resulting in Ca2+ overloading of the muscle fibers as in Duchenne dystrophy, and extensive degeneration of the junctional folds (Fig. 4F), all attributable to lack of cytoskeletal support.14
Figure 4.
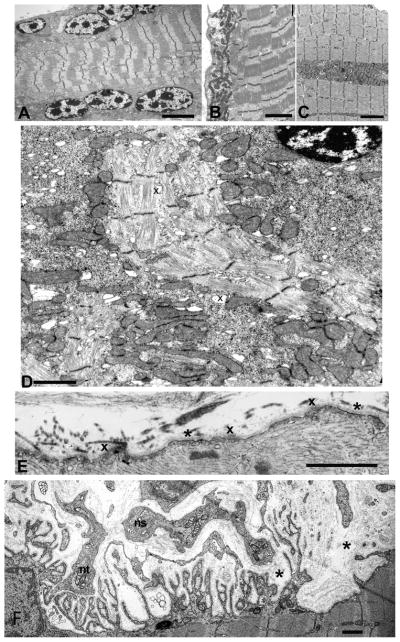
Ultrastructural findings in plectin deficient muscle fiber. (A) Note subsarcolemmal rows of large nuclei harboring multiple prominent chromatin bodies. (B) and (C) Subsarcolemmal and intrafiber clusters of mitochondria surrounded by fiber regions devoid of mitochondria. (D) Aberrant and disrupted myofibrils surrounded by clusters of mitochondria intermingled with glycogen, ribosomes, and dilated vesicles (x). Note pre-apoptotic nucleus at upper right. (E) Focal sarcolemma defects due to gaps in the plasma membrane. Where the plasma membrane is absent, the overlying basal lamina is thickened (x). Small vesicles underlie the thickened basal lamina. Asterisks indicate segments of the preserved plasma membrane. (F) Degenerating EP in plectin deficiency. On the left, the postsynaptic region is only partially occupied by the nerve terminal (nt). The center of the panel shows a nerve sprout (ns) and a nerve terminal are separated by a widened synaptic space from the underlying junctional folds. Many folds are atrophic and degenerating. Streaks of collapsed basal lamina and small globular residues represent remnants of preexisting folds (asterisks). Bars = 4 μm in (A), 3 μm in (B), (C), 1.4 μm in (D), 1 μm in (E) and (F). (Reprinted from Reference 14, by permission.)
CMS in centronuclear myopathy
Centronuclear myopathy (CNM) is a clinically and genetically heterogeneous congenital disorder diagnosed by presence of central nuclei in the majority of muscle fibers with only few or no necrotic or regenerating fibers. The disease genes identified to date are myotubularin (MTM1), dynamin 2 (DNM2), amphiphysin 2 (BIN1) and skeletal muscle ryanodine receptor (RYR1) but several patients diagnosed by pathologic criteria have no identified mutations. Apart from limb weakness, ptosis and ophthalmoparesis often occur in all genetically-identified CNM subtypes and some CNM patients fatigue abnormally. One CNM patient with myasthenic features had a homozygous missense mutation in BIN1.15 Four CNM patients with myasthenic features responding to pyridostigmine with no known mutation were also reported but EP structure and parameters of neuromuscular transmission were not evaluated.16
We investigated an adult CNM patient with myasthenic features.17 His early motor development was normal but he never ran well. At age 13 years, he had fatigable limb-muscle weakness and mild eyelid ptosis.. At age 39 years, he had a 19–35% EMG decrement. He responded well to pyridostigmine. Mutation analysis revealed no mutations in currently identified CNM disease genes.
EP studies revealed pre- and postsynaptic abnormalities. Synaptic contacts visualized by the cholinesterase reaction showed multiple EP regions on single fibers suggesting remodeling. Quantitative EM revealed simplified postsynaptic regions, normal nerve terminal size, normal synaptic vesicle density, and mild AChR deficiency. The amplitude of the miniature EP potential (MEPP) was reduced to 60% of normal. The MEPP decay time was 1.5+-fold prolonged pointing to presence of fetal AChR at the EPs. Quantal release by nerve impulse (m) was reduced to 40% of normal due to decreased number of quanta available for release. The decreased MEPP amplitude could be due to a combination of factors: simplification of the postsynaptic region reduces the input resistance of the muscle fiber and hence the MEPP amplitude; the decreased number of AChRs per EP; and presence of immature AChRs at the EP that generate lower amplitude MEEPs than adult AChRs. The decreased quantal release by nerve impulse could be due to decreased synaptic contacts per muscle fiber or to a defect in the synaptic vesicle cycle. Because the cholinesterase reactive synaptic contact areas of the fibers were not appreciably reduced, the decrease in m likely signals a defect in the synaptic vesicle cycle.
CMS caused congenital defect in glycosylation
Glycosylation of nascent peptides increases their solubility, folding, assembly, and intracellular transport.18,19,20 Two enzymes involved in glycosylation, glutamine fructose-6-phosphate transaminase (GFPT1) and dolichol phospate-GlycNac-1-phosphotransferase (DPAGT1) are now known to be associated with CMS. GFPT1 controls the flux of glucose into the hexosamine pathway, and hence the biosynthesis of N-acetylglucosamine, a key component of glycosyl residues for N- and O-linked glycosylation of proteins. DPAGT1 catalyzes the first committed step in the path of N-glycosylation.
CMS caused by defects in GFPT1
This CMS was identified in 2011 by Senderek and co-workers 21 by linkage and homozygosity analysis studies of multiplex kinships with limb-girdle CMS associated with tubular aggregates in skeletal muscle. GFPT1 is expressed in many tissues and has a long muscle-specific isoform (GFPTL1). We investigate 11 CMS patients with GFPT1 deficiency. Ten had a childhood onset, slowly progressive limb-girdle weakness responsive to pyridostigmine. In contrast, one (Patient 6) did not move in utero and was apneic and floppy at birth with multiple joint contractures. At age 9 years, she has marked weakness of all except the ocular muscles, requires ventilatory support and gastrostomy feedings, and responds only slightly to pyridostigmine and 3,4-diaminopyridine. Interestingly, her mother has dermatomyositis and a high titer of AChR antibodies but no clinical myasthenia. The patient never had antibodies against adult AChR and tests by Angela Vincent detected no antibodies against fetal AChR in the patient or her mother.
Mutation analysis of each patient revealed two heteroallelic mutations in GFPT1 in each patient and 12 of these are novel. The most severely affected Patient 6 harbors a nonsense mutation and an intronic deletion that causes skipping of the muscle specific exon of GFPT1.
Conventional histochemical studies revealed tubular aggregates in type 2 fibers in 6 of 7 patients, and rimmed vacuoles in 3 patients. The vacuoles in Patient 6 had autophagic features. Six patients had abnormal EM studies; 5 had abnormally small nerve terminals and simplified postsynaptic regions; Patient 6 had numerous fibers harboring dilated and degenerating vesicular profiles, multiple autophagic vacuoles, and bizarre apoptotic nuclei.
Parameters of neuromuscular transmission were evaluated by microelectrode studies in 5 patients. In 3, the MEPP amplitude was significantly reduced and this was most marked in Patient 6. Quantal release by nerve impulse was normal in 4 patients, but was markedly reduced in one patient.
CMS caused by defects in DPAGT1
We recently identified two CMS patients harboring mutations in DPAGT1.22 While our work was in progress, this CMS was reported by Belaya and co-workers.23 One of our patients is a 16-year-old mentally retarded girl with severe generalized CMS since infancy. One similarly affected sibling also has autistic features. The second patient is a 14-year-old girl with mild cognitive deficits and progressive limb-girdle CMS since infancy. Both responded poorly to anti-AChE therapy. Intercostal muscle specimens in both show small tubular aggregates in type 2 fibers, type1 fiber atrophy, and a vacuolar myopathy with autophagic features. Endplate studies revealed that quantal release, postsynaptic response to acetylcholine quanta, and EP AChR content are reduced to ~50% of normal. Quantitative EM of 65 EP regions shows hypoplastic endplates, very small nerve terminals, and poorly differentiated postsynaptic regions. Neither patient harbors mutations in currently recognized CMS disease genes but exome sequencing in each identified two heteroallelic mutations in DPAGT1. Immunoblots of muscle extracts probed by two different antibodies demonstrated reduced to absent glycosylation of ~70 kDa proteins. We hypothesize that hypoglycosylation of synapse-specific proteins causes defects in motor as well as central synapses.
Acknowledgments
Work done in our laboratories was supported by a grant from the National Institutes of Health (NS-6277, Drs. Engel, Shen, and Selcen; NS-031744, Dr. Sine) and by a research grant from the Muscular Dystrophy Association (Dr. Engel).
Reference List
- 1.Ohno K, Tsujino A, Shen XM, Brengman J, Harper CM, Bajzer Z, Udd B, Beyring R, Robb S, Kirham FJ, et al. Choline acetyltransferase mutations cause myasthenic syndrome associated with episodic apnea in humans. Proc Natl Acad Sci USA. 2001;98(4):2017–2022. doi: 10.1073/pnas.98.4.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shen XM, Crawford TO, Brengman J, Acsadi G, Iannaconne S, Karaca E, Khoury C, Mah JK, Edvardsons S, Bajzer Z, et al. Functional consequences and structural interpretation of mutations in human choline acetyltransferase. Hum Mutat. 2011;32:1259–1267. doi: 10.1002/humu.21560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim AR, Dobransky T, Rylett RJ, Shilton BH. Surface-entropy reduction used in the crystallization of human choline acetyltransferase. Acta Crystallogr Sect D. 2005;61:1306–1310. doi: 10.1107/S0907444905018822. [DOI] [PubMed] [Google Scholar]
- 4.Kim AR, Rylett RJ, Shilton BH. Substrate binding and catalytic mechanism of human choline acetyltransferase. Biochemistry. 2006;45:14621–14631. doi: 10.1021/bi061536l. [DOI] [PubMed] [Google Scholar]
- 5.Engel AG, Brengman J, Edvardson S, Shen XM, Sine SM. Highly fatal fast-channel congenital syndrome caused by AChR ε subunit mutation at the agonist binding site. Neurology. 2011;79:449–454. doi: 10.1212/WNL.0b013e31825b5bda. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Celie PH, van Rossum-Fikkert SE, van Dijk WJ, Brejc K, Smit AB, Sixma TK. Nicotine and carbamycholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron. 2004;41(6):907–914. doi: 10.1016/s0896-6273(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 7.Cheng X, Wang H, Grant B, Sine SM, McCammon A. Targeted molecular dynamics study of C-loop closure and channel gating in nicotinic receptors. PLOS Comput Biol. 2006;2(9):e134, 1173–1184. doi: 10.1371/journal.pcbi.0020134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang HL, Toghraee R, Papke D, Cheng XL, McCammon A, Ravaioli U, Sine SM. Single-channel current through nicotinic receptor produced by closure of binding C-loop. Biophys J. 2009;96(9):3582–3590. doi: 10.1016/j.bpj.2009.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li SX, Huang S, Bren N, Noridomi K, Dellisanti CD, Sine SM, Chen L. Ligand-binding domain of an α7-nicotinic receptor chimera and its complex with agonist. Nat Neurosci. 2011;14:1253–1259. doi: 10.1038/nn.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen XM, Brengman J, Sine SM, Engel AG. Myasthenic syndrome AChRα C-loop mutant disrupts initiation of channel gating. J Clin Invest. 2012;122:2613–2621. doi: 10.1172/JCI63415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horovitz A, Fersht A. Strategy for analyzing the co-operativity of intramolecular interactions in peptides and proteins. J Mol Biol. 1990;214(3):613–617. doi: 10.1016/0022-2836(90)90275-Q. [DOI] [PubMed] [Google Scholar]
- 12.Mukhtasimova N, Free C, Sine SM. Initial coupling of binding to gating mediated by conserved residues in muscle nicotinic receptor. J Gen Physiol. 2005;126(1):23–39. doi: 10.1085/jgp.200509283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Banwell BL, Russel J, Fukudome T, Shen XM, Stilling G, Engel AG. Myopathy, myasthenic syndrome, and epidermolysis bullosa simplex due to plectin deficiency. J Neuropathol Exp Neurol. 1999;58:832–846. doi: 10.1097/00005072-199908000-00006. [DOI] [PubMed] [Google Scholar]
- 14.Selcen D, Juel VC, Hobson-Webb LD, Smith EC, Stickler DE, Bite AV, Ohno K, Engel AG. Myasthenic syndrome caused by plectinopathy. Neurology. 2011;76(4):327–336. doi: 10.1212/WNL.0b013e31820882bd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Claeys KG, Maisonobe T, Bohm J, et al. Phenotype of a patient with recessive centronuclear myopathy and a novel BIN1 mutation. Neurology. 2010;74:519–521. doi: 10.1212/WNL.0b013e3181cef7f9. [DOI] [PubMed] [Google Scholar]
- 16.Robb SA, Sewry CA, Dowling JJ, Feng L, Cullup S, Lillis S, Abbs MM, Laporte J, Manzur AY, Knight RK, et al. Impaired neuromuscular transmission and response to aceylcholinesterase inhibitors in centronuclear myopathy. Neuromuscul Disord. 2011;21:379–386. doi: 10.1016/j.nmd.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 17.Liewluck T, Shen XM, Milone M, Engel AG. Endplate structure and parameters of neuromuscular transmission in sporadic centronuclear myopathy associated with myasthenia. Neuromuscul Disord. 2011;21:387–395. doi: 10.1016/j.nmd.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haeuptle MA, Hennet T. Congenital disorders of glycosylation: An update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutation. 2009;30:1628–1641. doi: 10.1002/humu.21126. [DOI] [PubMed] [Google Scholar]
- 19.Freeze HH, Sharma V. Metabolic manipulation of glycosylation disorders in humans and animal models. Semin Cell Dev Biol. 2010;21:655–662. doi: 10.1016/j.semcdb.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freeze HH, Eklund EA, NGBG, Patterson MC. Neurology of inherited glycosylation disordrs. Lancet Neurology. 2012;11:453–466. doi: 10.1016/S1474-4422(12)70040-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Senderek J, Muller JS, Dusl M, Strom TM, Guerggueltcheva V, Diepolder I, Laval SH, Maxwell S, Cossins J, Krause S, et al. Hexosamine biosynthetic pathway mutations cause neuromuscular transmission defect. Am J Hum Genet. 2011;88:162–172. doi: 10.1016/j.ajhg.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Selcen D, Shen X-M, Li Y, Wiben E, Engel AG. Congenital myashenic syndrome, autophagic myopathy, and cognitive dysfunction caused by mutaions in DPAGT1. Ann Neurol. 2012 (Abstr.) [Google Scholar]
- 23.Belaya K, Finlayson S, Slater C, Cossins J, Liu WW, Maxwell S, McGowan SJ, Maslau S, Twigg SRF, Walls TJ, et al. Mutations in DPAGT1 cause a limb-girdle congenital myasthenic syndrome with tubular aggregates. Am J Hum Genet. 2012;91:1–9. doi: 10.1016/j.ajhg.2012.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]