

Abstract
DNA mismatch repair proteins (MMR) maintain genetic stability by recognizing and repairing mismatched bases and insertion/deletion loops mistakenly incorporated during DNA replication, and initiate cellular response to certain types of DNA damage. Loss of MMR in mammalian cells has been linked to resistance to certain DNA damaging chemotherapeutic agents, as well as to increase risk of cancer. Mismatch repair pathway is considered to involve the concerted action of at least 20 proteins. The most abundant MMR mismatch-binding factor in eukaryotes, MutSα, recognizes and initiates the repair of base-base mismatches and small insertion/deletion. We performed molecular dynamics simulations on mismatched and damaged MutSα-DNA complexes. A comprehensive DNA binding site analysis of relevant conformations shows that MutSα proteins recognize the mismatched and platinum cross-linked DNA substrates in significantly different modes. Distinctive conformational changes associated with MutSα binding to mismatched and damaged DNA have been identified and they provide insight into the involvement of MMR proteins in DNA-repair and DNA-damage pathways. Stability and allosteric interactions at the heterodimer interface associated with the mismatch and damage recognition step allow for prediction of key residues in MMR cancer-causing mutations. A rigorous hydrogen bonding analysis for ADP molecules at the ATPase binding sites is also presented. Due to extended number of known MMR cancer causing mutations among the residues proved to make specific contacts with ADP molecules, recommendations for further studies on similar mutagenic effects were made.
Keywords: DNA repair, MMR proteins, MutSα, MMR-dependent apoptosis, Mismatch recognition, MMR cancer causing mutations
Introduction
DNA mismatch repair proteins (MMR) maintain genetic stability in both prokaryotes and eukaryotes by detecting and repairing mismatched bases and insertion/deletion loops mistakenly incorporated during DNA replication. The MMR machinery first recognizes the base-base mismatch and insertion/deletion loops and second, directs the repair assembly to the newly synthesized DNA strand carrying the defective genetic information. The steps of mismatch recognition process were first elucidated in E.coli culminating with the reconstruction of its MMR system from individual purified components (1). The human mismatch repair process was recently reconstructed (2, 3) and three molecular mechanisms of mammalian mismatch repair have been proposed: the molecular-switch model (4), the active-translocation model (5, 6), and the DNA bending/verification model (7, 8).
In addition, MMR proteins initiate cellular response to certain types of DNA damage, such as those produced by chemotherapeutic agents, including cisplatin adducts (cis-diammine-dichloro-platinum(II),CDDP). Mammalian cells with defective MMR show partial or complete failure to undergo apoptosis following specific types of DNA damage and the human mismatch-binding factor MutSα (MSH2/MSH6) promotes apoptosis in normal cells (9). In a proposed molecular mechanism for MMR-dependent damage response, altered DNA flexibility and changes in protein-DNA interactions are transmitted across the MSH2/MSH6 complex via distinct conformational changes (10-12). A different model for MutSα function suggests that all lesions are recognized in a similar manner, and the diversity of MMR-dependent responses to DNA mismatch or damage is generated in events downstream of the recognition step (13). The identification of mutations in MutSα that eliminate the repair functions without reducing the MMR-dependent damage response supports the former model (14).
Loss of MMR in mammalian cells has been linked to resistance to certain DNA damaging agents including clinically important cytotoxic chemotherapeutics, as well as to increased risk of cancer. It is considered that cells deficient in MMR proteins have a “mutator phenotype” in which the rate of spontaneous mutations is greatly elevated causing a predisposition to cancer (15). Inactivation of MMR in human cells is associated with hereditary and sporadic cancers, with hereditary non-polyposis colon cancer (HNPCC) being the most highlighted (16, 17).
The mismatch recognition in a newly synthesized human DNA is mediated by one of the two heterodimers of MutS homologues. The most abundant MMR mismatch-binding factor, MutSα, which is a heterodimer of MSH2 and MSH6, initiates the repair of base-base mismatches and small insertion/deletion, IDLs, of one or two extra-helical nucleotides, whereas the recognition and repair of larger IDLs is initiated by MutSβ, which is a heterodimer of MSH2 and MSH3 (18-20). The homodimeric prokaryotic MutS (21), which is a heterodimer at the structural level, and the heterodimeric eukaryotic MutSα (13) are made of two polypeptide chains. Each subunit has at least 800 amino acids in a modular architecture divided into five domains: the mismatch binding domain, which contains the highly conserved amino-terminal motif GXFYE that is required for mismatch recognition, the connector domain, the lever domain, the clamp domain, and the ATPase domain, which is the carboxy-terminal domain. The prokaryotic MutS proteins share only limited sequence identity with human MutSα, and the homodimer is about 600 amino acids smaller than the heterodimer. In the crystal structures of human MutSα bound to different DNA substrates (13): only MSH6 makes specific contacts with the mis-paired bases and MSH2 is considered the non-mismatch binding monomer; the connector domain is well positioned to be involved in inter-domains allosteric signaling; the long α-helical lever domain displays a conserved loop likely involved in signaling transduction between ATPase and DNA binding domains; the small clamp domains make significant nonspecific DNA contacts; the ATPase domains consist of the highly conserved Walker motifs, typical of ABC transporters.
Nucleotide mis-incorporation during the DNA synthesis generates DNA base-base mismatches. DNA damage accumulates in cells over time as a result of both normal metabolic activities and environmental chemical and physical agents. It affects the primary structure of the double helix in which the bases are chemically modified by introducing non-native bonds or bulky adducts, such as the case of cisplatin. If unrepaired, the damaged DNA alters or eliminates the cell’s ability to transcribe the gene that the affected DNA encodes, or has the potential to generate mutations in the cell’s genome, which cannot be repaired, causing dysfunction and disease. Cells possess mechanisms to correct DNA mismatches safeguarding the integrity of the genome, and to repair DNA damage, and thus prevent mutations.
DNA mismatch repair pathway corrects base mismatches generated during DNA replication, thereby preventing mutations becoming permanent in dividing cells. It also inhibits recombination between non-identical DNA sequences (22) and plays a role in DNA damage response pathways that eliminates severally damaged cells by initiating both checkpoint and apoptotic cellular responses following certain types of DNA damage (23). Recent studies (10, 23) have revealed that the role of MMR proteins in mismatch repair can be uncoupled from the MMR-dependent damage response. Mismatch repair pathway is considered to involve the concerted action of at least 20 polypeptides (24).
Since early 1990s, the MMR system has been intensively studied primarily due to its link with the HNPCC, one of the most common inherited cancer-predisposition syndromes. To date, about 180 mutations in the MMR genes that code for MSH2 and MSH6 components of MutSα have been reported to cause HNPCC, with more than 85% of them in msh2 (InSight database: http://www.insight-group.org/). Mutations in the msh2 and msh6 genes have also been associated with a large variety of non-HNPCC and non-colonic tumors. These include endometrial, ovarian, gastric, cervical, breast, brain, and bladder tumors as well as leukemia and lymphoma.
Most of the insights into features of mismatch recognition are based on structures of prokaryotic MutS. However, given its low homology, analysis of the dynamics of the DNA binding site and the inter-unit communication within the eukaryotic MutSα structures will bring a better understanding not only of the conformational changes associated with the mismatch recognition step, but also of the effects of cancer-causing mutations on mammalian MMR proteins.
We have performed molecular dynamics simulations on mismatched and damaged MutSα-DNA complexes and here we present a comprehensive DNA binding site analysis in search for an answer to the question how MMR identifies the repair site. Conformational differences associated with MutSα binding to mismatched and damaged DNA have been identified and they provide insight into the involvement of MMR proteins in DNA-repair and DNA-damage responses. Allosteric interactions within the heterodimer associated with the mismatched and damaged recognition step allow for identification of key residues in MMR cancer-causing mutations.
Methods
Molecular Dynamics Simulations
The simulations of the G-T mismatch are performed using the same basic protocol as in our previous work (10, 11) based on the X-ray structure of human MSH2/MSH6 protein complex with heteroduplex DNA (13). This structure has a truncated N-terminus, the missing residues are largely unstructured and while they are involved in nuclear transport (25), they appear to have no other function. Hydrogen atoms were added using the hbuild facility of CHARMM (26). The CHARMM force field was used for the entire complex with additional parameters based on pre-existing cisplatin parameters (27-29). This force field has been extensively parameterized for a wide range of biologically important molecules, including nucleic acids, amino acids, lipids and some small-molecule ligands. The platinum cross-linked DNA structure was built using the mismatch as a template. The cross-linked structure was fitted into the binding pocket to maximize the structural overlap with the mismatched DNA structure, followed by rotations and translations to minimize the energy of the unrelaxed structure using the coordinate manipulation and energy minimization facilities of CHARMM (26). The platinum atom cross-links two adjacent guanines. The structure was fully solvated with TIP3P water (30) in a cubic box using the visual molecular dynamics (VMD) package (31). There are 855 residues in MSH2, 974 residues in MSH6, 30 nucleotides in the DNA fragment, and two ADP molecules, a total of 300048 atoms in the platinum cross-linked complex and 30039 in the mismatched system. Although there are increasingly accurate implicit-solvent models, e.g., (32-34), they have yet to be thoroughly vetted on large DNA/protein complexes such as the ones simulated herein. The water molecules were briefly minimized for 100 cycles of conjugate gradient minimization with a small harmonic force constant on all protein atoms. The entire system then underwent 250 ps of molecular dynamics simulation to achieve a thermal equilibration using Berendsen pressure regulation with isotropic position scaling (35). The temperature was reassigned from a Boltzmann distribution every 1000 cycles, in 25 K increments, from an initial temperature of 0 K to a target temperature of 300 K. Following the equilibration, a 10 ns production simulation was performed in NAMD package (36), under NVE ensemble, using standard parameters: a 2.0 fs time step using SHAKE on all bonds to hydrogen atoms (37), a 12 Å cutoff, Particle Mesh Ewald with a 128 grid points on a side (38), Berendsen’s constant pressure algorithm with a target pressure of 1.01325 bar, a compressibility of 45.7 mbar, a relaxation time of 1 ps, and a pressure frequency of 40 fs, and a coordinate save frequency of 200 fs; all as implemented in NAMD. A total of ten simulations were performed, five for the mismatched and five for the platinum cross-linked systems. The same protocol was employed with different initial conditions. The initial coordinates, velocities, and system dimensions were taken from the final state of the corresponding equilibration simulation. Note that residue 1 of MSH6 in our system corresponds to residue 362 in the solved structure (13).
Cα root mean square deviations and total energies are provided in Supplementary Material, Figures S5 and S6. These data show there are two different relaxation timescales, a fast one on the 10s-100s of picosecond time scale, and a slow one on the nanoscale. The Figures show that most of the relaxation to equilibrium occurs within the first 2 ns, and that while there may be additional long-time relaxation, starting the simulation analysis at 5 ns allows for a conservative removal of the majority of the non-equilibrium effects. Since our different simulations started from different initial conditions, it is expected they to show different pathways to equilibration, and they show the expected variation in relaxation.
Choice of Using Multiple Simulations versus a Single Simulation
A standard question in performing molecular dynamics simulation is whether or not to perform a single long simulation or to perform multiple shorter simulations. The ergodic hypothesis from statistical physics as can be found in standard textbooks such as (39) tells us that running a single simulation versus running multiple simulations from different initial conditions will in principle provide the same results. However, this is strictly speaking only true in the infinite time-limit, in practice, two serious issues need to be considered: whether the simulations are sufficient long enough that processes of interest can happen, i.e., that sufficient portions of phase space can be sampled, and secondly that any initial equilibration period be discarded, not because the conformations sampled are unphysical, but rather because they would be over-sampled. The first issue is essentially one of whether or not the simulations are of the correct time-scale for the physical process considered. In this manuscript, fluctuations about the native-state and perturbations due to binding cisplatin are considered; so the nanosecond time-scale should be considered. The time-scale required would be very different if processes of folding, assembly or diffusion were being considered. The latter question is one of efficiency. However, since we are considering only fluctuations about the native state, and we wish to sample phase-space well within the region addressable by nanosecond timescale fluctuations, we adopt the multiple MD simulation method (40, 41) on which several long trajectories are started from the same initial configuration but with different initial atomic velocities. While the question is not completely closed as to whether or not it is better to perform single simulations or multiple simulation, multiple MD simulations have been shown to not just reduce the computational time required, due to the “perfect” parallelism of running multiple simulations, but also to improve the statistics of sampling, and to minimize force-field induced artifacts (42). We can also address the quality of individual trajectories if necessary (42), although each trajectory is expected to be somewhat different as each trajectory is necessarily finite and has different initial conditions in phase space; it is only when the set of trajectories together is considered that we expect to have reasonable phase-space coverage as multiple simulations can allow for the sampling of different regions of phase-space (43). Ultimately though the question of sufficiency can only be addressed by making predictions that can be addressed experimentally, thus far (10, 11, 14) 2-10 nanosecond scale simulations have been sufficient to examine the experimental questions addressed, however, more experiments naturally will be necessary to address the predictions raised in this paper.
Clustering Analysis
To determine the occupancy of different conformations during the time course of the simulations clustering analysis was performed. The clustering was performed based on the root-mean-square distances between conformations comprising the trajectory, calculated across all residues. The conformations were grouped together using distances in dihedral angel space, backbone dihedral angels in our case, as a measure of conformational similarity, using the ART-2′ algorithm (44), as implemented in CHARMM (26). The conformation closest to the cluster’s center was considered as being representative of the class of structures making up the cluster, and was further analyzed. The conformations generated in this fashion, 12 for the platinum cross-linked and eight for the mismatched systems were weighted in the overall trajectory (Supplementary Material, Figure S2), and were further investigated for similarities.
Their hierarchical clustering dendrogram constructed using the agglomerative method of average linkage implemented in Matlab (45) is presented in Supplementary Material, Figure S1. The affinity for clusters is the relative RMSD between the conformations and at each particular stage the method joins together the two clusters that are the most similar, or have the highest affinity.
Hydrogen Bonding Analysis
Hydrogen bond analysis was performed using the hydrogen bond analysis tool, HBAT (46), based on (47). Strong hydrogen bonds (O-H…O, N-H…O, O-H…N, N-H…N) in protein-ligand complexes are characterized by small deviations from linearity, while weak hydrogen bonds (C-H…O, C-H…N) have variable geometries (48). The criterion for hydrogen bonding interactions was defined as having a separation distance ≤3.0 Å between the hydrogen atom and acceptor (oxygen, nitrogen or sulfur) atom and the cutoff for the angle between the donor-H…acceptor was set at 90°, 90° < θ ≤ 180°. The second half of the each of the 10 ns production simulation, a total of 25 ns production simulation for each system, was considered for the hydrogen bond analysis. While hydrogen bonds formed by water molecules are considered relevant to the protein-ligand binding (47), our structural model contains no water molecules, and therefore such interactions could not be considered in our analysis.
Results and Discussion
Mapping the DNA Binding Site
The prokaryotic MMR pathway has been extensively studied and characterized both biochemically and genetically. Due to the conservation of this complex pathway, E. coli MMR provided much of the understanding of the mammalian MMR. However, while eukaryotic MSH2 and MSH6 share common domain architecture with their prokaryotic homologues, they differ in length and sequence. The differences are particularly noteworthy at the level of the mismatch binding domain. The protein-DNA interface in MutSα-DNA complexes differs from that in the prokaryotic structures, in the sense that in the latter, the mismatch binding domain of the equivalent MSH2 makes extensive contacts with the DNA’s backbone (21). In contrast, the mismatch binding domain of MSH2 in MutSα is rotated up and away from the DNA’s backbone and makes, reportedly, only one contact with DNA (13). In addition, an extended strand at the N terminus of MSH2 blocks its DNA binding face and packs against the mismatch binding domain of MSH6. It is also known that the mismatch binding domains in prokaryotic structures do not interact with one another (21). Thus, the situation in eukaryotes is much more complex and the understanding of MMR implications in human disease can only be appreciated in the mammalian system. In this work the focus will be on the understanding of the human mismatch-binding factor MutSα interaction with mismatched and cisplatin damaged DNAs, as well as, on the identifying the conformational changes and allosteric interactions within the heterodimer associated with the repair site recognition step and MMR-dependent DNA damage signaling process. The findings will be compared with experimental results, when available, and discussed in comparison with the E.coli MMR pathway.
In our simulations, the structural model for DNA mismatch recognition by the MMR machinery was the solved structure of MutSα with two ADP molecules bound in the ATPase sites of the heterodimer, bound to a 15 base pair duplex DNA containing a central G-T mispair causing distortion in the orientation of the bases (13). The bases at the mismatch remain in the DNA helix but do not stack, and as a result a bending of ~60° occurs in this region. The structural model for DNA damage recognition was based on the crystal structure of DNA containing a (1, 2) GpG cisplatin intra-strand crosslink (49, 50) built into the crystal structure of MutSα (13). Coordination of the cis-{Pt(NH3)2}2+ fragment to DNA, in which platinum atom links N7 atoms of two adjacent guanine residues, Gua8 and Gua9, alters the duplex DNA. The structural alterations induced in DNA by forming adducts with platinum have been extensively studied (51). NMR studies led to the conclusion that the (1, 2) GpG cisplatin intra-strand adduct bends the DNA helix by 78° towards the major groove and also unwinds it by 25° at the site of the platination, resulting in de-stacking of the bases (52). (1, 2) GpG cisplatin intra-strand DNA adduct is the most prevalent of those made by cisplatin, the widely used anticancer drug accidently discovered in 1965 (53), and it is recognized by human mismatch repair proteins (54, 55). Since MutSα purified from cell extracts carries ADP (13, 15), the model with two ADP molecules was chosen in both cases. The mismatched or the damaged DNA fragment is fully encircled by the mismatch binding (red) and clamp (purple) domains from both MutSα subunits (Figure 1). Note that in our system, residue 1 of MSH6 corresponds to residue 362 in the solved structure (13). Residues sequence and numbering for MSH2 and MSH6 are presented in Supplementary Material, Figures S7 and S8.
Figure 1.
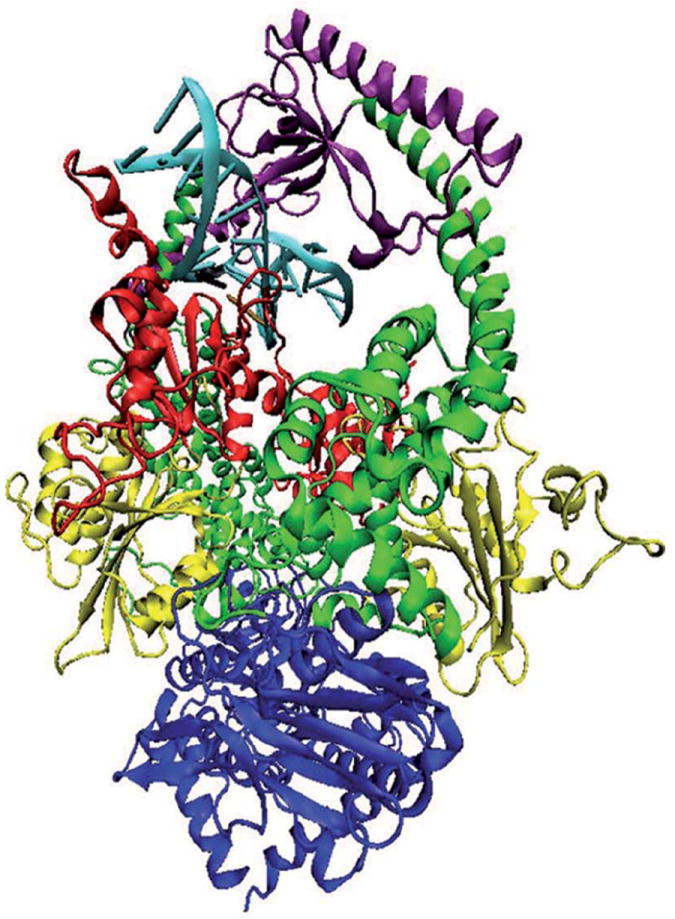
Structural model of MutSα in complex with a 15 base pair duplex DNA containing a central G-T mismatch. DNA is shown in light blue with the mismatch pair marked: black for guanine and ochre for thymine. The color code for the heterodimer domains is: red for the mismatch binding domain, residues 1 to 124 in MSH2 and 1 to 157 in MSH6; yellow for the connector domain, residues 125 to 297 in MSH2 and 158 to 356 in MSH6; green for the lever domain, residues 300 to 456 and 554 to 619 in MSH2, and 357 to 573 and 648 to 714 in MSH6; purple for the clamp domain, residues 457 to 553 in MSH2 and 574 to 647 in MSH6; blue for the ATP-ase domain, residues 620 to 855 in MSH2 and 715 to 974 in MSH6. Note that in our system, residue 1 of MSH6 corresponds to residue 362 in the solved structure.
Before discussing the recognition of the mismatched DNA by MutSα proteins it should be pointed out that it is well established that biomolecular recognition is mediated by weak chemical interactions, such as hydrogen bonds (56). Hydrogen bonding directionality and reversibility make it so important in biological processes. Protein-ligand binding is determined by multi-centered interactions and multipoint recognition. An acceptor furcation, which is the approach of many donors toward an acceptor, is preferred rather than a donor furcation (48). Strong hydrogen bonds (O-H…O, N-H…O, O-H…N, N-H…N) in protein-ligand complexes are characterized by small deviations from linearity, while weak hydrogen bonds (C-H…O, C-H…N) have variable geometries (48). In addition, it was substantiated that the presence of C-H…O bonding, whose existence was unequivocally established in the early 80’s (57), amongst the protein-ligand interactions is significant.
Recognition of the Mismatched DNA by MSH6
Multiple strong (O-H…O, N-H…O, N-H…N) and weak (C-H…O, C-H…N) hydrogen bonds are observed between the mismatched DNA and MSH6 subunit, in general agreement with the reported (13) hydrogen bond patterns. There are weak C-H…O hydrogen bonds made by Glu73 of the conserved Phe-X-Glu motif of MSH6 to the mispaired thymine and a strong N-H…O bond made between the backbone carbonyl of Val68 and mispaired guanine. The side chain of Tyr484 of the lever domain of the mismatch recognition monomer (MSH6) strongly hydrogen bonds (O-H…O) to the phosphate group of the mispaired guanine (Figure 2, panel A) about 83% of the simulation time (Table I). During the entire simulation time, the side chain of Lys70 makes strong (N-H…O, N-H…N) and/or weak (C-H…O, C-H…N) contacts with the mispaired guanine. In addition, strong (N-H…N) and weak (C-H…O or C-H…N) hydrogen bonds are made by Phe71 and Met91 with the mismatched G-T pair (Table I). Notice the orientation of Glu73 away from the mismatched site, which is prevalent more than 80% of the simulation time (Figure 2, panel A).
Figure 2.
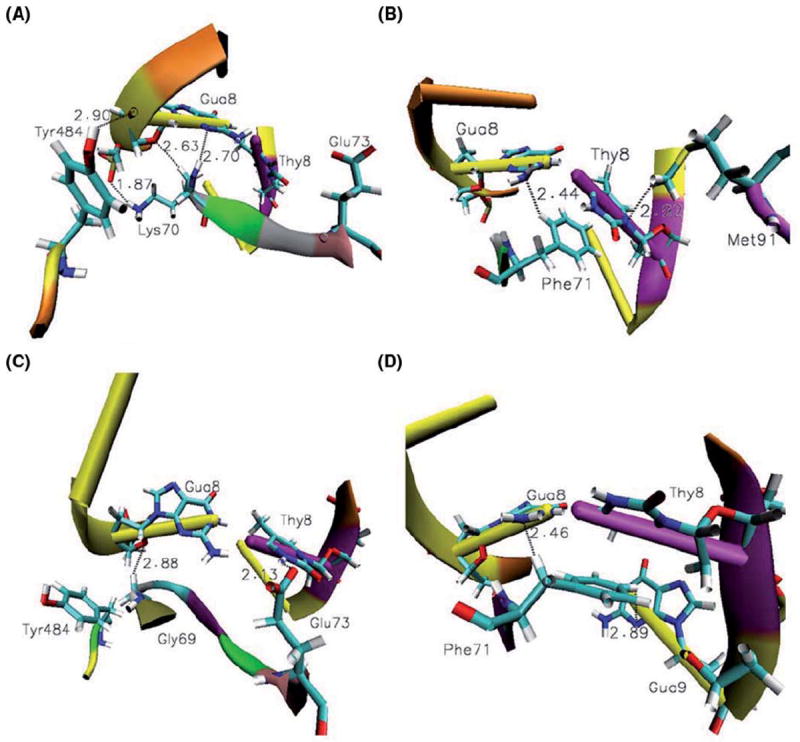
Recognition of the mismatched DNA by MutSα. (A) Tyr484 makes strong N-H…O hydrogen bonds with the mispaired guanine more than 83% of the simulation time. The side chain of Lys70 makes strong N-H…O and /or weak C-H…O, C-H…N hydrogen bonds with the mispaired guanine. Notice the orientation of the Glu73 away from the mismatched site which is prevalent more than 80% of the simulation time. (B) Phe71 and Met91 also probe the mismatched pair by strong and weak interactions present more than 80% of the simulation time. Recognition of cisplatin-DNA by MutSα. (C) The side chain of Glu73 makes strong N-H…O bonds with the mispaired thymine. Notice the orientation of Tyr484 away from the cross-linked DNA fragment, which is common almost half of the simulation time. Gly69 stabilizes the complex by weak C-H…O bonding with mispaired guanine. (D) Weak C-H…N bonds made by Phe71 with mismatched, platinum cross-linked guanine, Gua8, and the base adjacent to the mismatched thymine, Gua9.
Table I.
Recognition of the mismatched (mis) and platinum cross-linked (plat) Gua8-Thy8 in MutSα-DNA complexes by MSH6.
| Residue | GUA8
|
THY8
|
|||||
|---|---|---|---|---|---|---|---|
| %Conf.
|
%Conf.
|
||||||
| Interactions type | Mis | Plat | Residue | Interactions type | Mis | Plat | |
| Arg613 | N-H…O2c, C-H…O(N) | 29.86 | 27.17 | Glu73 | N-H…O, C-H…O | 15.63 | 68.94 |
| Glu732a | N-H…O | 7.56 | 12.58 | Gly99 | C-H…O(N) | 37.20 | 46.98 |
| Gly69 | C-H…O(N) | 40.19 | 79.63 | Met91 | C-H…O(N, S) | 91.94 | 76.06 |
| Lys70 | N-H…O(N), C-H…O(N) | 100 | 67.03 | Phe71 | C-H…O(N) | 17.47 | 33.58 |
| Phe71 | N-H…N, C-H…N | 85.34 | 55.21 | Phe90 | C-H…O | 17.47 | 22.94 |
| Tyr484 | O-H…O | 82.79 | 22.94 | Val89 | C-H…O | 7.56 | 38.26 |
| Val682b | N-H…O | 100 | 39.32 | Ser98 | C-H…O | 0 | 24.92 |
| Lys92 | N-H…O, C-H…O | 0 | 6.47 | ||||
Glu73 being the acceptor;
Val68 being the acceptor;
Present only in the mismatched complex.
A complete analysis of protein-DNA hydrogen bounding for the mismatched structural model is included in Supplementary Material, Table S3. The method we used found all the reported (13) hydrogen bonds and others either unreported or undetected by their method.
Recognition of the Platinum Cross-Linked DNA by MSH6
While interactions involving Lys70, Phe71, Tyr484, Val68 and Met91 are suggested by simulations to be dominant (present more than 60% of the simulation time) for the recognition of the mismatched bases by MSH6, a different set of interactions involving Gly69 and Glu73, besides Lys70 and Met91, are predominant for the recognition of the damaged, platinum cross-linked mismatched bases. The lack of strong O-H…O and N-H…O interactions by Tyr484 and Val68, respectively, seen with the mismatched guanine, is compensated in the cisplatin adduct by the presence of strong N-H…O and/or weak C-H…O and C-H…N interactions by Glu73 and Gly69 with the mismatched thymine and guanine, respectively (Table I, Figure 2, panel C). Previously, Glu73 was identified and proved by experimental data as a key contact with cisplatin-DNA and required for cisplatin cytotoxicity (10). In addition, Phe71 weakly hydrogen bonds (C-H…O and C-H…N) the cross-linked guanine, Gua8, and the base adjacent to the mispaired thymine, Gua9 (Figure 2, panel D). Although these interactions are instrumental for MutSα proteins (Table II) in probing for weakened base stacking (notice the possibility for π-π interactions with the mispaired thymine in Figure 2, panel D) and susceptibility to kink, this residue seems to be secondary for cisplatin adduct recognition (Table I, Phe71 hydrogen bonds the mispaired guanine less than 60% of the simulation time). The latter prediction is supported by experimental data (10), and, similarly, MutS homologue of Phe71 is find to be instrumental and indispensable for mismatch recognition during repair, but it is not required for cisplatin cytotoxicity (10).
Table II.
Different binding modes for mismatched (mis) and platinum cross-linked (plat) DNA fragments by MutSα. Only hydrogen bonding prevalent more than 60% of the simulation time is presented1
| Residue | Interactions type | % | Residue | Interactions type | % | ||
|---|---|---|---|---|---|---|---|
| 1. Thr46 | Mis | O-H..O, C-H…O; Cyt11 | 32.11 | 14. Val148 | Mis | N-H…O; Gua10; | 100 |
| Plat | O-H…O, C-H…O; Cyt11; | 68.61 | Plat | N-H…O; Gua10; | 79.14 | ||
| 2. Pro47 | Mis | C-H…O; Cyt11; | 100 | 15. Tyr484 | Mis | O-H…O, C-H…O; Gua8, Cyt9; | 100 |
| Plat | C-H…O; Cyt11; | 90.79 | Plat | O-H…O; Gua8, Gua9; | 56.80 | ||
| 3. Val68 | Mis | N-H…O; Gua8; C-H…O; Cyt9; | 100 | 16. Trp609 | Mis | N-H…O, C-H…O; Thy13, Thy14; C-H…N; Thy13; N-H…O; Thy13, Thy14; | 100 |
| Plat | N-H…O; Gua8; C-H…O; Gua10; | 63.28 | Plat | C-H…O; Gua10, Thy13; | 42.42 | ||
| 4. Gly69 | Mis | C-H…N, C-H…O; Cyt9; Gua8; | 100 | 17. Arg613 | Mis | N-H…O, C-H…O; Cyt7, Gua8; C-H…N; Cyt7; | 65.39 |
| Plat | C-H…N, C-H…O; Gua8, Gua9; | 63.28 | Plat | C-H…O(N); Gua8; | 27.17 | ||
| 5. Lys70 | Mis | N-H…O; Cyt7, Gua8; N-H…N; Gua8; C-H…O; Cyt7, Gua8; C-H…N; Gua8, Gua9; | 44.52 | 18. Lys636 | Mis | N-H…O, C-H…O; Cyt5, Gua6; C-H…N; Gua6; | 76.63 |
| Plat | N-H…O, C-H…O; Cyt7, Gua8; N-H…N; Gua8; C-H…N; Gua8; | 74.23 | Plat | N-H…O, C-H…O; Cyt5, Gua6; | 29.03 | ||
| 6. Phe71 | Mis | N-H…N; Gua8; C-H…O; Gua9; C-H…N; Gua8, Gua9; | 100 | 19. Lys639 | Mis | N-H…O; Cyt4, Thy14; C-H…O; Thy14, Cyt15; C-H…N, Ade3, Cyt4; | 64.47 |
| Plat | C-H…O(N); Gua8, Gua9; | 75.32 | Plat | N-H…O, C-H…O; Cyt15, Thy14; | 63.46 | ||
| 7. Glu73 | Mis | N-H…O; Gua8; Thy8; C-H…O; Thy8; | 100 | 20. Lys6* | Mis | N-H…O; Gua11; C-H…O; Cyt10, Gua11; | 76.63 |
| Plat | N-H…O; Gua8, Thy8, Gua10; C-H…O(N); Thy8; | 93.53 | Plat | N-H…O, C-H…O; Cyt10, Gua11; | 77.11 | ||
| 8. Leu88 | Mis | C-H…O; Gua9; | 15.63 | 21. Lys512* | Mis | N-H…O, C-H…O; Gua5; | 56.90 |
| Plat | C-H…O; Gua9; | 82.52 | Plat | N-H…O; Ade4, Gua5; C-H…O(N); Gua5; | 63.04 | ||
| 9. Met91 | Mis | C-H…O; Gua7; Thy8; C-H…N; Cyt7; C-H…S; Thy8; | 100 | 22. Thr526* | Mis | O-H…O; Ade4; | 61.00 |
| Plat | C-H….O; Cyt7, Thy8; | 92.83 | Plat | O-H…O; Ade4; | 35.78 | ||
| 10. Lys92 | Mis | N-H…O; Gua7; Cyt11; C-H…O; Gua7; | 40.19 | 23. Lys528* | Mis | N-H…O, C-H…O; Thy3, Ade4; | 84.37 |
| Plat | N-H…O, C-H…O; Cyt6, Cyt7, Thy8; N-H…N; Cyt7; C-H…N; Cyt6, Cyt7; | 67.34 | Plat | N-H…O, C-H…O(N); Thy3, Ade4; | 75.35 | ||
| 11. Pro101 | Mis | C-H…O(N); Gua9; | 100 | 24. Gln545* | Mis | C-H…O; Cyt5, Gua6; C-H…N; Cyt5; C-H…O; Cyt5, Gua6; | 83.52 |
| Plat | C-H…O(N); Gua9; | 100 | Plat | C-H…N; Cyt5; | 68.64 | ||
| 12. Tyr108 | Mis | O-H…O; C-H…O; Gua9; Cyt10; | 67.86 | 25. Lys546* | Mis | N-H…O, C-H…O; Gua6, Cyt7; N-H…O; Gua6, Cyt7; C-H…O; Gua6; | 100 |
| Plat | O-H…O, C-H…O; Gua9, Cyt10; | 79.42 | Plat | C-H…N; Cyt7; | 90.79 | ||
| 13. Val147 | Mis | C-H…O; Gua10; | 100 | ||||
| Plat | C-H…O; Gua10; | 76.25 |
Marked with * are MSH2 residues involved in hydrogen bonding with mismatched and platinum cross-linked DNA fragments. A complete list of the hydrogen bonding for MutSα-DNA complexes is included in the Supplementary Material, Tables S1, S2 and S4.
MSH2 makes no contacts with the mismatched bases in both complexes, in agreement with experimental results (13), and in contrast with its MutS homologue (10).
Different Binding Modes for Mismatched and Platinum Cross-Linked MutSα Complexes
The interactions indicated in Figure 3 for the Val68–Gly69-Lys70-Phe71-X- Glu73 loop of MSH6 suggest that this loop indeed is responsible for probing duplex DNAs for weakened base stacking and susceptibility to bend (mainly these are the residues hydrogen bonding the mismatched pair, Table I). Strong O-H…O bonding by Tyr108 and weak C-H…O bonding by Met91 and Pro101 of the mismatch binding domain of MSH6 with the bases adjacent to the mismatched site (Cyt7, Gua9, Cyt9 and Gua7 in the mismatched substrate and Cyt7, Gua9, Gua8 and Cyt7 in the platinum cross-linked mismatched substrate) are likely to contribute to the protein-DNA complex stability in both cases (they are present more than 60% of the simulation time, Table II). Positions one to three relative to the mismatched pair towards 5′ ends, bases Cyt5, Gua6 and Cyt7, are stabilized by strong N-H…O and weak C-H…O, C-H…N contacts with Gln545 and Lys546, respectively, of the clamp domain of MSH2. In both cases, the pairs of these three bases make strong N-H…O contacts with Val148 and weak C-H…O contacts with Pro47 and Val147 of the mismatch binding domain of MSH6 on one side of the mismatched pair, and strong N-H…O and weak C-H…O contacts with Lys6 of the mismatch binding domain of MSH2, the only contact made by it with the DNA fragment, on the other side.
Figure 3.
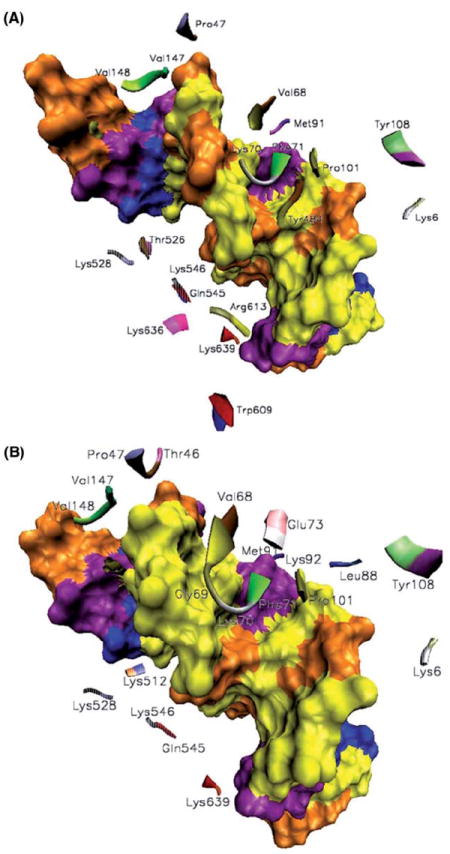
Different binding modes for mismatched (A) and platinum cross-linked (B) DNA fragments by MutSα. The color coding for the DNA bases on the surface representation is the following: Gua-Cyt, yellow-brown; Ade-Thy, blue-purple. MSH2-DNA contacts are depicted in translucent representations. Besides the common contacts, Tyr484, Arg613, Lys636 and Thr526, with the latter from MSH2, are essential for the binding of MutSα to mismatched G-T bases, and Thr46, Gly69, Glu73, Leu88, Lys92 and Lys512, with the latter from MSH2, are essential for the binding of MutSα to platinum cross-linked mismatched G-T bases.
The ends of DNA substrates are stabilized by contacts with the clamp domains: by strong N-H…O and weak C-H…O contacts with Lys639 from MSH6 at one end of the substrate and Lys528 from MSH2 at the other end.
However, overall, MutSα proteins binding modes to mismatched and platinum cross-linked DNA substrates are significantly different, and, as it will be discussed in the next section, this induces unique conformational changes and allosteric interactions.
Besides the above described differences in the mismatch recognition, molecular dynamics simulations suggest that additional strong N-H…O and weak C-H…O, C-H…N contacts made by Arg613 and Lys636 of the clamp domain of MSH6 are dominant in the protein-mismatched DNA binding mode (they are present more than 60% of the simulation time). Furthermore, strong O-H…O contacts made by Thr526 of the clamp domain of MSH2 with an adenine positioned four bases away from the mismatched thymine also occur in both systems but they are predominant only in the mismatch binding mode (present 61% of the simulation time versus 36% in the platinum cross-linked system, Table II). In addition, uncommon in the complex made by MutSα proteins with the platinum cross-linked DNA, strong N-H…O and C-H…O, C-H…N contacts made by Trp609 of the clamp domain of MSH6 with 3′ end of the thymine mismatched strand are dominant in the complex made by MutSα proteins with the mismatched DNA (present during the entire simulation time in the mismatched system versus 42% of it in the platinum cross-linked system, Table II).
In contrast, molecular dynamics simulations suggest that contacts uncommon (present less than 60% of the simulation time) for the protein-mismatched DNA binding mode may be critical for the protein-platinum cross-linked-DNA binding mode. In this regard, strong N-H…O, N-H…N and weak C-H…O contacts are made by Lys92 of the mismatch binding domain of MSH6 primarily with Cyt6 and Cyt7 bases adjacent to the mismatched thymine. It is also the case of weak C-H…O bonding made by Leu88 with the guanine adjacent to the mismatched thymine. Gua5-Cyt11 base pair makes contacts with MutSα’s clamp and mismatch binding domains: Lys512 (N-H…O, C-H…O) from MSH2 and Trp46 (O-H…O, C-H…O) from MSH6, respectively.
Stability, Allosteric Communications and Differences at the Heterodimer Interface
Contacts between Mismatch Binding Domains of MutSα: Molecular dynamics simulations reveal multiple contacts made by Arg107 of MSH6 with Lys6, Gln5 and Pro5 of the N-terminal of MSH2 (Table III; details in Figure 4, panels A and B). It is also the case of Ser111 from MSH6 and Ala2 from MSH2. In addition, the former is hydrogen bonding Met1 of MSH2 in the mismatched MutSα-DNA complex, while the latter interacts with Leu86 from MSH6 in the platinum cross-linked mismatched MutSα-DNA complex. Unique for the platinum cross-linked mismatched MutSα-DNA complex is also the propensity for multiple hydrogen bonding by Ser171 from the connector domain of MSH6 (Table III) with Gln61 and Thr60 from the mismatch binding domain of MSH2. While common interactions at the heterodimer interface can be considered responsible for the stability of the protein, specific contacts suggest distinctive conformational changes associated with the different binding modes.
Table III.
Summary of interactions at the heterodimer interface: common, and unique for the platinum cross-linked and mismatched MutSα-DNA complexes3.
| Common | %Conf. Plat | %Conf. Mis | Unique platinated | %Conf. | Unique mismatched | %Conf. | |
|---|---|---|---|---|---|---|---|
| 1 | Lys6-Arg107** | 69.56 | 88.12 | Ala2**-Leu86 | 60.41 | Met1**-Ser111 | 64.73 |
| 2 | *Arg107**-Gln4 | 70.47 | 100 | Asn311-Met823 | 60.00 | Gln61**-Ser171** | 60.00 |
| 3 | *Arg107**-Pro5** | 61.17 | 72.20 | *Gly388-Leu719 | 92.73 | *Ser171**-Thr60 | 82.53 |
| 4 | *Ser111-Ala2** | 100 | 87.84 | *Thr389-Lys720 | 89.21 | *Gly388-Lys720 | 75.22 |
| 5 | *Asn390-Gly721 | 66.12 | 82.53 | *ASN390-Lys720 | 79.13 | *Thr389-Gly721 | 60.00 |
| 6 | Ile544-Ser637 | 72.45 | 100 | Gln718**-Gly388 | 86.42 | *Thr389-Ser723** | 87.84 |
| 7 | *Lys636-Ile544 | 80.95 | 87.62 | Leu719-Asn390 | 60.19 | Ser717-Thr389 | 64.25 |
| 8 | *Ser637-Ile544 | 65.98 | 76.63 | Lys720-Gly391 | 93.31 | Gln718**-Thr389 | 76.63 |
| 9 | Asn671**-Gly857 | 68.46 | 83.50 | Ser755-His887** | 83.62 | Leu719-Thr389 | 60.99 |
| 10 | Gly761-Gly955 | 100 | 80.06 | Lys773-Glu950 | 70.75 | Lys720-Glu394 | 76.63 |
| 11 | *Ser827-Asn671** | 84.32 | 83.52 | His785-Thr860 | 61.20 | Lys720-Ser392 | 72.94 |
| 12 | Gly827-Asp862 | 80.77 | 88.10 | *Arg821-Asp716 | 67.32 | Gly721-Asn390 | 68.84 |
| 13 | Ile828-Asp862 | 100 | 83.30 | Met823-Asn311 | 60.00 | Gly753-Asn775 | 65.81 |
| 14 | *Thr828-Asn671** | 100 | 40.16 | Lys847-Asn868 | 64.24 | Tyr757-Asn966 | 60.78 |
| 15 | *Phe829-Asn671** | 86.11 | 68.62 | Lys847-Asp894 | 68.78 | Tyr757-Glu893** | 87.60 |
| 16 | Phe836-Leu833 | 55.30 | 60.00 | *His887**-Gly753 | 69.08 | Tyr769-Glu950 | 71.98 |
| 17 | *Ala865-Ala844 | 100 | 100 | *Ala945-Met729** | 62.02 | *Pro774-Gly753 | 60.29 |
| 18 | *Ala869-Ala844 | 52.45 | 77.26 | *Asn775-Gly753 | 82.53 | ||
| 19 | *Ala869-Val840 | 63.18 | 100 | *Ala837-Asn835** | 64.88 | ||
| 20 | *His887**-Thr754 | 53.21 | 75.44 | *Ala859-Ser825 | 62.29 | ||
| 21 | *Lys935-Asp758 | 100 | 95.68 | *Thr860-His783** | 61.28 | ||
| 22 | *Phe939-Asp758 | 77.37 | 82.53 | *Tyr888-Thr756 | 68.62 | ||
| 23 | *Lys954-Glu768 | 80.80 | 100 | *His889-Ser755 | 68.62 | ||
| 24 | *Gly955-Gly761 | 100 | 100 | *His889-Thr756 | 78.19 | ||
| 25 | *Ala959**-Tyr757 | 100 | 79.54 | *Ser890-Tyr757 | 83.50 | ||
| 26 | *Arg973**-Glu850 | 60.13 |
The hydrogen bonds are presented as donor-acceptor.
Denotes the cases in which the donor residue belongs to MSH6.
Denotes known MSH2/MSH6 residues associated with cancer when mutated. Only the hydrogen bonds with more than a 60% presence in either system were considered, and they are considered key residues for the heterodimer interface stability and allostery. A complete list is included in Supplementary Material Tables S5 and S6.
Figure 4.
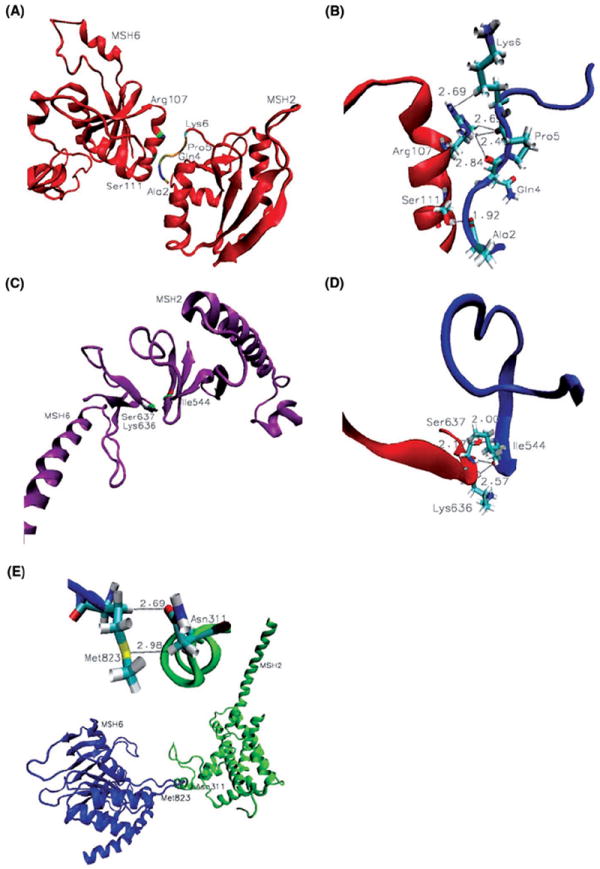
Stability and differences at the heterodimer interface. (A) Contacts between mismatch binding domains:Arg107 and Ser111 from MSH6 and residues at the N-terminal of MSH2; (B) Details of weak C-H…O and C-H…N contacts between mismatch binding domains of MSH6 (red) and MSH2 (blue); (C) Interactions between clamp domains; (D) Details of weak C-H…O and strong N-H…O contacts made by key residue Ile544 of MSH2 (blue) with Lys636 and Ser637 of MSH6(red); (E) Unique for the platinum cross-linked DNA complex is the C-H…S and C-H…O bonding between Met823 and Asn311 that allows for allosteric communication between the ATPase domain of MSH6 and the lever domain of MSH2 (Table III).
Interestingly enough, all of the above discussed contacts at the heterodimer interface involve residues that are among the known MSH2/MSH6 residues associated with cancer when mutated or deleted. They are denoted by ** in Table III. In this regard, mutations or deletions in the MMR genes that code for Met1, Ala2, Pro5 and Gln61 of MSH2, and Arg107 and Ser171 of MSH6 are reported to be associated with colon tumors (InSight database: http://www.insight-group.org/). Note that in our system, residue 1 of MSH6 corresponds to residue 362 in the solved structure (13).
Stability at MutSα-DNA Complexes Interface by Multiple Contacts of Clamp Domains Key Residues: In both complexes, multiple contacts between key residues of small, mainly β-strand clamp domains, Ile544 from MSH2, and Lys636 and Ser637 from MSH6, bring stability at the heterodimer interface (Table III; details in Figure 4, panels C and D). Remarkably, as described above, clamp domains make specific DNA contacts essentially on the side of the mispair opposite to the contacts made by the mismatch binding domains, underlining their role in the mismatch recognition by MutSα.
Allosteric Communications Between Lever and ATPase Domains
Different binding modes for mismatched and platinum cross-linked DNAs recognition by MutSα drive distinctive conformational changes within the protein, translated into unique inter-domain allosteric communications. The disordered, interconnected loops Gly388-Glu394 from the lever domain of MSH6 and Gln717-Lys723 from the ATPase domain of MSH2 display peculiar changes in their relative position and shape, an allosteric response to the conformational changes associated with the different binding modes. Multiple contacts, and wide and contracted loops in the mismatch complex are replaced by fewer contacts, and narrow and elongated loops in the platinum cross-linked complex (Table III; details of the changes in the relative position and shape of the interconnected loops are presented in Figure 5, panels A, mismatch, and b, platinum cross-linked complexes). Changes in the shape of the disordered, interconnected loops in the overall context of the protein structure are presented in Supplementary Material, Figure S4.
Figure 5.
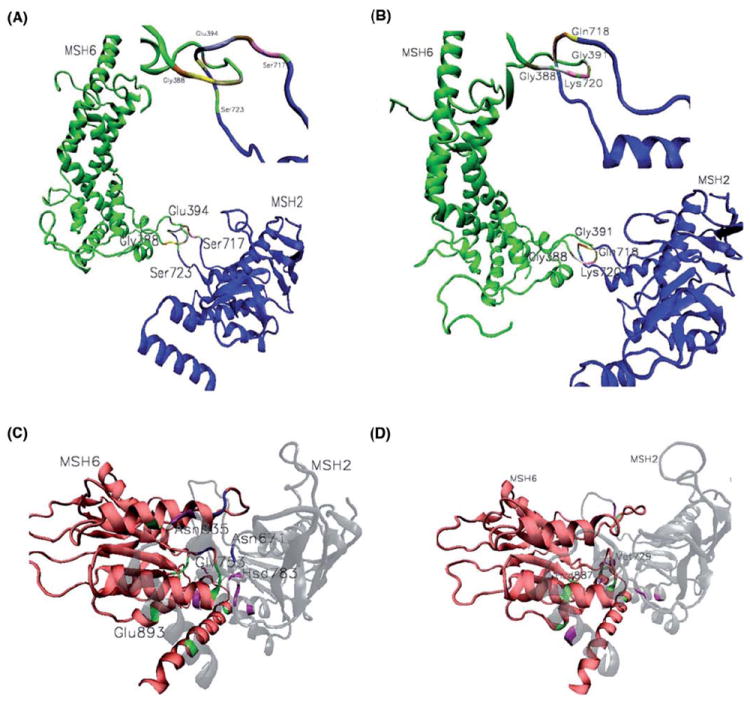
Allosteric communications. The disordered, interconnected loops Gly388-Glu394 from lever domain of MSH and Gln717-Lys723 from the ATPase domain of MSH2 undergo peculiar changes in their relative position and shape, in response to the conformational changes associate with the different binding modes. Multiple contacts and, wide and contracted loops in the mismatch complex (A) are replaced by fewer contacts and, narrow and elongated loops in the platinum cross-linked complex (B). Key residues at the ATPase interface. Unique contacts for mismatched (C) and platinum cross-linked (D) complexes are depicted in green and magenta for MSH6 and MSH2, respectively. Exception is the case of Asn671 of MSH2 and its MSH6 contacts, marked in blue, which in both complexes makes specific contacts with four residues at the ATPase domains interface, and when mutated causes cancerous tumors. On the same list are included, Met729, His783 and Asn835 from MSH2, and His887, a key residue for the cisplatin complex stability by making multiple specific contacts at the ATPase interface, and Glu893 from MSH6. Gly753 from MSH2 is also making multiple contacts at the ATPase interface, recommending it as a key residue for the mismatched complex stability (Table III). Hsd denotes neutral histidine.
It is noteworthy the presence of residues Gln718 and Ser723 of MSH2 from the lever-ATPase interconnected loops, denoted by ** on Table III, on the list of known MMR’s HNPCC causing mutations (InSight database: http://www.insight-group.org/).
Unique for the platinum cross-linked DNA complex is the C-H…S and C-H…O bonding between Met823 and Asn311 that allows for allosteric communication between the ATPase domain of MSH6 and the lever domain of MSH2 (Table III; details are presented in Figure 4, panel E).
Interactions at the ATPase Interface
The ATPase domain is the most highly conserved region of MutSα (13) and, like in prokaryotes (21), the MutSα heterodimer interface in the ATPase domain is extensive (Table III). Unique contacts for mismatched and platinum cross-linked complexes are also depicted in Figure 5, panels C and D; in green and magenta are marked the contacts from MSH6 and MSH2, respectively, with one exception, which is the case of Asn671 and its contacts, marked in blue.
In both complexes, Asn671 from the Walker P-loop of MSH2 makes specific contacts with four residues at the ATPase domains interface: Ser827, Thr828, Phe829 and Gly857, thus its presence on the MMR’s cancer causing mutations provides no surprise. On the same list, His887 from MSH6 is indicated here as a key residue for the cisplatin complex stability by making multiple specific contacts at the ATPase interface. Gly753 from MSH2 is also making multiple contacts at the ATPase interface, making it a key residue for mismatched complex stability and possible candidate for tests on MMR related cancerous tumors.
In response to the different binding modes, distinctive conformational changes in MutSα allow for a shorter ATPase interface in the platinum cross-linked complex.
Nucleotide Binding Sites
In our structural model two molecules of adenosine diphosphate bound in the ATPase sites of the heterodimer (13). MutSα binds ADP in the P-loop mode (Figure 6 and Figure S3 in the Supplementary Material). Unlike in prokaryotes where the nucleobase adenine stacks between a phenylalanine and a histidine (21), at the MutSα nucleotide-binding sites the nucleobase adenine stacks between a phenylalanine and a tyrosine, Phe747-Tyr926 from MSH6 and Phe650-Tyr815 from MSH2. The nucleobase adenine makes specific hydrogen bonds with the main-chain atoms of Ala649-Ile651 (Table IV).
Figure 6.
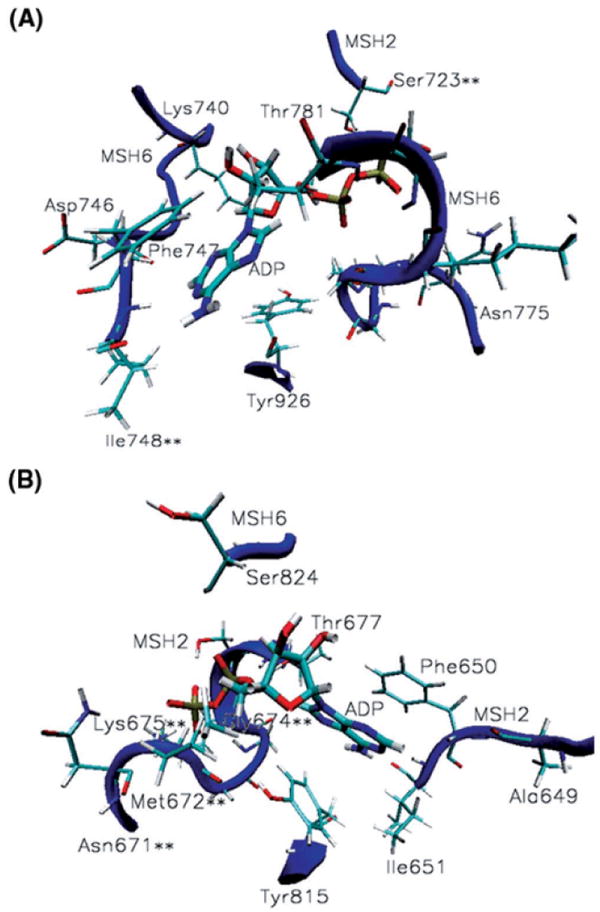
MutSα nucleotide binding sites. (A) ADP binding contacts at the ATPase domain of MSH6: adenine stacks between Phe747 and Tyr926 and makes specific contacts with Lys740, Asn746-Ile748; the phosphate group binds at the P-loop Asn775-Thr781 and hydrogen bonds Ser723 from the alternate binding site in the mismatched complex only. (B) ADP binding contacts at the ATPase domain of MSH2: adenine stacks between Phe650 and Tyr815 and makes specific contacts with Ala649-Ile651; the phosphate group binds at the P-loop Asn671-Thr677 and hydrogen bonds Ser824 from the alternate binding site. Noteworthy is the significant presence of cancer causing mutations among the ADP binding contacts of both subunits (residues marked with **), 25% of them.
Table IV.
Hydrogen bonding for ADP molecules at the ATPase binding sites in the mismatched (mis) and platinum cross-linked (plat) MutSα-DNA complexes4.
| MSH2 | Interactions type | %Conf. Mis | %Conf. Plat | MSH6 | Interactions type | %Conf. mis | %Conf. plat |
|---|---|---|---|---|---|---|---|
| 1. Ala649 | C-H…O | 71.93 | 49.69 | Lys740 | N-H…O | 81.02 | 86.32 |
| 2. Phe650 | C-H…N | 91.94 | 70.75 | Asn746 | C-H…O | 70.35 | 29.84 |
| 3. Ile651 | N-H…N | 100 | 100 | Phe747 | C-H…N | 61.97 | 91.55 |
| 4. Asn671** | C-H…O | 91.94 | 29.58 | Ile748** | N-H…O(N), C-H…N | 100 | 100 |
| 5. Met672** | N-H…O, C-H…O | 100 | 100 | Asn775 | N-H…O, C-H…O | 23.37 | 72.47 |
| 6. Gly673 | N-H…O | 82.58 | 100 | Met776 | N-H…O, C-H…O | 84.38 | 100 |
| 7. Gly674** | N-H…O, C-H…N | 100 | 100 | Gly777 | N-H…O | 84.38 | 100 |
| 8. Lys675** | N-H…O, C-H…O | 100 | 100 | Gly778 | N-H…O, C-H…O | 100 | 100 |
| 9. Ser676 | O(N)-H…O, C-H…O | 100 | 100 | Lys779 | N-H…O, C-H…O | 91.94 | 86.11 |
| 10. Thr677 | O(N)-H…O, C-H…O(N) | 100 | 100 | Ser780 | O(N)-H…O, C-H…O | 100 | 100 |
| 11. Ser723** | O-H…O, C-H…O | 67.92 | 0.00 | Thr781 | O(N)-H…O, C-H…O(N) | 100 | 100 |
| 12. Tyr815 | N-H…O | 46.98 | 80.00 | Ser824 | O-H…O | 62.07 | 83.62 |
| Tyr926 | N-H…O, C-H…O(N) | 59.21 | 67.86 |
Only hydrogen bonds present more than 60% of the simulation time are presented and a complete list is included in the Supplementary Material, Tables S7 and S8. In italic are hydrogen bonds made by either MSH2 or MSH6 with the neighboring ADP.
Denotes residues on the list of known MutSα’s cancer causing mutations.
The phosphate group of ADP binds at the loop of the Walker A motif (a glycine rich loop preceded by a beta sheet and followed by an alpha helix), residues Asn671-Thr677 in MSH2 and Asn775-Thr781 in MSH6 (Table IV and Figure 6). Like in prokaryotes (21), in MutSα homologues, Ser824 from MSH6 and Ser723 from MSH2, are hydrogen bonding the phosphate group from the alternate binding site (data marked in italic on Table IV and depicted in Figure 6).
Hydrogen bonding analysis of relevant conformations generated by molecular dynamics simulations shows that 25% of MutSα’s residues (marked with ** in Table IV and Figure 6) hydrogen bonding ADP molecules are known to cause cancerous tumors when mutated, underlying the significance of ATPase activity in the MutSα reaction cycle and allowing for prediction of additional cancer causing mutations for MutSα.
Of particular interest is the case of Gly674 at the P-loop of MSH2, which hydrogen bonds the phosphate group of the nucleotide. It has been reported that its mutation has differential effects on the DNA repair and DNA damage response functions, uncoupling DNA mismatch repair and apoptosis pathways (58). It causes DNA repair deficiency, but does not affect the DNA damage response function. Our hydrogen bonding analysis (Table IV) indicates that, in both mismatched and damaged complexes, Gly674 may be critical to nucleotide binding by making strong N-H…O and weak C-H…N hydrogen bonds with its phosphate group the entire time of the simulation. This suggests that, besides being essential to nucleotide binding in the mismatched and damaged DNAs recognition, Gly674 may be instrumental to downstream events involved in DNA repair pathway.
Hydrogen bonding analysis for ADP molecules at the ATPase binding sites (Table IV) also suggests that Asn671, Ser723 and Asn746 may be required for ATPase activity in repair response (these residues are making specific contacts with ADP molecules more than 60% of the simulation time in the mismatched system, while significantly less than that in the platinum cross-linked system), while Asn775 is suggested to interfere with the ATPase activity in apoptosis response (it makes specific contacts with the ADP molecules more than 70% of the simulations time in the platinum cross-linked system versus about 23% of the simulation time in the mismatched system).
Conclusions and Predictions for MutSα Cancer Causing Mutations
Molecular dynamics simulations on X-ray crystallography models allow for a refined fingerprinting of protein-DNA interactions in mismatched MutSα-DNA complexes. They highlight established experimental results, such as identifying Glu73 as a key contact with cisplatin-DNA, and provide new insights, such as recommending Gly69, Tyr484 and Trp609 as key contacts in the mismatched DNA recognition, as well as Leu88 and Lys636 as key contacts in the cisplatin-DNA recognition (Figure 3; data presented in Table II for the simulations, as well as in Supplementary Material, Table S3 for the crystalline structure of the mismatched complex).
Rigorous hydrogen bonding analysis of relevant conformations provided by 50 ns molecular dynamics simulations shows that MutSα proteins recognize the mismatched and platinum cross-linked DNA substrates in significantly different modes. We show that this induces unique conformational changes and allosteric interactions associated with repair and apoptosis pathways.
In regard to the effects of the DNA shape on the mismatch recognition, one hypothesis is that MutSα bends the DNA, widening its minor groove in the vicinity of the mispair (13). Under the hypothesis that, despite varied shapes and hydrogen bonding potentials mispaired or unpaired G, A, T, and C bases share the similarity of weakened base staking and susceptibility to kink, and that MutSα recognizes a broad range of mismatches by its preferential binding to the flexible (un-stacked) and potentially kinked DNA (59), and which this study favors, the very significant changes in the shapes of the mismatched and platinum cross-linked DNA fragments might trigger the different recognition binding modes by the protein.
A refined protein-DNA binding mode, as well as identifying MutSα specific conformational changes induced by recognition of mismatched and damaged DNAs, provided by molecular dynamics simulations, allows for identification by subsequent structure-base virtual screening of conformational selective ligands for drug discovery development (11, 12, 60).
The observed MutSα mutations found among the essential contacts at the heterodimer interface (residues marked with ** in Table III) show their propensity for multiple specific hydrogen bonds (notice the above described cases of Arg107 from MSH6 at the interface of the mismatch binding domains, Asn671 of MSH2 at the interface of ATPase domains and His887 of MSH6 at the interface of ATPase domains). The analysis of the molecular dynamics simulations suggests the same propensity for multiple specific hydrogen bonds for: Ile544 of MSH2, predicted to be a key residue for heterodimer stability at the clamp domains interface; Asp758, Gly761 and Ala844 of MSH2, and Gly955 of MSH6, predicted to be key residues at the ATPase domains interface (Table III, common interactions at the heterodimer interface), making them suitable for testing of similar mutagenic effects. In addition, from the analysis of conformational changes associated with the apoptosis response (platinum cross-linked complex), the following residues are predicted to be among the key residues associated with the apoptosis pathway: Asn311 of MSH2 and Met823 of MSH6 at the lever/ATPase interface; Leu719 and Lys720 of MSH2 and Gly388 of MSH6 at the ATPase/lever interface; Lys847 of MSH2 at the ATPase domains interface (Table III, interactions at the heterodimer interface unique for the platinum cross-linked system). Furthermore, from the analysis of conformational changes associated with the repair response (mismatched complex) the following residues are predicted to be among the key residues associated with MMR repair pathway: Gly388 and Thr389 of MSH6, and Lys720 and Gly721 of MSH2 at the lever/ATPase interface; Gly753, Thr756 and Tyr757 of MSH2, and His889 and Glu950 of MSH6 at the ATPase domains interface (Table III, interactions at the heterodimer interface unique for the mismatched system).
At the nucleotide binding sites, the residues known to cause cancerous tumors when mutated, with two exceptions, Asn671 and Ser723, make specific contacts with ADP molecules over the entire simulation time (residues marked with ** in Table IV). Molecular dynamics simulations show that Ile651, Ser676 and Thr677 of MSH2, and Gly778, Ser780 and Thr781 of MSH6 are also making specific hydrogen bonds with the nucleotides during the entire simulation time, making them commendable for investigations regarding similar mutagenic effects.
Supplementary Material
Acknowledgments
The computations herein were performed on the WFU DEAC cluster; we thank WFU’s Provost’s office and Informations Systems Department for their generous support. FRS also acknowledges funding from the NIH through R01 CA12937 and P30 CA12197.
Footnotes
Supplementary material dealing with details of the clustering analysis, complete hydrogen bonding analysis of the protein-DNA binding site, heterodimer interface and nucleotide binding sites, as well as details of the allosteric communication at the lever-ATPase domains interface, is available at no charge from the authors directly; the supplementary data can also be purchased from Adenine Press for US $50.00. It can also be downloaded for free of charge from the author’s server at http://bob.olin.wfu.edu/~web/.
References
- 1.Lahue RS, Au KG, Modrich P. Science. 1989;245:160–164. doi: 10.1126/science.2665076. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Y, Yuan F, Presnell SR, Tian K, Gao Y, Tomkinson AE, Gu L, Li GM. Cell. 2005;122:693–705. doi: 10.1016/j.cell.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 3.Constantin N, Dzantiev L, Kadyrov FA, Modrich P. J Biol Chem. 2005;280:39752–39761. doi: 10.1074/jbc.M509701200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gradia S, Subramanian D, Wilson T, Acharaya S, Makhov A, Griffith J, Fishel R. Mol Cell. 1999;3:255–261. doi: 10.1016/s1097-2765(00)80316-0. [DOI] [PubMed] [Google Scholar]
- 5.Blackwell LJ, Martik D, Bjornson KP, Bjorson ES, Modrich P. J Biol Chem. 1998;273:32055–32062. doi: 10.1074/jbc.273.48.32055. [DOI] [PubMed] [Google Scholar]
- 6.Martik D, Baitinger C, Modrich P. J Biol Chem. 2004;279:28402–28410. doi: 10.1074/jbc.M312108200. [DOI] [PubMed] [Google Scholar]
- 7.Wang H, Hays JB. J Biol Chem. 2003;278:28686–28693. doi: 10.1074/jbc.M302844200. [DOI] [PubMed] [Google Scholar]
- 8.Wang H, Hays JB. EMBO J. 2004;23:2126–2133. doi: 10.1038/sj.emboj.7600153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young L, et al. J Invest Dermatol. 2003;121:876–880. doi: 10.1046/j.1523-1747.2003.12486.x. [DOI] [PubMed] [Google Scholar]
- 10.Salsbury FR, et al. Nucleic Acids Res. 2006;34:2173–2185. doi: 10.1093/nar/gkl238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vasilyeva A, Clodfelter JE, Gorczynski MJ, Gerardi AR, King SB, Salsbury FR, Scarpinato KD. J Nucleic Acids. 2010 doi: 10.4061/2010/162018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salsbury FR. Curr Opin Pharmacol. 2010;10:738–744. doi: 10.1016/j.coph.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Warren J, et al. Mol Cell. 2007;26:579–592. doi: 10.1016/j.molcel.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 14.Drotschmann K, Topping RP, Clodfelter JE, Salsbury FR. DNA Repair (Amst) 2004;3:729–742. doi: 10.1016/j.dnarep.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 15.Jiricny J. Nat Rev. 2006;7:335–346. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 16.Peltomaki P. J Clin Oncol. 2003;21:1174–1179. doi: 10.1200/JCO.2003.04.060. [DOI] [PubMed] [Google Scholar]
- 17.Peltomaki P. Hum Mol Genetics. 2001;10:735–740. doi: 10.1093/hmg/10.7.735. [DOI] [PubMed] [Google Scholar]
- 18.Palombo F, Gallinari P, Iaccarino I, Lettieri T, Hughes M, D’Arrigo A, Truong O, Hsuan JJ, Jiricny J. Science. 1995;30:1912–1914. doi: 10.1126/science.7604265. [DOI] [PubMed] [Google Scholar]
- 19.Drummond JT, Li GM, Longley MJ, Modrich P. Science. 1995;30:1909–1912. doi: 10.1126/science.7604264. [DOI] [PubMed] [Google Scholar]
- 20.Acharya S, Wilson T, Gradia S, Kane MF, Guerrette S, Marsischky GT, Kolodner R, Fishel R. Proc Natl Acad Sci U S A. 1996;93:13629–13634. doi: 10.1073/pnas.93.24.13629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lamers MH, Perrakis A, Enzlin JH, Winterwerp HHK, de Wind N, Sixma TK. Nature. 2000;407:711–717. doi: 10.1038/35037523. [DOI] [PubMed] [Google Scholar]
- 22.Westmoreland J, Porter G, Radman M, Resnick MA. Genetics. 1997;145:29–38. doi: 10.1093/genetics/145.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Brien V, Brown R. Carcinogenesis. 2006;27:682–692. doi: 10.1093/carcin/bgi298. [DOI] [PubMed] [Google Scholar]
- 24.Cannavo E, Gerrlts B, Marra G, Jiricny J. J Biol Chem. 2007;282:2976–2986. doi: 10.1074/jbc.M609989200. [DOI] [PubMed] [Google Scholar]
- 25.Gassman NR, Clodfelter JE, McCaulet AK, et al. PLoS One. 6:e17907. doi: 10.1371/journal.pone.0017907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 27.Scheeff ED, Briggs JM, Howell SB. Mol Pharmacol. 1999;56:633–643. [PubMed] [Google Scholar]
- 28.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fisher S, Gao J, Guo H, Ha S, et al. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 29.MacKerell DA, Jr, Banavali N, Foloppe N. Biopolymers. 2001;56:257–265. doi: 10.1002/1097-0282(2000)56:4<257::AID-BIP10029>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 30.Jorgensen WL, Chandsrasekhar J, Madura JD, Impey RW, Klein ML. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 31.Humphrey W, Dalke A, Schulten K. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 32.Lee MS, Salsbury FR, Jr, Brooks CL. J Chem Phys. 2002;116:10606–10615. [Google Scholar]
- 33.Lee MS, Salsbury FR, Jr, Olson MA. J Comp Chem. 2004;25:1967–1978. doi: 10.1002/jcc.20119. [DOI] [PubMed] [Google Scholar]
- 34.Salsbury FRJ. Mol Phys. 2006;104:1299–1309. [Google Scholar]
- 35.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 36.Kale L, Skeel R, Bhandarkar M, Brunner R, Gursoy A, Krawetz N, Phillips J, Shinozaki A, Varadarajan K, Schulten K. J Comp Phys. 1999;15:1283–1312. [Google Scholar]
- 37.van Gunsteren WF, Berendsen HJC. Mol Phys. 1977;34:1311–1327. [Google Scholar]
- 38.Darden T, York D, Pedersen L. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 39.Sethna JP. Statistical Mechanics: Entropy, Order parameters and Complexity. Clarendon Press; 2010. [Google Scholar]
- 40.Louise-May S, Auffinger P, Wsthof E. Curr Opin Struct Biol. 1996;6:289–298. doi: 10.1016/s0959-440x(96)80046-7. [DOI] [PubMed] [Google Scholar]
- 41.Auffinger P, Louise-May S, Westhof E. J Am Chem Soc. 1995;117:6720–6726. [Google Scholar]
- 42.Vaiana AC, Westhof E, Auffinger P. Biochimie. 2006;88:1061–1073. doi: 10.1016/j.biochi.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 43.Salsbury FRJ, Crowder MW, Kingsmore SF, et al. J Mol Modeling. 2009;15:133–145. doi: 10.1007/s00894-008-0410-0. [DOI] [PubMed] [Google Scholar]
- 44.Karpen ME, Tobias DJ, Brooks CL., III Biochemistry. 1993;32:412–420. doi: 10.1021/bi00053a005. [DOI] [PubMed] [Google Scholar]
- 45.Matlab v.7.10.0. The MathWorks Inc.; Natick, Massachusetts: 2010. [Google Scholar]
- 46.Tiwari A, Panigrahi SK. In Silico Biol. 2007;7:651–661. [PubMed] [Google Scholar]
- 47.Panigrahi SK, Desiraju GR. Proteins. 2007;67:128–142. doi: 10.1002/prot.21253. [DOI] [PubMed] [Google Scholar]
- 48.Sarkhel S, Desiraju GR. Proteins. 2004;54:247–259. doi: 10.1002/prot.10567. [DOI] [PubMed] [Google Scholar]
- 49.Takahara PM, Rosenzweig AC, Frederick CA, Lippard SJ. Nature. 1995;377:649–652. doi: 10.1038/377649a0. [DOI] [PubMed] [Google Scholar]
- 50.Takahara PM, Frederick CA, Lippard SJ. J Am Chem Soc. 1996;118:12309–12321. [Google Scholar]
- 51.Kartalou M, Essigmann JM. Mutat Res. 2001;478:1–21. doi: 10.1016/s0027-5107(01)00142-7. [DOI] [PubMed] [Google Scholar]
- 52.Gelasco A, Lippard SJ. Biochemistry. 1998;37:9230–9239. doi: 10.1021/bi973176v. [DOI] [PubMed] [Google Scholar]
- 53.Rosenberg B, Camp L, Van Krigas T. Nature. 1965;205:698–699. doi: 10.1038/205698a0. [DOI] [PubMed] [Google Scholar]
- 54.Duckett DR, Drummond JT, Murchie AI, Reardon JT, Sancar A, Lilley DM, Modrich P. Proc Natl Acad Sci U S A. 1996;93:6443–6447. doi: 10.1073/pnas.93.13.6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yamada M, O’Regan E, Brown R, Karran P. Nucleic Acids Res. 1997;25:491–496. doi: 10.1093/nar/25.3.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jeffrey GA, Saenger W. Hydrogen Bonding in Biological Structures. Springer Verlag; 1991. [Google Scholar]
- 57.Taylor R, Kennard O. J Am Chem Soc. 1982;104:5063–5070. [Google Scholar]
- 58.Lin PD, Wang Y, Scherer SJ, Clark AB, Yang K, Avdievich E, Jin B, et al. Cancer Res. 2004;64:517–522. doi: 10.1158/0008-5472.can-03-2957. [DOI] [PubMed] [Google Scholar]
- 59.Yang W. Cell Research. 2008;18:184–197. doi: 10.1038/cr.2007.116. [DOI] [PubMed] [Google Scholar]
- 60.Salsbury FR. Protein Pept Lett. 2010;17:744–750. doi: 10.2174/092986610791190318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.