

Abstract
Translation initiation of hepatitis C Virus (HCV) RNA is the initial obligatory step of the viral life cycle, mediated through the Internal Ribosome Entry Site (IRES) present in the 5′-untranslated region (UTR). Initiation on the HCV IRES is mediated by multiple structure-specific interactions between IRES RNA and host 40S ribosomal subunit. In the present study we demonstrate that the SLIIIef domain, in isolation from other structural elements of HCV IRES, retain the ability to interact with 40S ribosome subunit. A small RNA SLRef, mimicking the SLIIIef domain was found to interact specifically with human La protein and the ribosomal protein S5 and selectively inhibit HCV RNA translation. More importantly, SLRef RNA showed significant suppression of replication in HCV monocistronic replicon and decrease of negative strand synthesis in HCV cell culture system. Finally, using Sendai virus based virosome, the targeted delivery of SLRef RNA into mice liver succeeded in selectively inhibiting HCV IRES mediated translation in vivo.
Keywords: HCV IRES, Translation inhibition, Sendai virosome, La autoantigen, Ribosome assembly
Introduction
Hepatitis C virus (HCV) is a major etiological agent of liver diseases such as chronic hepatitis and often leads to hepatocellular carcinoma.1 Over 180 million people throughout the world are carriers of HCV.2 Currently available therapy for HCV is mostly interferon-α, either alone or in combination with ribavirin. However this therapy lacks efficacy due to HCV quasispecies generation, and also leads to adverse side effects in many patients.3 Hence, development of novel and effective therapeutics against HCV is the need of the hour.
HCV is a small, enveloped, single stranded (positive sense) RNA virus belonging to the family Flaviviridae. Its genome is ≈9.5 Kb in length consisting of 5′ untranslated region (UTR), a long open reading frame (ORF) encoding the viral polyprotein and a 3′- UTR.4,5 The initiation of HCV genomic RNA translation is mediated by the high affinity interaction of the HCV IRES element with 40S ribosomal subunits, which promotes the recruitment of eIF3 and the eIF2/GTP/Met-tRNAimet ternary complex to form the 48S initiation complex. This is subsequently followed by 60S ribosomal subunit binding and initiation of protein synthesis.6-8 The specific interaction between HCV IRES and 40S subunit which is independent of cap structure and initiation factors is mediated through multiple interactions of HCV IRES with the 40S subunit proteins, including ribosomal protein S5.9,10 This function is encoded in the conserved structural motifs of HCV IRES. From literature it is known that SLIIIc, SLIIId, SLIIIe and SLIIIf domains plays important role in IRES-40S complex formation.8 The recent reports on the crystal structure of 40S and HCV IRES revealed important insights on the SLIIIef and pseudoknot domain in the context of 40S recruitment.11,12 Along with RNA elements of HCV genome, host proteins also known to play crucial role in HCV translation. Many host factors like La, hnRNP D, PTB proteins interact with HCV genome and affect HCV translation and replication.5,13,14 So sequestering these host factors and thus stopping them from interacting with HCV genome will severely affect the HCV translation and replication.15
Since mechanism of HCV IRES mediated translation is unique and fundamentally different from the mode of cellular RNA translation, it might act as a good target for anti-HCV therapeutics with high specificity and low cytotoxicity. In the past decade, RNA molecules have been explored as possible candidates for novel HCV therapeutics including siRNAs, ribozymes, antisense RNAs, small RNA, and RNA aptamers, as these acts directly on the genome.16-18
In the present study we have studied the interaction of SLIIIef domain of HCV IRES with 40S subunit and host proteins to design small RNA molecules to inhibit HCV translation. We designed three different RNAs namely SLRe, SLRf and SLRef mimicking SLIIIe, SLIIIf and SLIIIef domains of HCV IRES and investigated their ability to interact with 40S ribosome subunit and host proteins. Only small RNA SLRef (34 nt) was found to interact with 40S and selectively block ribosome assembly. In addition SLRef showed significant interaction with La protein and ribosomal protein S5. Since SLRef RNA sequesters host factors important for HCV translation and replication, we characterized the anti HCV activity of SLRef RNA.10,19,20 SLRef RNA significantly inhibited HCV translation and replication. Furthermore the targeted delivery of this RNA in vivo using Sendai virosome demonstrated selective and significant inhibition of HCV IRES mediated translation in mice liver.
Results
SLRef RNA binds to 40S subunit and inhibits ribosome assembly on HCV IRES
Ability of short RNAs SLRef, SLRe and SLRf to interact with 40S subunit was investigated using sucrose gradient analysis and gel retardation assay. Here SLRef, SLRe and SLRf RNAs mimic SLIIIef, SLIIIe and SLIIIf domains of HCV IRES respectively (Fig. 1A). In order to study the direct interaction of SLRef RNA with the 40S subunits, 40S purified from Hela cells were pre-incubated with γ 32P ATP-labeled SLRef RNA, SLRe RNA and SLRf RNA and then analyzed by sucrose density gradient fractionation. The ribosome binding profile showed that, SLRef RNA binds to 40S subunit and forms SLRef-40S complex. But SLRe and SLRf RNAs were unable to interact with 40S (Fig. 1B). To investigate whether SLRef RNA would interfere with 40S- HCV IRES interactions, gel retardation assay was performed using 40S subunits. Pre-incubation of the ribosome subunits with unlabelled SLRef RNA reduced the binding of 40S with radio labeled HCV IRES RNA. However SLRf and SLRe RNAs did not show any significant effect on 40S binding to HCV IRES RNA (Fig. 1C).
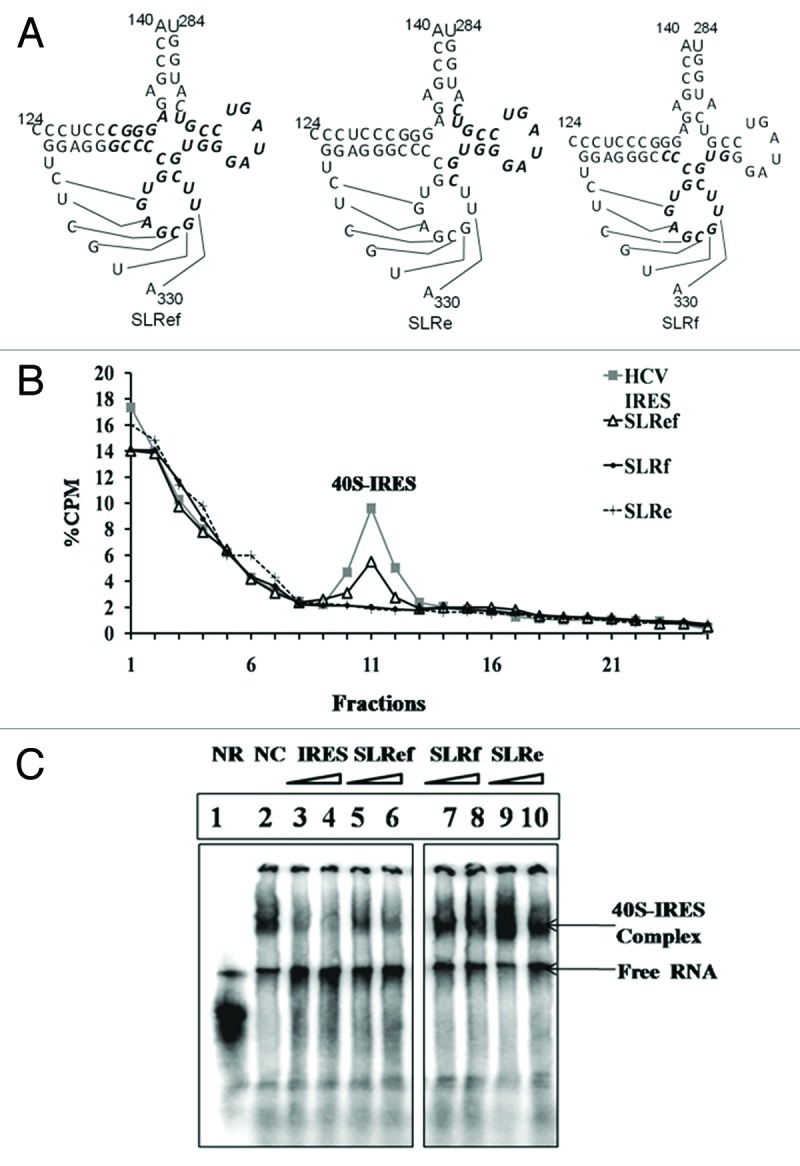
Figure 1. Interactions between short RNAs and 40S subunit. (A). Diagrammatic representation of short RNAs mimicking SLIIIef region of HCV IRES. (B) Representation of Sucrose gradient sedimentation profiles of γ32P ATP end labeled SLRef RNA, SLRe and SLRf RNA with HeLa 40S subunit. (C) Electrophoretic mobility shift assay (EMSA) reveals that SLRef interferes with HCV IRES and ribosome interaction. Lane 1 represents HCV IRES without ribosome subunits, lane 2 represents HCV IRES with ribosome subunits, as indicated in panel lane 3–10 includes 100 and 200 fold molar excess of unlabelled HCV IRES or 300 and 600 fold molar excess of either SLRef, SLRf and SLRe RNAs during incubation, respectively.
Further the effect of SLRef RNA on ribosome recruitment by the HCV IRES was examined. For this purpose ribosomal assembly reaction containing α 32P UTP-labeled HCV 5′ UTR were incubated with 200 fold molar excess of SLRef RNA and SLRf RNA and then fractionated by centrifugation. In the presence of SLRef RNA both 48S and 80S ribosomal complexes were reduced suggesting that SLRef RNA prevented the ribosomal complex assembly on the HCV IRES (Fig. 2A) without affecting the ribosome assembly on the capped RNA (Fig. 2B). But SLRf RNA had no effect on ribosome assembly (Fig. 2A). Ribosome assembly was performed in presence of GMP-PNP and cycloheximide to confirm the 48S and 80S peaks in the ribosome profile (Fig. 2C). GMP-PNP and cycloheximide blocks the translation initiation at 48S and 80S assembly stage respectively, which results in the increased height of the corresponding peak.
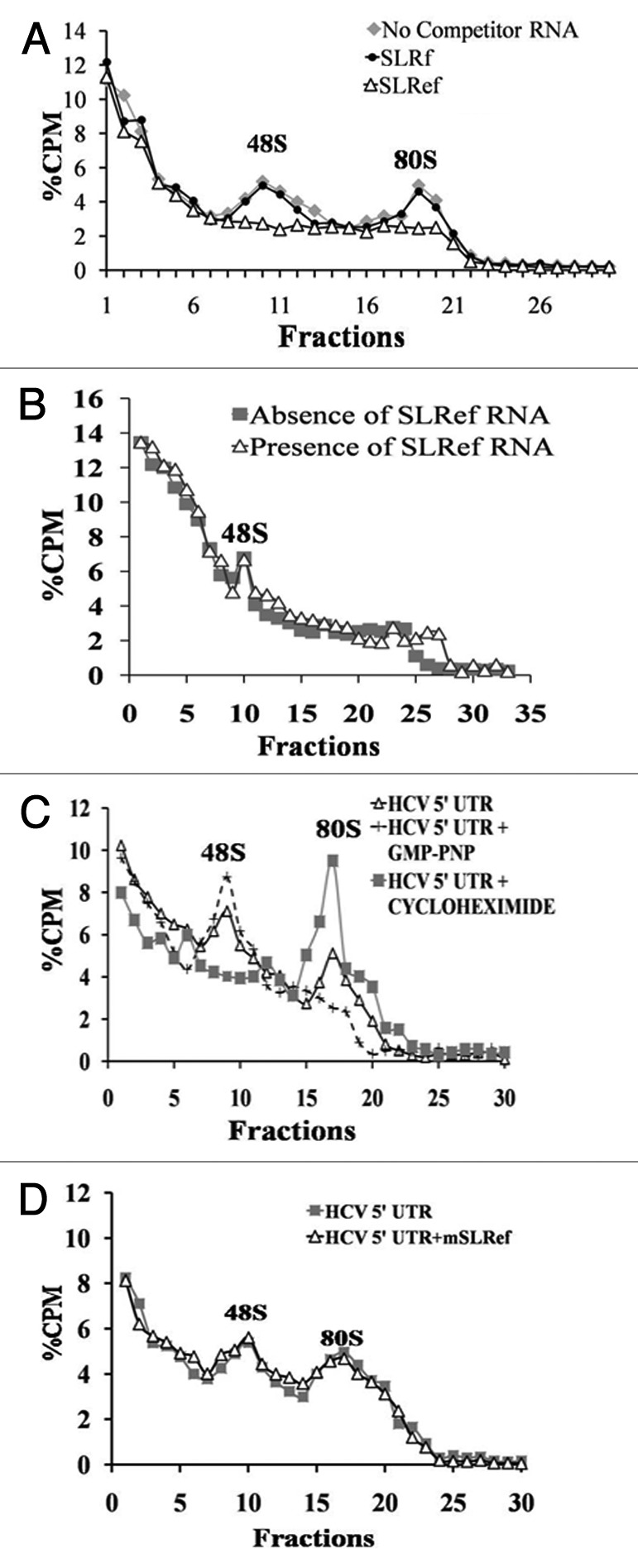
Figure 2. Prevention of ribosome assembly on HCV IRES by SLRef RNA. (A) Representation of Sucrose gradient sedimentation profiles of α 32P UTP-labeled HCV 5′ UTR RNA in the presence or absence of 200 fold molar excess of either SLRef RNA or SLRf RNA. Percentage CPM represents the percentage of total counts added to the reaction against the fraction number of gradient. The fractions were collected from top downwards. The 80S and 48S peaks were indicated. (B) Representation of sucrose gradient sedimentation profiles of α 32P UTP-labeled capped Luc RNA in the presence or absence of 200 fold molar excess of SLRef RNA. (C) Sucrose gradient sedimentation profiles of α 32P UTP-labeled HCV 5′ UTR RNA incubated in the RRL, in the presence of 2 mM GMP-PNP or 2 mM of cycloheximide. (D) Representation of sucrose gradient sedimentation profiles of α 32P UTP-labeled HCV 5′ UTR RNA in the presence or absence of 200 fold molar excess of mSLRef RNA
It was earlier reported that a single mutation in SLIIIef region of HCV IRES (A298G) drastically reduces the IRES activity and binding of this RNA to S5 ribosomal protein.10 Here, it was examined whether the similar mutation in SLRef RNA (A298G) would affect the ability of SLRef RNA to inhibit the ribosome assembly on HCV IRES. As expected SLRef mutant (mSLRef) RNA failed to prevent the ribosomal assembly on HCV IRES (Fig. 2D). These observations suggest that specific mutations in sequence might be preventing binding of mSLRef RNA with 40S subunit.
Interaction of SLRef RNA with host factors
Host factors play crucial role in HCV life cycle. Many host factors are reported to interact with HCV IRES to regulate translation. So we performed UV- cross linking experiments with Huh7 S10 cytoplasmic extract to study the interactions of SLRef RNA with host factors. α 32P UTP-labeled HCV IRES RNA was incubated with S10 extract in the presence or absence of 100 fold unlabeled SLRef and SLRf RNAs (Fig. 3A). Results suggested that SLRef RNA significantly competed outs the binding of HCV IRES with a protein of apparent molecular weight of 52 kDa. This protein was then confirmed as La protein after immunoprecipitation with La antiserum (Fig. 3B). Interestingly, SLRef also competed out the binding of HCV IRES RNA with another protein of apparent molecular weight of 25 kDa, which was reported as ribosomal protein S5 in literature.21 To further confirm we performed UV- cross linking experiments between α 32P UTP-labeled HCV IRES RNA and recombinant La (p52) and S5 (p25) proteins in presence of SLRef RNA, which showed that SLRef RNA competes with the HCV IRES RNA for binding with these proteins (Fig. 3C and 3E). To investigate the affinity of SLRef RNA toward La and rpS5, RNA-protein filter binding assay was performed. 32P labeled smaller RNAs (SLRe, SLRf and SLRef) were used in the filter binding assay. SLRef RNA showed significant binding with La (kd = 0.58 µM), rpS5 (Kd = 1.87 µM), whereas the other small RNAs did not show appreciable interaction (Fig. 3D and 3F). Graph was plotted from the average of three independent experiments.

Figure 3. Interaction of SLRef RNA with Cellular Proteins. (A). 32P-labeled HCV IRES RNA was UV cross-linked to Huh7 S10 extract in absence (lane C) or presence of 100 fold molar excess of SLRef and SLRf RNA (as indicated), digested with RNase A and resolved by SDS-10% PAGE followed by phosphor imaging. (B). 32P-labeled HCV IRES RNA in absence (lane 2) or presence (lane 3) of SLRef RNA was UV-cross linked with Huh 7 S10 extracts and the complexes were immunoprecipitated using La antiserum (lane 2, 3) or using pre-immune serum (Lane 1) as indicated. (C). α 32P-labeled HCV IRES RNA was UV cross-linked to recombinant purified La protein, with 100 and 200 fold molar excess of either unlabeled SLRef or SLRf RNA (as described in the panel) as competitor RNAs.(D) γ 32P-labeled small RNAs were incubated with increasing concentration of La protein and then analyzed by filter binding assay.(E) α 32P-labeled HCV IRES RNA was UV cross-linked to recombinant purified S5 protein, with 100 and 200 fold molar excess of either unlabeled HCV IRES RNA, SLRef, mSLRef, SLRf RNA (as described in the panel) as competitor RNAs.(F) γ 32P-labeled small RNAs were incubated with increasing concentration of S5 ribosomal protein and then analyzed by filter binding assay.
Inhibition of HCV IRES mediated translation by SLRef RNA
Above results suggests that SLRef RNA binds to La and 40S subunit and thus stops them from interacting with HCV RNA. Since both La and 40S are critical for HCV translation, we examined the effect of small RNAs (SLRef, SLRe and SLRf) on HCV translation. To investigate the effect of the small RNAs on HCV translation, small RNAs were co- transfected with HCV bicistronic RNA into Huh 7 cells (Fig. 4B). SLRef RNA showed dose dependent inhibition of HCV IRES mediated translation at increasing concentrations (50, 100 and 200 pmol) without inhibiting cap-dependent translation (For SLRef RNA p = 0.000015 with HCV IRES mediated translation and p = 0.386 with cap-dependent translation at 200pmol). Other small RNAs showed comparatively less inhibition of HCV translation. Huh7 cells transfected with HCV bicistronic RNA alone was considered as control. Four independent experiments were performed in duplicate. Effect of Small RNAs on HCV translation was also analyzed by in vito translation experiments. Increasing concentrations (ranging from 0.5 to 100 pmol) of the small RNAs were exogenously added to in vitro translation reactions of an HCV IRES containing bicistronic RNA in rabbit reticulocyte lysate (RRL) system. Among these, SLRef RNA showed significant inhibition of the HCV IRES mediated translation without inhibiting cap-dependent translation, which supports ex vivo results (Figs. S2A,S2B).We also performed in vitro translation with mutant SLRef RNA and we found that mSLRef RNA don’t have appreciable effect on HCV translation (Fig. S4). SLRef RNA (34nt) inhibited HCV translation more efficiently than SLIIIe+f RNA (65nt) which was shown to inhibit HCV translation in earlier studies (Fig. S2C, D).10
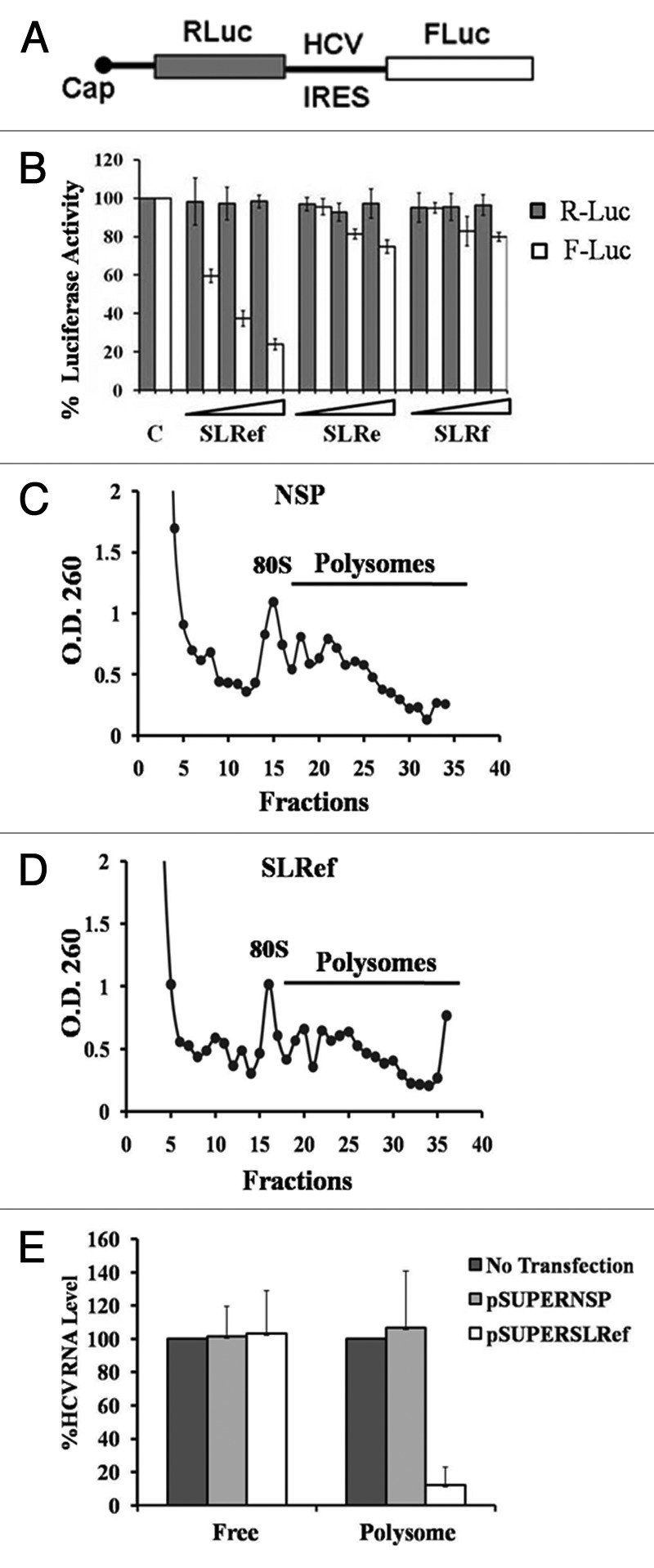
Figure 4. Inhibition of HCV IRES mediated translation by SLRef RNA. (A) Schematic representation of HCV bicistronic RNA. (B) Small RNAs were co-transfected with HCV bicistronic RNA to the Huh 7 cells. 12h post transfection lysate was collected and analyzed for luciferase activity. FLuc activity represented in the white bar indicates the effect of small RNAs on HCV RNA translation. RLuc activity represented in the black bars indicates the effect of small RNAs on cap-dependent translation. Luciferase activity of control was expressed as 100%. Data from the co-transfection experiments are expressed as means ± SD of four independent replicates. (C) and (D) Representative polysome profile of Rep2a cells after transfecting with pSUPER NSP and pSUPER SLRef. (E) Quantifications of HCV genome in the polysomes and free fraction by qRT-PCR. HCV RNA levels were normalized with GAPDH RNA level.
We also performed polysomal analysis to investigate the association of HCV RNA with the polysomes, which represents the actively translating HCV RNA population (Fig. 4C and D). Huh7 cells harboring the HCV monocistronic replicon (Rep2a) were transfected with pSUPERSLRef and pSUPERNSP DNA constructs which are able to efficiently express the short RNAs inside the transfected cells.18,22 Here pSUPERNSP expresses non specific RNA sequence.18 After 36hrs of post transfection cells were treated with cycloheximide and mRNAs were separated by velocity sedimentation on a sucrose gradient. RNA was isolated from free and polysome fractions (Fig. 4C, 4D). HCV RNA level in free and polysome fractions was quantified by real time PCR (Fig. 4E). Polysome associated HCV RNA level in pSUPERSLRef and pSUPERNSP transfected Rep2a cells were compared with untransfected Rep2a cells (Fig. S3). HCV RNA level was normalized with GAPDH RNA. These results suggested that HCV RNA association with polysome was significantly reduced on pSUPERSLRef transfection, but no appreciable effect was observed in case of pSUPERNSP (p = 0.0050 for pSUPERSLRef and p = 0.773 for pSUPERNSP).
Effect of SLRef RNA on HCV RNA replication
The effect of SLRef RNA on HCV RNA replication was investigated by transfecting the small RNAs into Huh7 cells harboring the HCV monocistronic replicon.36 Cells were maintained in the absence of hygromycin during and after transfection. SLRef RNA showed significant reduction of HCV negative strand RNA levels without inhibiting the levels of cellular GAPDH RNA, whereas SLRf RNAs did not cause significant reduction of HCV negative strand RNA (Fig. 5B). Untransfected Huh7 cells harboring the HCV monocistronic replicon were considered as 100%. Additionally, colony forming assay was performed to demonstrate the efficacy of SLRef RNA for curing of replicating HCV RNA in Huh7 cells at the single cell level. Cells were maintained in the presence of hygromycin after transfection. Results showed that most of the cells were free of HCV RNA in the presence of SLRef RNA (Fig. 5C). The efficacy of SLRef RNA on infectious HCV replication was evaluated further by HCV JFH1 cell culture system. HCV JFH1 RNA was generated from a cell culture adapted infectious JFH1 cDNA construct (a generous gift from Takaji Wakita, National Institute of Infectious Diseases, Japan). Increasing concentrations (250pmol, 500pmol, 750pmol and 1000 pmol) short RNAs were co-transfected with the HCV JFH1 RNA.35 qRT-PCR analysis demonstrated that SLRef RNA significantly inhibited negative strand RNA synthesis of HCV JFH1 (~90%) in the cell culture without affecting the cellular RNA level (Fig. 5E)(p. = 0.00069 for SLRef at 1nmol concentration). Cells transfected with JFH1 RNA alone was considered as 100%. The results represent the average of three independent experiments.

Figure 5. Effect of SLRef RNA on HCV replication ex vivo. (A) Schematic representation of HCV monocistronic replicon RNA (adapted from Frese et al. 2003).33 (B) SLRef, SLRf, SLRe RNAs (500pmol) were transfected in to the Huh7 cells harboring HCV monocistronic replicon construct. 48 h post transfection RNA was isolated and subjected to RT PCR. cDNA synthesized from RT-PCR was then subjected to qRT-PCR for analyzing the relative HCV RNA level. The white bar indicates the HCV mRNA level after transfection with small RNAs. Percentage of RNA level calculated considering untranfceted Huh7 cells harboring HCV monocistronic replicon construct . (C) Curing assay of replicon cells in the presence or absence of SLRef RNA. (D) Schematic representations of HCV JFH1 construct(Adapted from ref. 34) . (E) in vitro transcribed HCV RNA from HCV JFH1 construct was transiently co-transfected with SLRef RNA and SLRf RNA on the Huh7 cells. 48 h post transfection RNA was isolated and then analyzed by qRT-PCR for detecting the HCV RNA level. The bar indicates the HCV mRNA level. Percentage RNA level represents the effect of SLRef RNA on HCV RNA level. (F) Schematic representation of pSGR-JFH1/luc RNA (adapted from ref. 23).(G)pSGR-JFH1/luc RNA was co transfected with SLRef RNA (250pmol and 500pmol) into Huh7 cells and after 24hrs of post transfection RNA was isolated and subjected to RT PCR. cDNA synthesized from RT-PCR was then subjected to quantitative real time PCR for analyzing the relative HCV RNA level and lysate was analyzed for Luciferase activity, which indicates the HCV IRES activity.
To investigate the effect of SLRef directly on HCV replication process, Huh7 cells were co transfected with pSGR-JFH1/luc RNA and SLRef RNA (Fig. 5G).23 In pSGR-JFH1/luc construct, luciferase translation is mediated by HCV IRES where as HCV polyprotein synthesis is mediated by EMCV IRES.23 Luciferase activity and HCV RNA level was quantified after 24 h of transfection. We found that Luciferase activity was significantly inhibited (p = 0.00065 at 500pmol) and HCV RNA level was reduced to a small extent (p = 0.044 at 500pmol), suggesting that SLRef RNA might affect both translation and replication. But major effect was on translation. Figure was plotted from the average of three independent experiments.
Membrane fusion mediated delivery of SLRef to cells in culture
Finally, to investigate the effect of SLRef RNA on HCV IRES mediated translation in liver of live animal; we have taken help of Sendai virus based virosomes (Fig. S6). Essentially, the HCV bicistronic DNA construct was co-entrapped with either the pSUPER SLRef or a non specific control (pSUPER NSP) construct (Fig. S5) for delivery into animal liver or cells through reconstituted Sendai viral envelopes (F-virosomes).18 pSUPER SLRef DNA construct expresses SLRef RNA whereas pSUPER NSP produces nonspecific RNA. Initially, to evaluate the efficiency of gene expression mediated by F-virosomes and to test the ability of pSUPER SLRef to inhibit HCV IRES mediated gene expression, Huh7 cells in culture were transfected with F- virosomes loaded with HCV IRES bicistronic construct together with pSUPER SLRef or pSUPER NSP. The pSUPER SLRef strongly inhibited HCV IRES mediated FLuc expression. Compared with those of control (HCV bicistronic construct alone), firefly luciferase activity, measured 24 h post transfection, was reduced up to 75%; whereas the pSUPER NSP showed only marginal reduction of FLuc expression (Fig. 6A). Cells incubated with virosomes containing HCV bicistronic construct alone was considered as 100%.Two independent experiments were performed in duplicates.
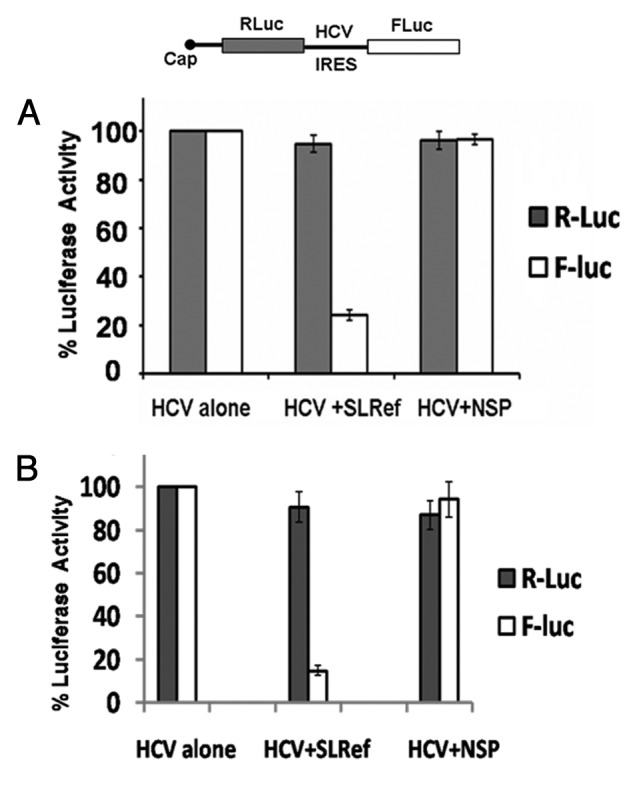
Figure 6. Effect of SLRef RNA on HCV IRES mediated translation in vivo in BALB/c mice. (A) Relative Luciferase activity of Huh7 cells harvested 24 h post transfection of F-virosome co-entrapped with HCV bicistronic construct and plasmid DNAs encoding either SLRef or NSP RNA. (B) Relative luciferase activities of liver parenchymal cells from BALB/c mice injected i.v. with F-virosomes loaded with the HCV bicistronic constructs along with the plasmid DNAs encoding either SLRef or NSP RNA.
Effect of SLRef on HCV IRES mediated gene expression in vivo.
To assess the target specificity of F-virosomes to deliver SLRef and inhibit target gene expression in vivo, F-virosomes containing HCV IRES bicistronic construct and pSUPER SLRef was delivered to female BALB/c mouse by injection through tail vein. Livers from injected animals were harvested two days after injection and assayed for dual luciferase expression. Mice treated with pSUPER SLRef along with the HCV IRES bicistronic plasmid showed significant reduction (~85%) in firefly luciferase (FLuc) expression (p = 0.000075) as compared with animal receiving virosomes containing the bicistronic plasmid only. Absolute values are shown in Figure S8. Also the preferential expression of luciferase gene was found to be mostly in the parenchymal cell-types (Hepatocytes) of mouse liver as compared with non-hepatocytes (data not shown). Control animals injected with buffer or the empty pSUPER plasmid showed no inhibition. Similarly, only marginal decrease in luciferase activity was observed in animals treated with pSUPER Nsp (p = 0.027) (Fig. 6B). Cells incubated with virosomes containing HCV bicistronic construct alone was considered as 100%. Three mice per group in duplicate set were used for each DNA construct.
Discussion
Conserved domains of HCV IRES perform crucial and distinct functions during translation initiation. Recruitment of 40S subunit to iAUG is independent of initiation factors and mediated by interactions of IRES elements in the 5′UTR with the 40S ribosomal subunit, thereby promoting the formation of 48S and subsequent 80S ribosome assembly. Particularly the four SLIII domains (IIIcdef) provide the core 40S subunit recruitment site. This domain also contains a conserved region (IIIef), which participates in pseudoknot formation and plays a crucial role in HCV IRES activity.8,24 Our studies revealed that short RNA mimicking SLIIIef domains retains the ability to interact with 40S subunit, even in the absence of other structural elements of the HCV IRES in our assay condition. But SLRe and SLRf RNAs did not show appreciable interactions with 40S subunit, suggesting that both IIIe and IIIf domains are necessary for its interaction with 40S subunit. However, we do not rule out the possibility that the SLRef RNA structure could be different from the SLIIIef domain which is part of the full length HCV-IRES. The salt concentration used in the study might contribute to the change in RNA structure.
In our ribosome assembly experiments SLRef RNA inhibited both 48S and 80S assembly. This clearly suggested that SLRef RNA is acting at a very initial stage of ribosome assembly. This supports our observations with gel retardation assays, where SLRef RNA inhibited HCV IRES-40S interactions.
UV cross-linking experiments revealed interactions between SLRef RNA and two host proteins namely rpS5 and La protein. In competition UV- cross linking experiments, SLRef RNA inhibited the interactions between host proteins (rpS5 and La protein) and HCV IRES. Both La and rpS5 proteins are very essential for HCV translation and replication. So sequestration of host proteins by SLRef RNA may be responsible for anti-HCV activity of SLRef RNA (Fig. 3C, 3E). Also SLRf RNA which failed to interact with rpS5 and La protein had no anti-HCV activity supporting our hypothesis.
In present study rpS5 was interacting with SLRef RNA which is derived from HCV IRES SLIIIef domain. These observations suggest that SLRef RNA interaction with 40S may be majorly through rpS5 protein, which is present in the exit site of the ribosome. A model based on cryo-electron microscopic structure of HCV IRES bound to the 40S ribosomal subunit clearly locates the S5 protein in close proximity to the basal part of SLII and the pseudoknot composed by SL IIIef domains.25,26 Thus it is possible that both SLII and SLIIIef influence the interaction with rpS5.
SLRef RNA inhibited HCV IRES mediated translation without affecting cap dependent translation. So it suggests that binding of SLRef RNA to rpS5 protein of 40S subunit does not affect the cap-dependent translation. This suggests that S5 protein bound to SLRef RNA can still function normally during cap-dependent translation unlike in HCV IRES mediated translation. Ribosomal protein S25 knock down is reported to inhibit HCV translation without affecting host translation.27 So all the ribosomal proteins may not be critical for cap-dependent translation.
Also SLRef found to interact with La protein and stop La from interacting with HCV IRES. La protein is reported to interact with HCV IRES and favor the HCV translation. La protein once binds to HCV IRES; it changes the HCV RNA structure to assist 40S subunit recruitment.5 Interestingly, peptides derived from La protein is reported to inhibit HCV translation and replication.28 So sequestering the La protein may be one of the major mechanisms responsible for anti-HCV activity of SLRef RNA.
These information’s, together with the observations made in this study, allow the proposition of a model for the inhibition of HCV IRES-mediated translation by the SLRef RNA. According to this model, when SLRef RNA, a structural mimic of this region of the HCV RNA, is introduced in trans, it interacts with the S5 protein of the 40S subunit and blocks the binding site of the HCV IRES. As this interaction is crucial for binding of the HCV IRES to the 40S subunit, abrogation of this interaction leads to failure of ribosome binding to the IRES and the inhibition of translation initiation (Fig. S7).
SLRef RNA showed significant inhibition of HCV RNA synthesis in both HCV subgenomic replicon and infectious virus cell culture experiments. Experiments performed with pSGR-JFH1/luc construct showed that, SLRef inhibits translation of HCV significantly and affects replication only to a small extent. So anti-HCV activity of SLRef RNA is a combined effect of translation and replication inhibition.
Various groups have demonstrated the inhibition of HCV IRES mediated translation in vitro and ex vivo by a wide range of nucleic acid molecules such as siRNA, DNAzymes, ribozymes and oligonucleotides.16-18 However these approaches are sequence specific, which can lead to the exertion of selection pressure on the genome for the generation of escape mutants. Hence, SLRef targets the interaction of a host protein with a conserved structural element in the viral IRES will be more efficacious as mutations in the viral genome are less likely to affect such interactions. Using RNA molecules as therapeutic agents has increased in the past decade as it is target-specific and elicits low immune response, and is generally free from many adverse side effects associated with chemotherapeutic agents.5 Intravenous administration of locked nucleic acid (LNA)–modified oligonucleotide (SPC3649) complementary to miR-122 was reported to reduce HCV load significantly in chimpanzee which is a animal model of HCV suggesting the potential of RNA based therapeutics.29
Targeted delivery of DNA/RNA molecules to the liver pose a major challenge in developing nucleic acid based therapy against HCV. Several methods available for nucleic acid delivery were not selective for hepatocytes. Recently we demonstrated the targeted delivery of anti HCV molecules to mice liver through Sendai virosome approach.30 In the present study we used the similar approach for SLRef delivery into the mice liver cells and demonstrated inhibition of HCV IRES-mediated translation in vivo.
The above results demonstrate that SLIIIef domains retain the ability to interact with 40S subunit, independent of other structural elements of HCV IRES. Studies also suggests that both IIIe and f domains of HCV IRES is necessary for its interactions with 40S subunit. It showed anti –HCV activity in infectious HCV cell culture system and also in mice experiments. More importantly, the small size of the SLRef RNA (34nt), allow the development of small molecule analogs which will be more effective therapeutically. In summary this study provides the insights about the interactions of SLIIIef domains of the IRES with the components of translation machinery involved in HCV translation, which can be exploited as novel target for developing potential anti-HCV molecules.
Materials and Methods
Plasmid Constructs
Three small RNAs were designed based on the SLIIIef sequence (Fig. 1A, Table 1). These RNAs corresponding to the various regions of SL IIIef (SLRef, SLRe, SLRf) were synthesized (custom made from Dharmacon). The bicistronic plasmid constructs (RLuc-HCV-IRES-FLuc) containing HCV IRES in between renilla luciferase and firefly luciferase gene was cloned into the pCDNA3.1 vector.5 PCDHCV-Luc construct was used to synthesize HCV IRES (18–383) by in vitro transcription.19 A cell culture-adapted infectious JFH1–cDNA construct (a generous gift from Takaji Wakita, National Institute of Infectious Diseases, Japan) was used to generate JFH1 RNA after linearization with XbaI followed by run-off transcription.31,35 For virosome experiments DNA sequences corresponding to SLRef, SLRe, SLRf and NSP RNAs were cloned in the polylinker region of the pSUPER vector (Fig. S5).18 Sequences of respective RNAs are mentioned in Table 1.
Table 1. Nucleotide Sequence of different Short RNAs.
| RNA | Sequences (5′-3′) |
|---|---|
| SLRe |
CUGCCUGAUAGGGUGC |
| SLRf |
GUGCUUGCGAGUGCCC |
| SLRef |
CGGGAUGCCUGAUAGGGUGCUUGCGAGUGCCCCG |
| NSP | CAGUCGCGUUUGCGACUGG |
Cell culture
Huh-7 and Huh7.5 cell monolayers were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) at 37°C in 5% CO2. The HCV monocistronic replicon (a kind gift from Dr. Ralf Bartenschlager, Heidelberg University), was passaged with 25 μg of hygromycin B per ml.18 Sendai virus (Z strain) was propagated in the allantoic sac of 10–11 d old embryonated chicken eggs as described earlier.30
In-vitro transcription and translation
RNAs were transcribed in vitro from different linearized plasmid constructs under T7 promoters in run-off transcription reactions as described before.19 Capped bicistronic RNAs were synthesized using Ribomax large scale RNA preparation system T7 (Promega) under standard conditions. Radiolabeled mRNAs were transcribed in vitro using T7 RNA polymerase (Promega) and [32P] Uridine triphosphate (BRIT). The transcription reaction was performed under standard conditions (Promega protocol). pcDNA3.1Luc construct was digested with PmeI. Capped luciferase RNA was synthesized by runoff transcription using pcDNA3.1Luc construct. The product was resolved on 8M Urea-6% PAGE and then gel-eluted. The shorter RNAs were labeled using [32P] adenosine triphosphate (BRIT) by end-labeling method and then gel-eluted.
In vitro translation of the capped bicistronic mRNAs were carried as described earlier. The reaction mixtures were incubated at 30°C for 90 min and assayed for both Renilla and Firefly luciferase activity.19
Purification of S5 ribosomal protein
Human S5 protein was expressed in bacteria and purified as mentioned earlier.10 In brief, Escherichia coli BL21 cells were transformed with the plasmid pET28a‐S5 (a gift from Dr S. Fukushi, Biomedical Laboratories, Japan) expressing the poly (His)‐tagged human S5 protein. Protein expression in bacterial culture was induced by 0.4 mM IPTG and purified using Ni2+–nitrilotriacetic acid–agarose (Qiagen, Hilden, Germany) under non‐denaturing conditions and eluted with 250mM imidazole.
Filter binding assay
The 32P labeled smaller RNAs were incubated along with the full length S5 or La protein at 30°C for 15 min in RNA binding buffer (containing 5mM HEPES pH 7.6, 25 mM KCl, 2 mM MgCl2, 3.8% glycerol, 2 mM DTT and 0.1 mM EDTA) and loaded onto nitrocellulose filters. The counts retained were measured in liquid scintillation counter.5 Kd values were calculated by using nonlinear regression applied by Graph pad Prism.
UV Cross-linking Experiment
α 32P UTP-labeled HCV IRES RNA were incubated with the purified proteins or Huh7 S10 extract at 30°C for 15 min in RNA binding buffer and then irradiated with a hand-held UV lamp for 10 min. UV- cross linking experiment was done using 2pmol - La protein, 15pmol - rpS5 proteins and 30µg of S10 extract.
The mixture was treated with 30 µg of RNase A (Sigma) at 37°C for 30 min. The protein-nucleotidyl complexes were electrophoresed on SDS-10% PAGE and analyzed by phosphor imaging.10
Immunoprecipitation
UV-crosslinking followed by Immunoprecipitation with HeLa S10 extracts was performed as described earlier.32 Briefly, 50 µg of protein (10 µg in each of five reactions) were incubated with α 32P UTP-labeled HCV IRES RNA in RNA binding buffer. After UV crosslinking and RNase digestion, the reactions were pooled together and the volume made up to 200 µl using 1x RIPA buffer (5 mM Tris–Cl pH 7.4, 150 mM NaCl, 1% Triton-X100, 0.1% SDS, 1% sodium deoxycholate). Anti-La polyclonal antibody or rabbit pre-immune serum was added and incubated on ice for 4 h. The immunocomplexes were precipitated by Protein A–CL Sepharose beads (Sigma-Aldrich) for 2 h at 4°C. The beads were washed three times with RIPA buffer. The bound proteins were analyzed by SDS–10% PAGE (PAGE) followed by phosphorimaging.
Transfection
An amount of 3 x 105 cells were seeded into 6 well plates. Cells were tranfected after 16 h. Lipfectamine 2000 (Invitrogen) was used in 1:3 (Nucleic acid in µg: Lipofectamine in µL) ratio. Huh 7 cells harboring the HCV monocistronic replicon were transfected with the varying concentration of small RNAs and DNA constructs using transfection reagent lipofectamine 2000 (Invitrogen). An amount of 500 ng of DNA and 1.5 µL lipofectamine 2000 was used for tranfection in 6 well plates. And Huh7 cells are cotranfected with HCV bicistronic RNA and short RNAs. Cells were harvested 12 h post-transfection using passive lysis buffer (Promega) and assayed for RLuc and FLuc activities according to Promega protocol.10
qRT-PCR analysis
Total RNA was isolated from cells using TRIzol reagent. HCV negative strand RNA was quantified by two–step qRT PCR using DyNAmo™ HS SYBR® Green qPCR Kit (Finzyumes). cDNA was synthesized with Moloney murine leukemia virus (M-MLV) reverse transcriptase at 42°C for 1 h (Promega) using 1µg of total RNA by adding primers targeting HCV negative strand and GAPDH mRNA in same reaction. Quantitative reverse transcription PCR (qRT PCR) was done using 100ng of cDNA in a 10µL reaction according to manufacturer’s instructions. 40 cycle number was used. Comparative threshold cycle (CT) method was used to calculate percentage of HCV RNA level normalized with glyceraldehyde 3- phosphate dehydrogenase (GAPDH). Percentage of HCV RNA level was calculated using the formula mentioned below. Primer sequences are available in Table 2.
Table 2. qRT PCR primer sequences.
| Primer sequences | Forward primer | Reverse primer |
|---|---|---|
|
HCV |
5′TGCGGAACCGGTGAGTACA 3′ |
5′CTTAAGGTTTAGGATTCGTGCTCAT3′ |
| GAPDH | 5′CATGAGAAGTATGACAACAGCCT 3′ | 5′ AGTCCTTCCACGATACCAAAGT 3′ |
% HCV RNA Level = 100-[(1–1/2n) × 100] when n is a positive value
% HCV RNA Level = 100+ [(1–1/2-n) × 100] when n is a negative value
Where ‘n’ is a ΔΔCT value
Colony formation assay (Curing Assay)
Small RNAs were transfected into Huh7 cells harboring monocistronic replicon. Twenty-four hours post transfection, cells were trypsinized and 4 X 103 cells were seeded in 35 mm dishes, incubated for 10 d in presence of 25 µg/ml of hygromycin B. The cells were fixed with 90% acetone, stained with Brilliant Blue G-250 in 40% methanol and 10% acetic acid and colonies were counted and approximate percentage CFU was calculated.18
Ribosomal assembly assay
α 32P-labeled HCV 5′ UTR RNA (3X 105 c.p.m.) was added to 25 µl of translation reaction containing 17.5 µl of RRL in the presence or absence of 200 fold molar excess of Short RNAs (both wild type and mutant). The reactions were incubated at 30°C for 15 min diluted to 150 µl with gradient buffer (20 mM TRIS-HCl pH 7.5, 100 mM KCl, 3 mM MgCl2, 1 mM DTT) and overlaid in 5–30% sucrose gradient. The ribosomal complex was sedimented by ultracentrifuge for 3 h at 30000 rpm at 4°C. Fractions (500 μl) were collected using fraction collecting system (Isco Teledyne) and the radioactivity was measured in a liquid scintillation counter.10
Purification of ribosomal subunits
40S subunits were isolated from Hela S3 cells. In brief the cells were homogenized in 4 volumes of buffer(20 mM Tris-HC1 pH 7.5, 100 mM KCl,5 mM MgC12, 5 mM 2-mercaptoethanol)containing 0.5% of non-ionic detergent NP-40 using a Dounce homogenizer. Lysate was centrifuged for 15 min at 30000 g, 4°C. 40S subunits were isolated from the supernatant as described earlier.9 Purity of 40S subunit preparation was analyzed by sqRT-PCR (Fig. S1).
Electrophoretic Mobility Shift Assay (EMSA)
Conditions for EMSA have been previously described.34 α 32P-labeled HCV IRES (17 fmol) was mixed with 0.135 A260 U of 40S subunits. After incubation at 30°C for 30 min, loading dye was added and the protein-nucleotidyl complex was resolved on a 4% (60:1) polyacrylamide gel at 4°C.
Sucrose gradient polysome fractionation
Cells were treated with 100 µg/ml cycloheximide for 10min at 37°C. After 10min incubation media was removed and cell culture dishes (15 cm diameter) were kept on ice. Cells were washed in 25 ml ice cold PBS containing 100µg/ml cycloheximide and then washed with 7.5 ml of 1X hypotonic buffer (5 mM TRIS-HCl pH-7.5, 1.5 mM KCl and 5 mM MgCl2) containing 100 µg/ml cycloheximide. Cells were lysed by scrapping in 350µL ice cold lysis buffer(5 mM TRIS-HCl pH-7.5, 1.5 mM KCl,5 mM MgCl2, 100 µg/ml cycloheximide, 1mM DTT, 200 U/ml RNase in from Promega, 200µg t- RNA, 0.5% Triton X -100, 0.5% Sodiumdeoxycholate, 1X protease inhibitor cocktail) and lysate was incubated for 15 min on ice. After incubation the KCl concentration in the lysate was adjusted to 150 mM. lyaste was spun for 8min at 3000 g, 4°C. 500 µL of supernatant was loaded on 15–50% sucrose gradient containing 100µg/ml cycloheximide and gradients were centrifuged at 36,000 rpm for 2 h at 4°C, 300-μl fractions were collected and OD 260 was determined using the NanoDrop ND-2000 spectrophotometer (Thermo Scientific). Polysomes fractions were pooled to isolate RNA using Trizol (Sigma). RNA level was quantified using real time PCR.
Membrane fusion mediated delivery of plasmids to cells in culture
Monolayer Huh7 cells were incubated with loaded F-virosomes (0.3 mg F-protein) containing 3–4 µg of HCV bicistronic construct(1–383) in presence or absence of pSUPER SLRef or pSUPER NSP in serum free medium at 37°C in 5% CO2. Two hour post fusion, 10% serum containing DMEM was added and cells were further incubated for 24 h.30 Cells were harvested after 24 h with passive lysis buffer (Promega) and luciferase assay was performed.
Administration of plasmid loaded virosomes to mice
HCV bicistronic construct (1–383) with pSUPER SLRef or pSUPER Nsp was loaded inside F-virosomes as described earlier.18In brief Female BALB/c mice (12 weeks old) weighing ~20 g were injected i.v. into the tail vein with 4 µg each of plasmid loaded virosomes. After 2 d post-injection, animals were sacrificed and hepatocytes were isolated (Figs. S6A and S6B).
Isolation and gene expression in isolated hepatocytes
Liver parenchymal cells were isolated by collagenase perfusion of the liver as described earlier.30 In brief, the perfused liver was excised from the animal, crushed into small pieces and then washed thoroughly with phosphate buffer, pH 7.4. The liver pieces were incubated at 37 °C for 30 min in a buffer containing collagenase A (hepatocyte qualified; Gibco) followed by filtration through a nylon mesh. The filtrate was centrifuged at 500 g for 10 min at 4 °C to pellet down the hepatocytes. The non-hepatocyte pellet was seperated by centrifuging the supernatant at 1500 g for 10 min at 4 °C. Both cell pellets were washed three times with ice-cold PBS, pH 7.4, lysed with passive lysis buffer (Promega) and the cell extracts were then subjected to a dual luciferase assay.
Statistical analysis
t-test was used for all the statistical analysis. All graphs represent mean ± SD. p-values were determined by using a paired t-test. P-values of less than 0.01 were considered to be statistically significant.
Supplementary Material
Acknowledgment
We thank our lab members for helpful discussion. This work was supported by grant from the Department of Biotechnology; Government of India and Life Science Research Board (LSRB) of DRDO, Govt. of India to SD and DPS.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental material can be found at: www.landesbioscience.com/journals/rnabiology/article/21208
Footnotes
Previously published online: www.landesbioscience.com/journals/rnabiology/article/21208
References
- 1.Saito I, Miyamura T, Ohbayashi A, Harada H, Katayama T, Kikuchi S, et al. Hepatitis C virus infection is associated with the development of hepatocellular carcinoma. Proc Natl Acad Sci U S A. 1990;87:6547–9. doi: 10.1073/pnas.87.17.6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoofnagle JH. Hepatitis C: the clinical spectrum of disease. Hepatology. 1997;26(Suppl 1):15S–20S. doi: 10.1002/hep.510260703. [DOI] [PubMed] [Google Scholar]
- 3.Iwasaki Y, Ikeda H, Araki Y, Osawa T, Kita K, Ando M, et al. Limitation of combination therapy of interferon and ribavirin for older patients with chronic hepatitis C. Hepatology. 2006;43:54–63. doi: 10.1002/hep.20984. [DOI] [PubMed] [Google Scholar]
- 4.Kato N, Hijikata M, Ootsuyama Y, Nakagawa M, Ohkoshi S, Sugimura T, et al. Molecular cloning of the human hepatitis C virus genome from Japanese patients with non-A, non-B hepatitis. Proc Natl Acad Sci U S A. 1990;87:9524–8. doi: 10.1073/pnas.87.24.9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pudi R, Abhiman S, Srinivasan N, Das S. Hepatitis C virus internal ribosome entry site-mediated translation is stimulated by specific interaction of independent regions of human La autoantigen. J Biol Chem. 2003;278:12231–40. doi: 10.1074/jbc.M210287200. [DOI] [PubMed] [Google Scholar]
- 6.Otto GA, Puglisi JD. The pathway of HCV IRES-mediated translation initiation. Cell. 2004;119:369–80. doi: 10.1016/j.cell.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 7.Kieft JS, Zhou K, Jubin R, Doudna JA. Mechanism of ribosome recruitment by hepatitis C IRES RNA. RNA. 2001;7:194–206. doi: 10.1017/S1355838201001790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lukavsky PJ. Structure and function of HCV IRES domains. Virus Res. 2009;139:166–71. doi: 10.1016/j.virusres.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pestova TV, Hellen CU, Shatsky IN. Canonical eukaryotic initiation factors determine initiation of translation by internal ribosomal entry. Mol Cell Biol. 1996;16:6859–69. doi: 10.1128/mcb.16.12.6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ray PS, Das S. Inhibition of hepatitis C virus IRES-mediated translation by small RNAs analogous to stem-loop structures of the 5′-untranslated region. Nucleic Acids Res. 2004;32:1678–87. doi: 10.1093/nar/gkh328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rabl J, Leibundgut M, Ataide SF, Haag A, Ban N. Crystal structure of the eukaryotic 40S ribosomal subunit in complex with initiation factor 1. Science. 2011;331:730–6. doi: 10.1126/science.1198308. [DOI] [PubMed] [Google Scholar]
- 12.Berry KE, Waghray S, Mortimer SA, Bai Y, Doudna JA. Crystal structure of the HCV IRES central domain reveals strategy for start-codon positioning. Structure. 2011;19:1456–66. doi: 10.1016/j.str.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Domitrovich AM, Diebel KW, Ali N, Sarker S, Siddiqui A. Role of La autoantigen and polypyrimidine tract-binding protein in HCV replication. Virology. 2005;335:72–86. doi: 10.1016/j.virol.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 14.Paek KY, Kim CS, Park SM, Kim JH, Jang SK. RNA-binding protein hnRNP D modulates internal ribosome entry site-dependent translation of hepatitis C virus RNA. J Virol. 2008;82:12082–93. doi: 10.1128/JVI.01405-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fontanes V, Raychaudhuri S, Dasgupta A. A cell-permeable peptide inhibits hepatitis C virus replication by sequestering IRES transacting factors. Virology. 2009;394:82–90. doi: 10.1016/j.virol.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Romero-López C, Sánchez-Luque FJ, Berzal-Herranz A. Targets and tools: recent advances in the development of anti-HCV nucleic acids. Infect Disord Drug Targets. 2006;6:121–45. doi: 10.2174/187152606784112182. [DOI] [PubMed] [Google Scholar]
- 17.Kikuchi K, Umehara T, Fukuda K, Hwang J, Kuno A, Hasegawa T, et al. RNA aptamers targeted to domain II of hepatitis C virus IRES that bind to its apical loop region. J Biochem. 2003;133:263–70. doi: 10.1093/jb/mvg036. [DOI] [PubMed] [Google Scholar]
- 18.Subramanian N, Mani P, Roy S, Gnanasundram SV, Sarkar DP, Das S. Targeted delivery of hepatitis C virus-specific short hairpin RNA in mouse liver using Sendai virosomes. J Gen Virol. 2009;90:1812–9. doi: 10.1099/vir.0.010579-0. [DOI] [PubMed] [Google Scholar]
- 19.Pudi R, Abhiman S, Srinivasan N, Das S. Hepatitis C virus internal ribosome entry site-mediated translation is stimulated by specific interaction of independent regions of human La autoantigen. J Biol Chem. 2003;278:12231–40. doi: 10.1074/jbc.M210287200. [DOI] [PubMed] [Google Scholar]
- 20.Domitrovich AM, Diebel KW, Ali N, Sarker S, Siddiqui A. Role of La autoantigen and polypyrimidine tract-binding protein in HCV replication. Virology. 2005;335:72–86. doi: 10.1016/j.virol.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 21.Fukushi S, Okada M, Stahl J, Kageyama T, Hoshino FB, Katayama K. Ribosomal protein S5 interacts with the internal ribosomal entry site of hepatitis C virus. J Biol Chem. 2001;276:20824–6. doi: 10.1074/jbc.C100206200. [DOI] [PubMed] [Google Scholar]
- 22.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–3. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 23.Kato T, Date T, Miyamoto M, Sugiyama M, Tanaka Y, Orito E, et al. Detection of anti-hepatitis C virus effects of interferon and ribavirin by a sensitive replicon system. J Clin Microbiol. 2005;43:5679–84. doi: 10.1128/JCM.43.11.5679-5684.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang C, Le SY, Ali N, Siddiqui A. An RNA pseudoknot is an essential structural element of the internal ribosome entry site located within the hepatitis C virus 5′ noncoding region. RNA. 1995;1:526–37. [PMC free article] [PubMed] [Google Scholar]
- 25.Spahn CM, Kieft JS, Grassucci RA, Penczek PA, Zhou K, Doudna JA, et al. Hepatitis C virus IRES RNA-induced changes in the conformation of the 40s ribosomal subunit. Science. 2001;291:1959–62. doi: 10.1126/science.1058409. [DOI] [PubMed] [Google Scholar]
- 26.Boehringer D, Thermann R, Ostareck-Lederer A, Lewis JD, Stark H. Structure of the hepatitis C virus IRES bound to the human 80S ribosome: remodeling of the HCV IRES. Structure. 2005;13:1695–706. doi: 10.1016/j.str.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 27.Landry DM, Hertz MI, Thompson SR. RPS25 is essential for translation initiation by the Dicistroviridae and hepatitis C viral IRESs. Genes Dev. 2009;23:2753–64. doi: 10.1101/gad.1832209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pudi R, Ramamurthy SS, Das S. A peptide derived from RNA recognition motif 2 of human la protein binds to hepatitis C virus internal ribosome entry site, prevents ribosomal assembly, and inhibits internal initiation of translation. J Virol. 2005;79:9842–53. doi: 10.1128/JVI.79.15.9842-9853.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327:198–201. doi: 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramani K, Hassan Q, Venkaiah B, Hasnain SE, Sarkar DP. Site-specific gene delivery in vivo through engineered Sendai viral envelopes. Proc Natl Acad Sci U S A. 1998;95:11886–90. doi: 10.1073/pnas.95.20.11886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yi M, Ma Y, Yates J, Lemon SM. Compensatory mutations in E1, p7, NS2, and NS3 enhance yields of cell culture-infectious intergenotypic chimeric hepatitis C virus. J Virol. 2007;81:629–38. doi: 10.1128/JVI.01890-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ray PS, Das S. La autoantigen is required for the internal ribosome entry site-mediated translation of Coxsackievirus B3 RNA. Nucleic Acids Res. 2002;30:4500–8. doi: 10.1093/nar/gkf583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frese M, Barth K, Kaul A, Lohmann V, Schwärzle V, Bartenschlager R. Hepatitis C virus RNA replication is resistant to tumour necrosis factor-alpha. J Gen Virol. 2003;84:1253–9. doi: 10.1099/vir.0.18997-0. [DOI] [PubMed] [Google Scholar]
- 34.Hellman LM, Fried MG. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat Protoc. 2007;2:1849–61. doi: 10.1038/nprot.2007.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–6. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frese M, Barth K, Kaul A, Lohmann V, Schwärzle V, Bartenschlager R. Hepatitis C virus RNA replication is resistant to tumour necrosis factor-alpha. J Gen Virol. 2003;84:1253–9. doi: 10.1099/vir.0.18997-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.